2. Results and Discussion
In a previous study, we explored potential routes for the preparation of derivatives of the heteroaromatic substituted oxoalkanenitrile
3, starting with electron rich aromatic compounds
1. The current efforts were targeted at exploring the potential use of a new rearrangement reaction we observed very recently. We noted that reactions of pyrazolone
1a and aniline derivative
1b with the cyanoanhydride
2, generated by condensation of cyanoacetic acid with acetic anhydride, result in the formation of the corresponding heteroaroyl and aryl substituted 3-oxoalkanenitriles
3a and
3b in excellent yields (
Scheme 1). The structure of
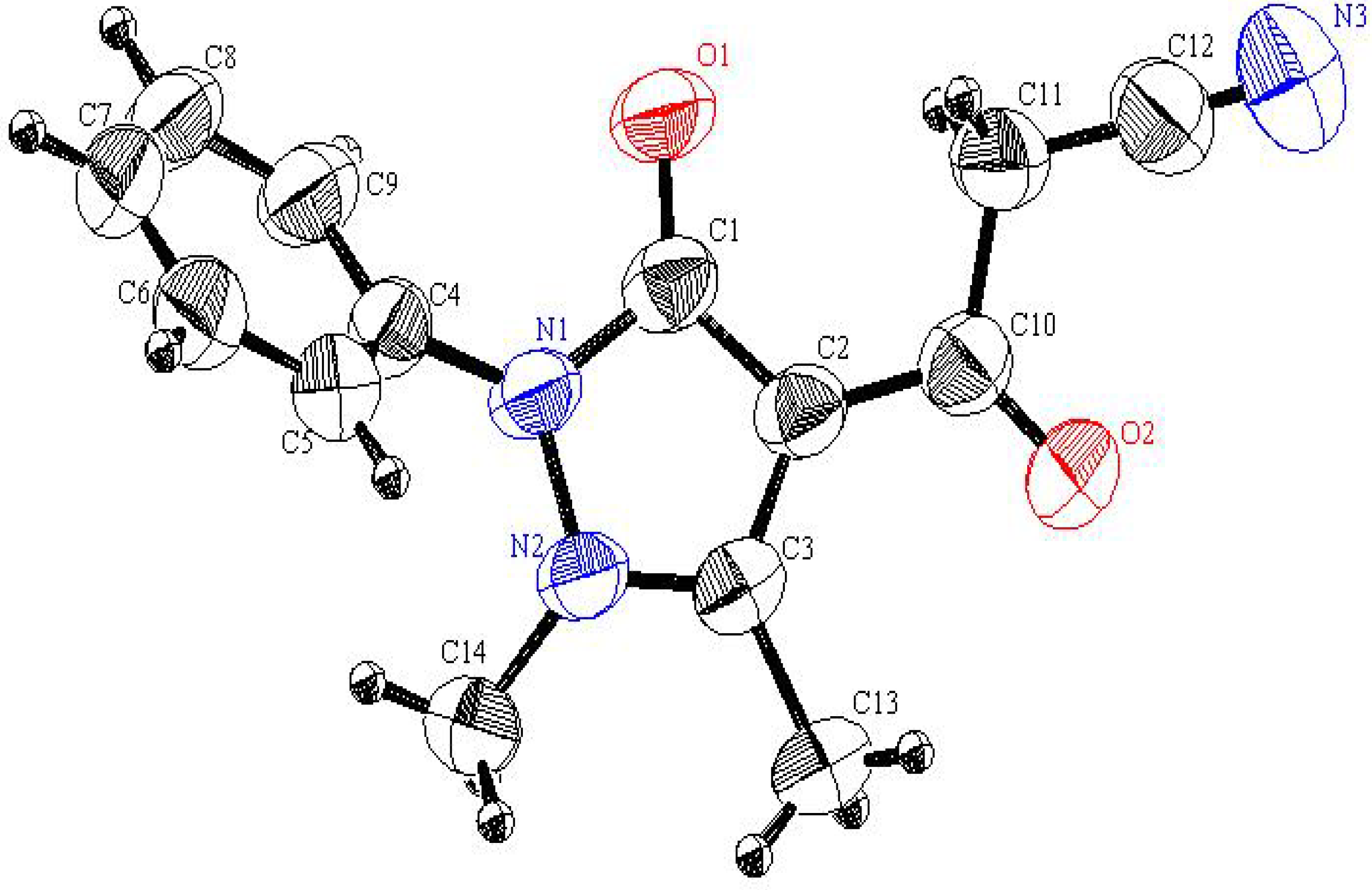
3a was assigned by using X-ray crystallographic analysis (
Figure 1) (CCDC 2011) [
11].
In contrast,
N-methylimidazole
1c failed to react with
2 under conditions identical to those used in the synthesis of
3a and
3b. However these substances do react when InCl
3 is present in the mixture to afford a product resulting from monocyanoacetylation of
1c. Preliminary inspection of the analytical data for this product suggested that it could be either one of the regioisomeric products
3c and
4. The results of NOE difference experiments, and in particular the fact that irradiation of methyl protons at 2.4 ppm enhanced the intensities of the imidazole CH protons at 7.29 and 8.19 ppm and
vice versa, revealed a close spacial proximity of the
N-methyl group and imidazole ring CH and suggested that the structure of the product is best represented by
1c (
Scheme 1).
