Amplification and Re-Generation of LNA-Modified Libraries

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
2.1. Library Amplification and Re-Generation
- Our starting DNA library encodes 40 randomized positions. The flanking primer-binding sites do not contain adenosines.
- The library is amplified by Phusion HF DNA polymerase under standard PCR reaction conditions to generate the corresponding double-stranded all-DNA library.
- We then add a shorter forward primer and perform 15 rounds of primer extension using KOD XL DNA polymerase in the presence of dGTP, dCTP, dTTP, and LNA ATP. This (re-)generates the LNA-modified pool.
- The full-length LNA-containing products are purified from a denaturing acrylamide gel. This step exploits the dissimilar lengths of the extension products and the PCR product.
- The LNA-modified strands can then be subjected to in vitro selection and eventually used as PCR template for the next round.
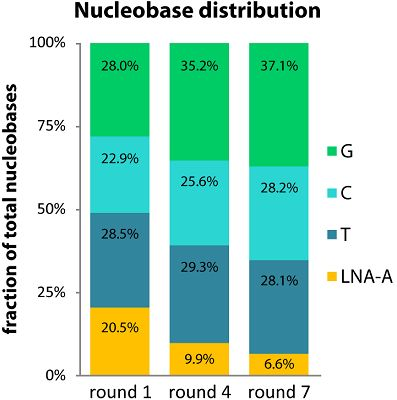
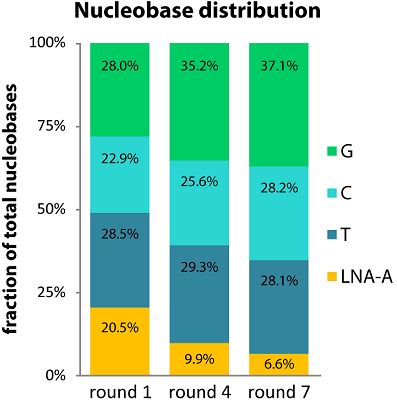
2.2. LNA-A Levels Decrease over Seven Rounds
2.3. Single LNA-As are Favored over Successive LNA-As

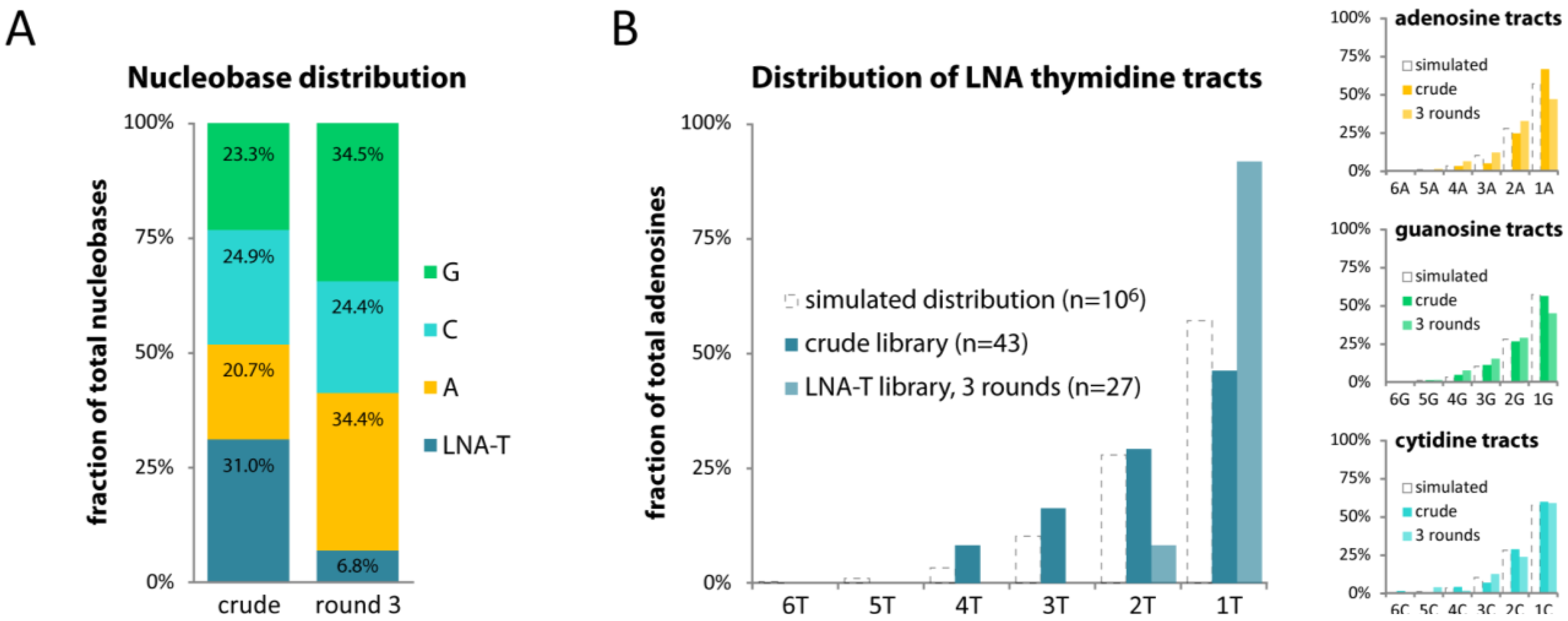
2.4. LNA-T also Skews Library Composition



3. Experimental
3.1. Library and Primers
3.2. Polymerase Chain Reaction (PCR)
3.3. Primer Extension with LNA
3.4. Sequencing and Analysis
3.5. Alternate strand Isolation
3.6. Lambda Exonuclease Digestion
3.7. Oligonucleotide Capture on Beads
3.8. Digestion Assay
3.9. Sanger Sequencing
4. Conclusions
Acknowledgments
- Sample Availability: Not available.
References
- Bunka, D.H.; Stockley, P.G. Aptamers come of age—At last. Nat. Rev. Microbiol. 2006, 4, 588–596. [Google Scholar] [CrossRef]
- Syed, M.A.; Pervaiz, S. Advances in aptamers. Oligonucleotides 2010, 20, 215–224. [Google Scholar] [CrossRef]
- Tuerk, C.; Gold, L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science 1990, 249, 505–510. [Google Scholar]
- Robertson, D.L.; Joyce, G.F. Selection in vitro of an RNA enzyme that specifically cleaves single-stranded DNA. Nature 1990, 344, 467–468. [Google Scholar] [CrossRef]
- Koshkin, A.; Singh, S.K.; Nielsen, P.; Rajwanshi, V.K.; Kumar, R.; Meldgaard, M.; Olsen, C.E.; Wengel, J. LNA (Locked Nucleic Acids): Synthesis of the Adenine, Cytosine, Guanine, 5-Methylcytosine, Thymine and Uracil Bicyclonucleoside Monomers, Oligomerisation, and Unprecedented Nucleic Acid Recognition. Tetrahedron 1998, 54, 3607–3630. [Google Scholar] [CrossRef]