Interestingly, reaction of
N-methylbenzamidazole
1d with cyanoanhydride
2 results in the formation of an unexpected productwith a molecular formula of C
13H
13N
3O
2 in 50% yield (
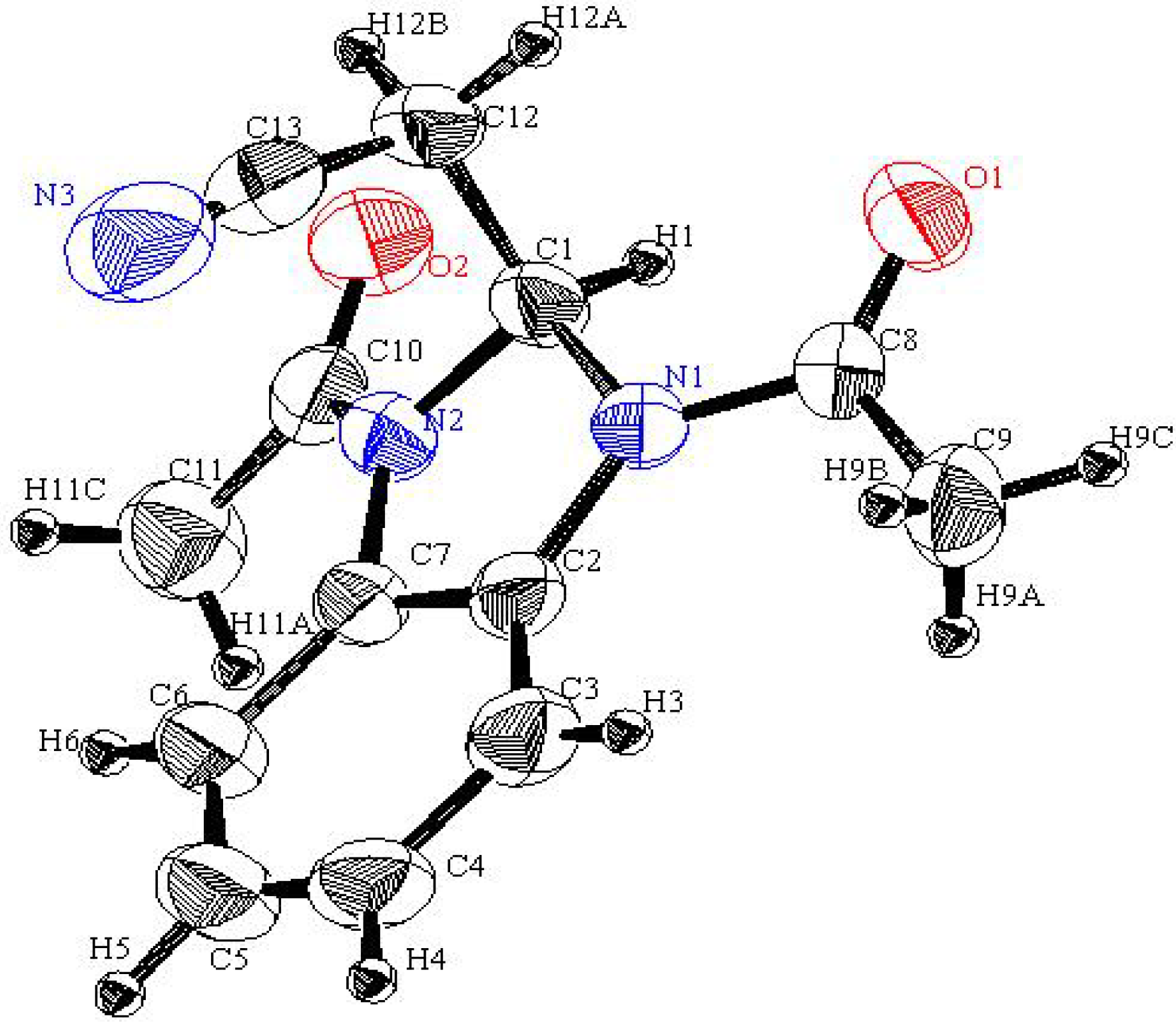
Scheme 1). X-ray crystallographic analysis (
Figure 2) was used to unambiguously assign the structure of this substance as the unusual bis-acetylbenzimidazolidine
5 (CCDC 2011) [
12]. It is believed that
5 is produced via the mechanistic route displayed in
Scheme 2, involving intermediates
6–8.
Scheme 1.
Cyanoacetylation of some π-excessive systems.
Scheme 1.
Cyanoacetylation of some π-excessive systems.
Figure 1.
An ORTEP plot of the X-ray crystal structure of 3a.
Figure 1.
An ORTEP plot of the X-ray crystal structure of 3a.
Figure 2.
An ORTEP plot of the X-ray crystal structure of 5.
Figure 2.
An ORTEP plot of the X-ray crystal structure of 5.
Scheme 2.
Suggested mechanism for formation of 5.
Scheme 2.
Suggested mechanism for formation of 5.
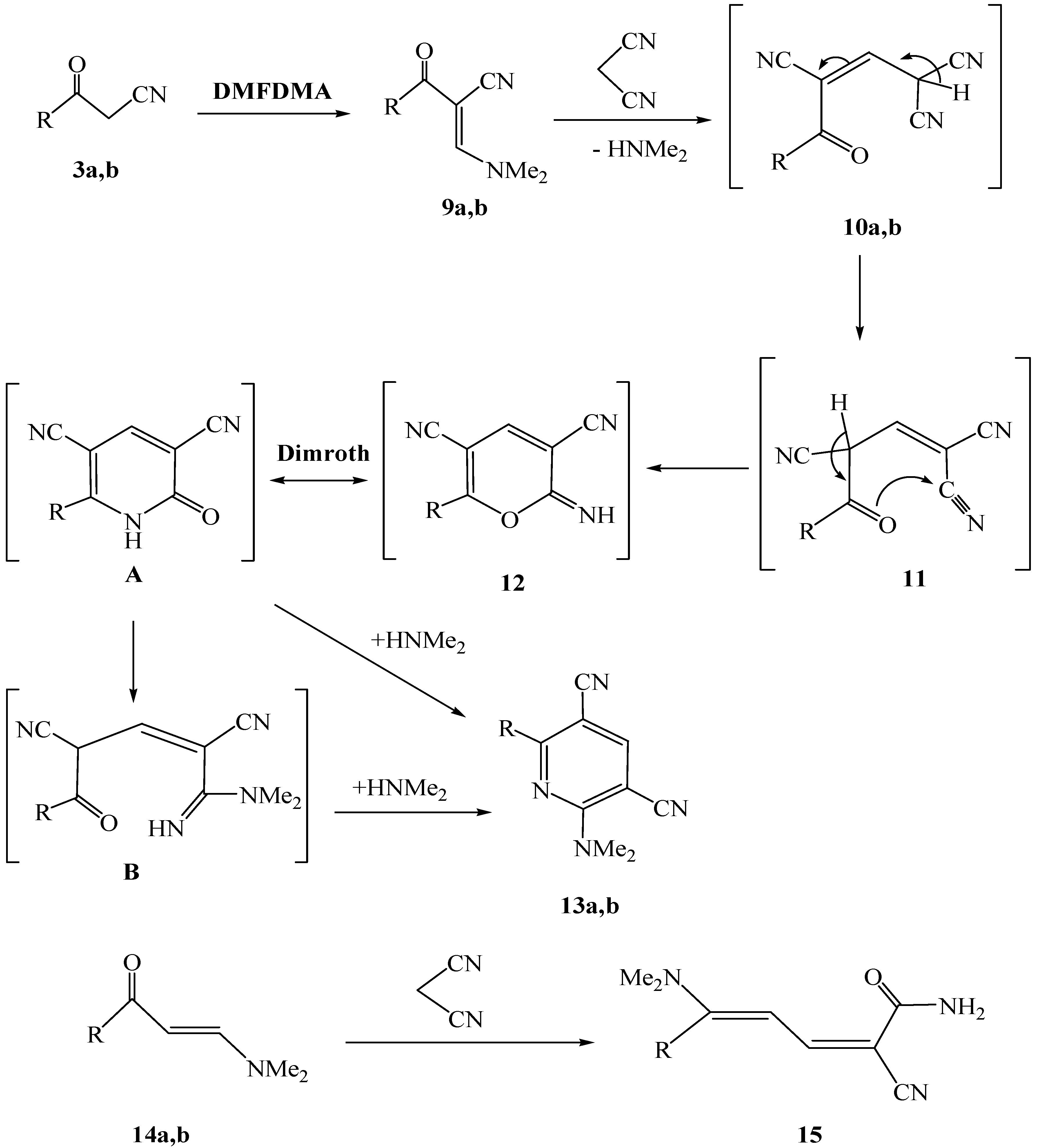
The heteroaroyl and aryl substituted 3-oxoalkanenitriles
3a and
3b were observed to undergo condensation reactions with dimethylformamide dimethylacetal (DMFDMA) to yield the corresponding enaminonitriles
9a,
b (
Scheme 3). The products have
trans-stereochemistry, as indicated by the results of NOE difference experiments that show the close spacial proximity of their respective olefinic methine protons and the methyl protons in
9a and aryl protons in
9b.
Further studies demonstrated that the enaminonitriles
9a and
9b react with malononitrile in refluxing ethanol containing piperidine to yield the corresponding dialkylaminopyridine-3,5-dicarbonitriles
13a and
13b (cf.
Scheme 3).
Scheme 3.
Formation of dialkylaminopyridine derivatives 13a and 13b.
Scheme 3.
Formation of dialkylaminopyridine derivatives 13a and 13b.