- Wengel, J. Synthesis of 3'-C- and 4'-C-branched oligonucleotides and the development of locked nucleic acid (LNA). Acc. Chem. Res. 1998, 32, 301–310. [Google Scholar] [CrossRef]
- Braasch, D.A.; Corey, D.R. Locked nucleic acid (LNA): Fine-tuning the recognition of DNA and RNA. Chem. Biol. 2001, 8, 1–7. [Google Scholar] [CrossRef]
- Kurreck, J.; Wyszko, E.; Gillen, C.; Erdmann, V.A. Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids. Res. 2002, 30, 1911–1918. [Google Scholar] [CrossRef]
- Elmen, J.; Thonberg, H.; Ljungberg, K.; Frieden, M.; Westergaard, M.; Xu, Y.; Wahren, B.; Liang, Z.; Orum, H.; Koch, T.; et al. Locked nucleic acid (LNA) mediated improvements in siRNA stability and functionality. Nucleic Acids Res. 2005, 33, 439–447. [Google Scholar] [CrossRef]
- Mook, O.R.; Baas, F.; de Wissel, M.B.; Fluiter, K. Evaluation of locked nucleic acid-modified small interfering RNA in vitro and in vivo. Mol. Cancer Ther. 2007, 6, 833–843. [Google Scholar] [CrossRef]
- Gao, S.; Dagnaes-Hansen, F.; Nielsen, E.J.; Wengel, J.; Besenbacher, F.; Howard, K.A.; Kjems, J. The effect of chemical modification and nanoparticle formulation on stability and biodistribution of siRNA in mice. Mol. Ther. 2009, 17, 1225–1233. [Google Scholar] [CrossRef]
- Wahlestedt, C.; Salmi, P.; Good, L.; Kela, J.; Johnsson, T.; Hokfelt, T.; Broberger, C.; Porreca, F.; Lai, J.; Ren, K.; et al. Potent and nontoxic antisense oligonucleotides containing locked nucleic acids. Proc. Natl. Acad. Sci. USA 2000, 97, 5633–5638. [Google Scholar]
- Di Primo, C.; Rudloff, I.; Reigadas, S.; Arzumanov, A.A.; Gait, M.J.; Toulme, J.J. Systematic screening of LNA/2'-O-methyl chimeric derivatives of a TAR RNA aptamer. FEBS Lett. 2007, 581, 771–774. [Google Scholar] [CrossRef]
- Schmidt, K.S.; Borkowski, S.; Kurreck, J.; Stephens, A.W.; Bald, R.; Hecht, M.; Friebe, M.; Dinkelborg, L.; Erdmann, V.A. Application of locked nucleic acids to improve aptamer in vivo stability and targeting function. Nucleic Acids Res. 2004, 32, 5757–5765. [Google Scholar]
- Virno, A.; Randazzo, A.; Giancola, C.; Bucci, M.; Cirino, G.; Mayol, L. A novel thrombin binding aptamer containing a G-LNA residue. Bioorg. Med. Chem. 2007, 15, 5710–5718. [Google Scholar]
- Hernandez, F.J.; Kalra, N.; Wengel, J.; Vester, B. Aptamers as a model for functional evaluation of LNA and 2'-amino LNA. Bioorg. Med. Chem. Lett. 2009, 19, 6585–6587. [Google Scholar]
- Mallikaratchy, P.R.; Ruggiero, A.; Gardner, J.R.; Kuryavyi, V.; Maguire, W.F.; Heaney, M.L.; McDevitt, M.R.; Patel, D.J.; Scheinberg, D.A. A multivalent DNA aptamer specific for the B-cell receptor on human lymphoma and leukemia. Nucleic Acids Res. 2010, 39, 2458–2469. [Google Scholar]
- Forster, C.; Zydek, M.; Rothkegel, M.; Wu, Z.; Gallin, C.; Gessner, R.; Lisdat, F.; Furste, J.P. Properties of an LNA-modified ricin RNA aptamer. Biochem. Biophys. Res. Commun. 2012, 419, 60–65. [Google Scholar]
- Kanwar, J.R.; Roy, K.; Kanwar, R.K. Chimericaptamers in cancer cell-targeted drug delivery. Crit. Rev. Biochem. Mol. Biol. 2011, 46, 459–477. [Google Scholar]
- Lebars, I.; Richard, T.; Di Primo, C.; Toulme, J.J. LNA derivatives of a kissing aptamer targeted to the trans-activating responsive RNA element of HIV-1. Blood Cells Mol. Dis. 2007, 38, 204–209. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. Enzymatic incorporation of LNA nucleotides into DNA strands. ChemBioChem 2007, 8, 490–492. [Google Scholar]
- Veedu, R.N.; Vester, B.; Wengel, J. Polymerase directed incorporation studies of LNA-G nucleoside 5'-triphosphate and primer extension involving all four LNA nucleotides. New J. Chem. 2010, 34, 877–879. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. Efficient enzymatic synthesis of LNA-modified DNA duplexes using KOD DNA polymerase. Org. Biomol. Chem. 2009, 7, 1404–1409. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. In vitro incorporation of LNA nucleotides. Nucleosides Nucleotides Nucleic Acids 2007, 26, 1207–1210. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. Novel applications of locked nucleic acids. Nucleic Acids Symp. Ser. (Oxf) 2007, 51, 29–30. [Google Scholar] [CrossRef]
- Veedu, R.N.; Vester, B.; Wengel, J. Polymerase chain reaction and transcription using locked nucleic acid nucleotide triphosphates. J. Am. Chem. Soc. 2008, 130, 8124–8125. [Google Scholar]
- Kuwahara, M.; Obika, S.; Nagashima, J.; Ohta, Y.; Suto, Y.; Ozaki, H.; Sawai, H.; Imanishi, T. Systematic analysis of enzymatic DNA polymerization using oligo-DNA templates and triphosphateanalogs involving 2',4'-bridged nucleosides. Nucleic Acids Res. 2008, 36, 4257–4265. [Google Scholar]