It is believed that in these processes malononitrile adds to the enaminonitriles to yield intermediate Michael adducts
10a,
b that readily cyclize to generate pyranimine
12, which undergo a Dimroth rearrangement to give pyridone
A. The latter undergoes ring opening to afford intermediate
B that then reacts with dimethylamine to yield the observed pyridines
13a and
13b. Although to our knowledge a rearrangement reaction of this type has not yet been reported, it involves a sequence of events that parallel those involved in the formation of dieneamide
15 in the reaction of
14 with malononitrile, described earlier by Al-Mousawi [
13] and Khalil [
14] (
Scheme 3). We reported in a recent related work the X-ray crystal structure of 2-(1-methyl-1
H-pyrrol-2-yl)-6-(piperidin-1-yl)pyridine-3,5-dicarbonitrile, that was prepared by reaction of enaminonitrile of 2-cyanoacetyl-1-methylpyrrole with malononitrile in presence of piperidine, with leaving no doubt about its structure [
9].
Scheme 4.
Formation of pyridinecarbonitrile 18a and 18b and pyranonecarbonitrile 20a and 20b.
Scheme 4.
Formation of pyridinecarbonitrile 18a and 18b and pyranonecarbonitrile 20a and 20b.
Additional studies revealed that heteroaroyl and aryl substituted 3-oxoalkanenitriles
3a and
3b condense with acetylacetone in acetic acid in the presence of ammonium acetate to yield the dimethylpyridine derivatives
17a and
17b (
Scheme 4). These oxoalkanenitriles also react with ethyl acetoacetate via water and ethanol elimination. Although it seemed reasonable that the products formed in these processes would be the pyranones
21 that would result from a bimolecular condensation that generates
18 (
Scheme 4), nevertheless, taking into consideration the possibility that a skeletal rearrangement similar to one observed in our recent studies [
9] could take place in this process, detailed NMR experiments were performed. Evidence supporting
20a and
20b as the structures of these products came from the results of NOE difference experiments, in which irradiation of the pyran ring CH protons at 5.9 ppm enhanced the intensities of the methyl proton resonances at 1.54 ppm. A plausible pathway for the formation of
20a and
20b from respective reactions of the heteroaroyl and aryl substituted 3-oxoalkanenitriles
3a and
3b involves initial production of intermediate
18 that then undergoes a documented [
15,
16,
17] 1,3-cyano shift to yield
19. Cyclization of
19 then yields
20a and
20b.
Further efforts showed that 3-oxoalkanenitriles
3b,c can be utilized as precursors for quinolinones. By employing a modification of the self-condensation reaction conditions reported earlier by Elnagdi
et al. [
18] (
Scheme 5) and Breil
et al. [
19], heating
3b and
3c over activated zeolite was found to lead to formation of the corresponding arenes
23b and
23c via a route possiblly involving the intermediacy of
22.
Scheme 5.
Self-condensation (trimerization) reaction of oxoalkanenitriles 3b and 3c.
Scheme 5.
Self-condensation (trimerization) reaction of oxoalkanenitriles 3b and 3c.
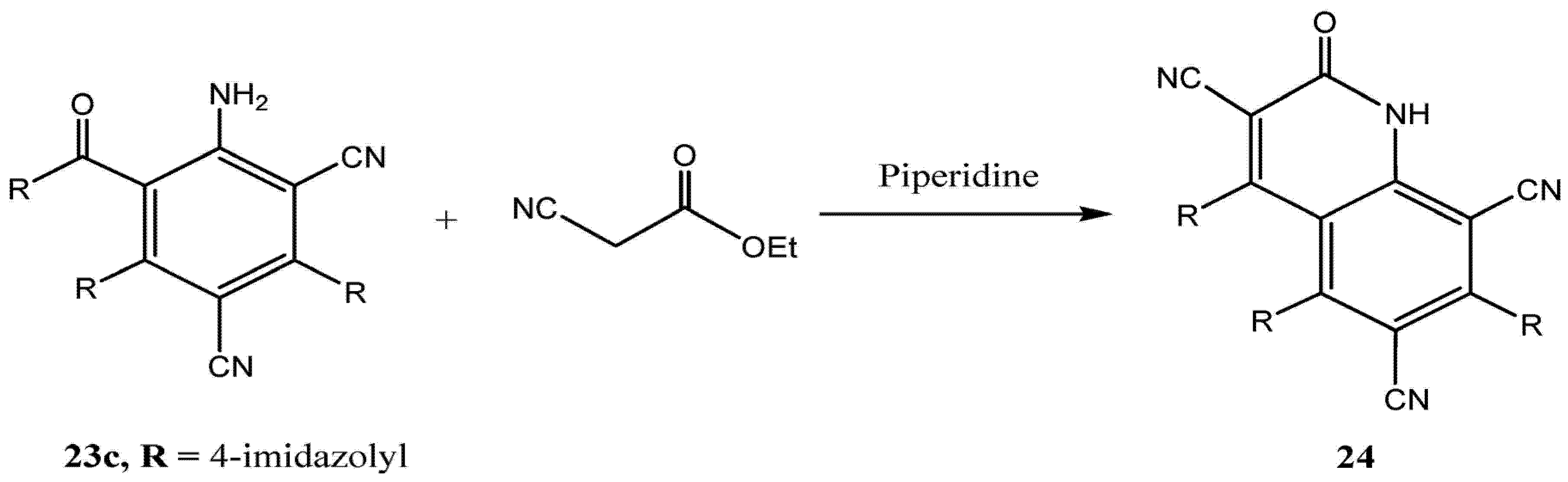
Finally, we observed that the aniline derivatives
23c (
Scheme 6) readily participate in condensation reactions with ethyl cyanoacetate in ethanolic piperidine to yield the corresponding quinolinones
24 (
Scheme 6).