- Thiel, W.H.; Bair, T.; Wyatt Thiel, K.; Dassie, J.P.; Rockey, W.M.; Howell, C.A.; Liu, X.Y.; Dupuy, A.J.; Huang, L.; Owczarzy, R.; et al. Nucleotide bias observed with a short SELEX RNA aptamer library. Nucleic Acid Ther. 2011, 21, 253–263. [Google Scholar]
- Zimmermann, B.; Gesell, T.; Chen, D.; Lorenz, C.; Schroeder, R. Monitoring genomic sequences during SELEX using high-throughput sequencing: Neutral SELEX. PLoS One 2010, 5, e9169. [Google Scholar]
- Veedu, R.N.; Burri, H.V.; Kumar, P.; Sharma, P.K.; Hrdlicka, P.J.; Vester, B.; Wengel, J. Polymerase-directed synthesis of C5-ethynyl locked nucleic acids. Bioorg. Med. Chem. Lett. 2010, 20, 6565–6568. [Google Scholar]
- Williams, R.; Peisajovich, G.S.; Miller, O.J.; Magdassi, S.; Tawfik, D.S.; Griffiths, A.D. Amplification of complex gene libraries by emulsion PCR. Nat. Methods 2006, 7, 545–550. [Google Scholar]
- Schütze, T.; Rubelt, F.; Repkow, J.; Greiner, N.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Glökler, J. A streamlined protocol for emulsion polymerase chain reaction and subsequent purification. Anal. Biochem. 2011, 410, 155–157. [Google Scholar]
- Shao, K.; Ding, W.; Wang, F.; Li, H.; Ma, D.; Wang, H. Emulsion PCR: A high efficient way of PCR amplification of random DNA libraries in aptamer selection. PLoS One 2011, 9, e24910. [Google Scholar]
- Drabovich, A.; Berezovski, M.; Krylov, S.N. Selection of smart aptamers by equilibrium capillary electrophoresis of equilibrium mixtures (ECEEM). J. Am. Chem. Soc. 2005, 127, 11224–11225. [Google Scholar] [CrossRef]
- Mendonsa, S.D.; Bowser, M.T. In vitro evolution of functional DNA using capillary electrophoresis. J. Am. Chem. Soc. 2004, 126, 20–21. [Google Scholar] [CrossRef]
- Mosing, R.K.; Mendonsa, S.D.; Bowser, M.T. Capillary electrophoresis-SELEX selection of aptamers with affinity for HIV-1 reverse transcriptase. Anal. Chem. 2005, 77, 6107–6112. [Google Scholar] [CrossRef]
- Mendonsa, S.D.; Bowser, M.T. In vitro selection of high-affinity DNA ligands for human IgE using capillary electrophoresis. Anal. Chem. 2004, 76, 5387–5392. [Google Scholar] [CrossRef]
- Berezovski, M.; Drabovich, A.; Krylova, S.M.; Musheev, M.; Okhonin, V.; Petrov, A.; Krylov, S.N. Nonequilibrium capillary electrophoresis of equilibrium mixtures: A universal tool for development of aptamers. J. Am. Chem. Soc. 2005, 127, 3165–3171. [Google Scholar]
- Qian, J.; Lou, X.; Zhang, Y.; Xiao, Y.; Soh, H.T. Generation of highly specific aptamers via micromagnetic selection. Anal. Chem. 2009, 81, 5490–5495. [Google Scholar] [CrossRef]