Scheme 6.
Synthesis of quinolinone derivative 24.
Scheme 6.
Synthesis of quinolinone derivative 24.
3. Experimental
3.1. General
Melting points were recorded on Gallenkamp apparatus and are reported uncorrected. Infrared spectra (KBr) were determined on a Perkin-Elmer 2000 FT-IR system. NMR measurements were determined using a Bruker DPX spectrometer, at 600 MHz for 1H-NMR and 125 MHz for 13C-NMR, in DMSO-d6 as solvent and using TMS as internal standard. Mass spectra were measured on MS 30 and MS 9 (AEI) spectrometers, with EI 70 eV. All fine chemicals and organic solvents were purchased from Aldrich Chemicals and used without any further purification. Also, zeolite (CAS Number: 1318-02-1) was purchased from Aldrich Chemicals. Elemental analyses were measured on a LECO CHNS-932 Elemental Analyzer. Copies of original data can be provided on request.
3.2. Synthesis of Acetic 2-Cyanoacetic Anhydride (2)
A mixture of acetic anhydride (1.02 g, 10.0 mmol) and (0.85 g, 10.0 mmol) of cyanoacetic acid in dry dioxane (15 mL) was stirred at reflux for 15 min. The formed mixture was filtered and the filtrate was used after cooling as the cyanoacetylating agent 2 for reactions of aromatic substrates in the presence of a catalytic amount (10% wt.) of InCl3 as a Lewis acid.
3.3. Cyanoacetylation Reactions of 1a–d with Acetic 2-Cyanoacetic Anhydride (2)
A mixture of pre-prepared acetic 2-cyanoacetic anhydride (2, 1.27 g, 10.0 mmol), the starting aromatic or heteroaromatic compound (10.0 mmol), and indium trichloride (0.12 g, 10% wt.) in case of 1-methylimidazole (1c) and 1-methylbenzimidazole (1d), in dry dioxane (15 mL) was stirred at reflux for 1 h. The reaction mixture was then poured into water. The formed solid product was then collected by filtration, washed with water and crystallized from ethanol. Cyanoacetylation of benzimidazole 1d afforded one isolable products 5 as the major product. The product was isolated by column chromatography using ethyl acetate-n-hexane (1:3) as eluent and its structure confirmed by X-ray determination.
3-(1,5-Dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-3-oxopropanenitrile (3a). Yellow crystals (85%, m.p. 156 °C); IR (KBr): υ = 2187 (CN), 1705, 1685 (two C = O) cm−1, 1H-NMR: δ = 2.62 (s, 3H, CH3), 3.36 (s, 3H, CH3), 4.32 (s, 2H, CH2), 7.36–7.59 (m, 5H, phenyl); 13C NMR: δ = 30.5 (CH3), 33.4 (CH3), 38.8 (CH2), 116.2 (CN), 101.8, 127.5, 128.9, 129.4, 133.2, 153.9 (aromatic carbons), 162.7 (amide C=O), 182.4 (ketonic C=O); MS, m/z (%), 255.1 (M+, 43), 215.1 (100%). Anal. Calcd. for C14H13N3O2: C, 65.87; H, 5.13; N, 16.46. Found: C, 65.79; H, 5.02; N, 16.34.
3-(4-(Dimethylamino)phenyl)-3-oxopropanenitrile (3b). Dark brown solid (90%, m.p. 164 °C); IR (KBr): υ = 2182 (CN), 1700 (C = O) cm−1, 1H-NMR: δ = 3.03 (s, 6H, two CH3), 4.55 (s, 2H, CH2), 7.21 (d, 2H, J = 8 Hz, benzene), 7.75 (d, 2H, J = 8 Hz, benzene); 13C-NMR: δ = 29.8 (Two CH3), 39.5 (CH2), 110.6, 115.9 (CN), 129.4, 130.5, 153.8 (aromatic carbons), 186.2 (C=O); MS, m/z (%), 188.1 (M+, 43), 148.1 (100%). Anal. Calcd. for C11H12N2O: C, 70.19; H, 6.43; N, 14.88. Found: C, 70.04; H, 6.28; N, 14.59.
3-(1-Methyl-1H-imidazol-4-yl)-3-oxo-propionitrile (3c). Brown solid (65%); IR (KBr): υ = 2180 (CN), 1704 (C = O) cm−1, 1H-NMR: δ = 2.46 (s, 3H, CH3), 4.27 (s, 2H, CH2), 7.29 (s, 1H, C-4 imidazole), 8.19 (s, 1H, C-2 imidazole); 13C-NMR: δ = 33.7 (CH2), 35.4 (CH3), 116.7 (CN), 109.6, 127.6, 133.3 (imidazole carbons), 183.3 (C=O); MS, m/z (%), 149.1 (M+, 43), 109.1 (100%). Anal. Calcd. for C7H7N3O: C, 56.37; H, 4.73; N, 28.17. Found: C, 56.11; H, 4.65; N, 27.76.
(1,3-Diacetyl-2,3-dihydro-1H-benzoimidazol-2-yl)-acetonitrile (5). Brown solid (50%); IR (KBr): υ = 2188 (CN), 1664, 1660 (C = O) cm−1, 1H-NMR (DMSO-d6): δ = 2.71 (s, 6H, two CH3), 4.13 (s, 2H, CH2), 4.89 (s, 1H, C-2 imidazole), 7.11–7.64 (m, 4H, benzene ring); 13C-NMR (DMSO-d6): δ = 30.9 (CH2), 34.6 (2 × CH3), 38.1 (CH imidazole), 117.2 (CN), 122.6, 126.3, 136.0 (benzene carbons), 168.8 (two C=O); MS, m/z (%), 243.1 (M+, 18), 157.1 (100%). Anal. Calcd. for C13H13N3O2: C, 64.19; H, 5.39; N, 17.27. Found: C, 64.13; H, 5.35; N, 17.02.
3.4. Reaction of Cyanoacetyl Derivatives 3a,b with DMFDMA
A mixture of 3a or 3b (10 mmol) and (DMFDMA) (15 mmol) was dissolved in dry xylene (50 mL) and the reaction mixture was refluxed while the reaction was followed to completion by TLC, using ethyl acetate-n-hexane (1:3) as eluent. The reaction mixture was concentrated under reduced pressure, cooled and the solid product, so formed, was then filtered and recrystallized from ethanol.
2-(1,5-Dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazole-4-carbonyl)-3-(dimethylamino)acrylonitrile (9a). Yellow solid (65%, m.p. 149 °C); IR (KBr): υ = 2180 (CN), 1690, 1682 (two C = O) cm−1; 1H-NMR: δ = 1.84 (s, 3H, CH3), 1.98 (s, 3H, CH3), 2.62 (s, 6H, two CH3), 6.83–7.42 (m, 5H, phenyl), 7.86 (s, 1H, enamine); 13C-NMR: δ = 31.8 (CH3), 32.1(CH3), 33.6 (Two CH3), 116.7 (CN), 96.0, 112.6, 122.4, 124.1, 127.9, 130.9, 136.5, 144.3 (aromatic carbons), 159.2 (amide C=O), 176.8 (ketonic C=O); MS, m/z (%), 310.1 (M+, 16), 215.1 (100). Anal. Calcd. for C17H18N4O2: C, 65.79; H, 5.85; N, 18.05. Found: C, 65.71; H, 5.62; N, 17.84.
3-(Dimethylamino)-2-(4-(dimethylamino)benzoyl)acrylonitrile (9b). Brown solid (60%, m.p. 206 °C); IR (KBr): υ = 2173 (CN), 1695 (C = O) cm−1; 1H-NMR: δ = 2.78 (s, 6H, two CH3), 2.90 (s, 6H, two CH3), 7.18 (d, 2H, J = 8 Hz, phenyl), 7.72 (d, 2H, J = 8 Hz, phenyl), 8.28 (s, 1H, enamine); 13C-NMR: δ = 31.7 (CH3), 34.0 (CH3), 37.6 (Two CH3), 116.3 (CN), 119.1, 126.0, 129.1, 131.2, 139.5, 144.3 (aromatic carbons), 183.2 (ketonic C=O); MS, m/z (%), 243.1 (M+, 16), 148.1 (100). Anal. Calcd. for C14H17N3O: C, 69.11; H, 7.04; N, 17.27. Found: C, 69.06; H, 6.99; N, 17.21.
3.5. Reaction of 9a,b with Malononitrile
A mixture of 9a or 9b (10 mmol) and malononitrile (0.66 g, 10 mmol) was dissolved in absolute ethanol (30 mL) and the reaction mixture was refluxed for 6 h, then concentrated under reduced pressure, cooled and the solid product, so formed, was then filtered and recrystallized from ethanol.
2-(1,5-Dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-6-(dimethylamino)pyridine-3,5-dicarbonitrile (13a). Yellow solid (60%, m.p. 172 °C); IR (KBr): υ = 2185, 2175 (two CN), 1665 (C=O) cm−1; 1H-NMR: δ = 2.45 (s, 3H, CH3), 2.61 (s, 3H, CH3), 2.84 (s, 6H, two CH3), 7.10–7.51 (m, 5H, phenyl), 8.48 (s, 1H, pyridine); 13C-NMR: δ = 29.7 (two CH3), 32.4 (two CH3), 116.4 (two CN), 94.5, 104.5, 126.9, 131.1, 135.2, 141.6, 143.1, 159.5, 162.6 (aromatic carbons), 169.8 (C=O); MS, m/z (%), 358.1 (M+, 16), 177.1 (100). Anal. Calcd. for C20H18N6O: C, 67.02; H, 5.06; N, 23.45. Found: C, 66.95; H, 4.98; N, 23.36.
2-(Dimethylamino)-6-(4-(dimethylamino)phenyl)pyridine-3,5-dicarbonitrile (13b). Dark brown solid (65%, m.p. 137 °C); IR (KBr): υ = 2190, 2182 (CN) cm−1; 1H-NMR: δ = 2.28 (s, 6H, two CH3), 2.98 (s, 6H, CH3), 2.90 (s, 6H, two CH3), 7.04 (d, 2H, J = 8 Hz, phenyl), 7.76 (d, 2H, J = 8 Hz, phenyl), 8.78 (s, 1H, pyridine); 13C-NMR: δ = 30.6 (2 × CH3), 32.4 (2 × CH3), 117.1, 117.6 (2 × CN), 122.2, 124.6, 129.6, 135.2, 137.7, 148.4, 149.7, 152.4, 168.3 (aromatic carbons); MS, m/z (%), 291.2 (M+, 16), 120.1 (100). Anal. Calcd. for C17H17N5: C, 70.08; H, 5.88; N, 24.04. Found: C, 69.94; H, 5.81; N, 23.93.
3.6. Reaction of 3a,b with Acetylacetone
A mixture of 3a or 3b (10 mmol), ammonium acetate (1.54 g, 20 mmol), and acetylacetone (1.00 g, 10 mmol) was dissolved in glacial acetic acid (20 mL) and the reaction mixture was refluxed for 24 h, then poured over an ice-water mixture and the solid product, so formed, was then filtered and recrystallized from ethanol.
2-(1,5-Dimethyl-3-oxo-2-phenyl-2,3-dihydro-1H-pyrazol-4-yl)-4,6-dimethylnicotinonitrile (17a). Yellow solid (68%, m.p. 163 °C); IR (KBr): υ = 2181 (CN), 1660 (C=O) cm−1; 1H-NMR: δ = 2.87 (s, 6H, two CH3), 3.15 (s, 6H, two CH3), 6.88–7.29 (m, 5H, phenyl), 8.24 (s, 1H, pyridine); 13C-NMR: δ = 35.8 (CH3), 37.2 (CH3), 47.1 (CH3), 117.1 (CN), 122.7, 128.9, 131.6, 132.3, 134.6, 150.2, 153.9, 155.0, 157.6 (aromatic carbons), 163.0 (C=O); MS, m/z (%), 318.1 (M+, 16), 215.1 (100). Anal. Calcd. for C19H18N4O: C, 71.68; H, 5.70; N, 17.60. Found: C, 71.54; H, 5.59; N, 17.28.
2-(4-(Dimethylamino)phenyl)-4,6-dimethylnicotinonitrile (17b). Dark brown solid (55%, m.p. 119 °C); IR (KBr): υ = 2176 (CN) cm−1; 1H-NMR: δ = 1.56 (s, 6H, two CH3), 2.12 (s, 3H, CH3), 2.28 (s, 3H, CH3), 6.73 (d, 2H, J = 8 Hz, phenyl), 7.84 (d, 2H, J = 8 Hz, phenyl), 8.34 (s, 1H, pyridine); 13C-NMR: δ = 34.7 (two CH3), 39.1 (CH3), 116.3 (CN), 104.6, 111.8, 121.7, 125.2, 131.6, 139.1, 146.7, 148.0, 158.2 (aromatic carbons); MS, m/z (%), 251.2 (M+, 16), 120.1 (100). Anal. Calcd. for C16H17N3: C, 76.46; H, 6.82; N, 16.72. Found: C, 76.13; H, 6.69; N, 16.52.
3.7. Reaction of 3a,b with Ethyl Acetoacetate
A mixture of 3a or 3b (10 mmol) and ethyl acetoacetate (1.30 g, 10 mmol) was dissolved in absolute ethanol (20 mL) in the presence of few drops of piperidine as catalyst. The reaction mixture was refluxed for 8 h, then concentrated under reduced pressure and the solid product, so formed, was then filtered and recrystallized from ethanol.
6-(2,3-Dimethyl-5-oxo-1-phenyl-2,5-dihydro-1H-pyrazol-4-yl)-4-methyl-2-oxo-2H-pyran-3-carbonitrile (20a). Yellow solid (58%, m.p. 141 °C); IR (KBr): υ = 2215 (CN), 1718, 1668 (C = O) cm−1; 1H-NMR: δ = 1.54 (s, 6H, two CH3), 2.51 (s, 6H, 2 × CH3), 5.98 (s, 1H, pyran), 6.74–7.77 (m, 5H, phenyl); 13C-NMR: δ = 34.1 (CH3), 35.2 (CH3), 116.2 (CN), 87.5, 104.5, 112.8, 115.1 (pyran), 125.9, 131.4, 135.2, 142.9, 150.2, 154.5 (aromatic carbons) 159.5 (C=O, antipyrine), 162.6 (C=O, pyran); MS, m/z (%), 321.1 (M+, 16), 215.1 (100). Anal. Calcd. for C18H15N3O3: C, 67.28; H, 4.71; N, 13.08. Found: C, 67.13; H, 4.66; N, 12.91.
6-(4-(Dimethylamino)phenyl)-4-methyl-2-oxo-2H-pyran-3-carbonitrile (20b). Brown solid (50%, m.p. 157 °C); IR (KBr): υ = 2185 (CN) cm−1; 1H-NMR: δ = 2.66 (s, 6H, two CH3), 3.37 (s, 3H, CH3), 5.97 (s, 1H, pyran), 6.52 (d, 2H, J = 8 Hz, phenyl), 7.01 (d, 2H, J = 8 Hz, phenyl); 13C-NMR: δ = 28.5 (2 × CH3), 33.4 (CH3), 117.1 (CN), 99.8, 103.6, 111.7, 115.5, 128.8, 134.2, 147.2, 149.9 (aromatic carbons), 162.4 (C=O); MS, m/z (%), 254.1 (M+, 16), 120.1 (100). Anal. Calcd. for C15H14N2O2: C, 70.85; H, 5.55; N, 11.02. Found: C, 70.59; H, 5.32; N, 10.87.
3.8. Trimerization of 3-Oxoalkanenitriles 3b,c
The proper 3-oxoalkanenitrile (3b or 3c, 10 mmol) was refluxed for 8 h in pyridine (20 mL). The reaction mixture was poured over HCl/H2O to neutralize the pyridine. The crude solid product was filtered and dried. The product 22 was dissolved in dry dioxane (15 mL) and refluxed again in the presence of a catalytic amount of zeolite (0.2 g, 20% wt.) for 12 h. The crude product was filtered, purified by DMF-EtOH to yield 23.
4-Amino-2,6-bis(4-(dimethylamino)phenyl)-5-(4-(dimethylamino)phenyl-4-carbonyl)-isophthalonitrile (23b). Dark brown solid (60%); IR (KBr): υ = 3350, 3330 (broad band, NH2), 2196, 2185 (CN), 1690 (C=O) cm−1; 1H-NMR: δ = 2.23 (s, 12H, CH3), 2.37 (s, 3H, CH3), 3.14 (s, 3H, two CH3), 6.24 (s, 2H, NH2), 6.84–7.61 (m, 12H, phenyl rings aromatic H); 13C-NMR: δ = 32.7, 35.1, 38.3 (six CH3), 116.3, 116.5 (2 × CN), 102.9, 110.2, 119.9, 122.0, 122.8, 127.2, 128.6, 135.0, 140.9, 146.2, 149.1, 151.2, 162.7, 164.3 (aromatic carbons), 181.2 (C=O); MS, m/z (%), 411.1 (M+, 18), 330.1 (100). Anal. Calcd. for C33H32N6O: C, 74.98; H, 6.10; N, 15.90. Found: C, 74.82; H, 5.93; N, 15.78.
4-Amino-2,6-bis(1-methyl-1H-imidazol-4-yl)-5-(1-methyl-1H-imidazole-4-carbonyl)isophthalonitrile (23c). Pale brown solid (51%); IR (KBr): υ = 3340, 3320 (broad band, NH2), 2187, 2176 (CN), 1658 (C=O) cm−1, 1H-NMR: δ = 2.91 (s, 3H, CH3), 3.24 (s, 6H, 2 × CH3), 5.81 (s, 2H, NH2), 7.41–7.90 (m, 6H, aromatic H, imidazole); 13C-NMR: δ = 33.7, 33.9 (three CH3), 116.8, 117.5 (2 × CN), 91.3, 104.2, 123.8, 124.1, 128.3, 133.0, 141.3, 145.9, 148.7, 148.9, 163.9, 167.5 (aromatic carbons), 183.7 (C=O); MS, m/z (%), 411.1 (M+, 18), 330.1 (100). Anal. Calcd. for C21H17N9O: C, 61.31; H, 4.16; N, 30.64. Found: C, 61.14; H, 4.08; N, 30.39.
3.9. Condensation Reaction of 23c with Ethyl Cyanoacetate
A mixture of 23c (10.0 mmol) and ethyl cyanoacetate (1.13 g, 10 mmol) in absolute ethanol (25 mL) containing a catalytic amount of piperidine was stirred at reflux for 24 h. The reaction mixture was cooled and the product was collected by filtration, and crystallized from ethanol to afford 4,5,7-tris(1-methyl-1H-imidazol-4-yl)-2-oxo-1,2-dihydroquinoline-3,6,8-tricarbonitrile (24). Dark brown solid (40%); IR (KBr): υ = 3335 (NH), 2181, 2198 (CN), 1656 (C=O) cm−1, 1H-NMR: δ = 2.89 (s, 6H, 2 × CH3), 3.13 (s, 3H, CH3), 7.13–7.78 (m, 6H, aromatic H, pyrrole), 8.22 (s, 2H, NH); 13C-NMR: δ = 31.1, 38.2 (three CH3), 116.9, 117.4 (2 × CN), 87.3, 105.4, 113.4, 115.0, 120.8, 123.2, 125.9, 128.5, 133.7, 136.2, 139.4, 141.6 (aromatic carbons), 164.1 (C=O); MS, m/z (%), 459.8 (M+, 18), 81.1 (100%). Anal. Calcd. for C24H16N10O: C, 62.60; H, 3.50; N, 30.42. Found: C, 62.58; H, 3.48; N, 30.37.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}