- Lou, X.; Qian, J.; Xiao, Y.; Viel, L.; Gerdon, A.E.; Lagally, E.T.; Atzberger, P.; Tarasow, T.M.; Heeger, A.J.; Soh, H.T. Micromagnetic selection of aptamers in microfluidic channels. Proc. Natl. Acad. Sci. USA 2009, 106, 2989–2994. [Google Scholar]
- Ahmad, K.M.; Oh, S.S.; Kim, S.; McClellen, F.M.; Xiao, Y.; Soh, H.T. Probing the limits of aptamer affinity with a microfluidic SELEX platform. PLoS One 2011, 6, e27051. [Google Scholar]
- Jing, M.; Bowser, M.T. Isolation of DNA aptamers using micro free flow electrophoresis. Lab Chip 2011, 11, 3703–3709. [Google Scholar] [CrossRef]
- Hoon, S.; Zhou, B.; Janda, K.D.; Brenner, S.; Scolnick, J. Aptamer selection by high-throughput sequencing and informatic analysis. Biotechniques 2011, 51, 413–416. [Google Scholar]
- Schutze, T.; Wilhelm, B.; Greiner, N.; Braun, H.; Peter, F.; Morl, M.; Erdmann, V.A.; Lehrach, H.; Konthur, Z.; Menger, M.; et al. Probing the SELEX process with next-generation sequencing. PLoS One 2011, 6, e29604. [Google Scholar]
- Kupakuwana, G.V.; Crill, J.E., II; McPike, M.P.; Borer, P.N. Acyclic identification of aptamers for human alpha-thrombin using over-represented libraries and deep sequencing. PLoS One 2011, 6, e19395. [Google Scholar]
- Berezhnoy, A.; Stewart, C.A.; McNamara, J.O., II; Thiel, W.; Giangrande, P.; Trinchieri, G.; Gilboa, E. Isolation and optimization of murine IL-10 receptor blocking oligonucleotide aptamers using high-throughput sequencing. Mol. Ther. 2012, 20, 1242–1250. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Doessing, H.; Hansen, L.H.; Veedu, R.N.; Wengel, J.; Vester, B. Amplification and Re-Generation of LNA-Modified Libraries. Molecules 2012, 17, 13087-13097. https://doi.org/10.3390/molecules171113087
Doessing H, Hansen LH, Veedu RN, Wengel J, Vester B. Amplification and Re-Generation of LNA-Modified Libraries. Molecules. 2012; 17(11):13087-13097. https://doi.org/10.3390/molecules171113087
Chicago/Turabian StyleDoessing, Holger, Lykke H. Hansen, Rakesh N. Veedu, Jesper Wengel, and Birte Vester. 2012. "Amplification and Re-Generation of LNA-Modified Libraries" Molecules 17, no. 11: 13087-13097. https://doi.org/10.3390/molecules171113087
APA StyleDoessing, H., Hansen, L. H., Veedu, R. N., Wengel, J., & Vester, B. (2012). Amplification and Re-Generation of LNA-Modified Libraries. Molecules, 17(11), 13087-13097. https://doi.org/10.3390/molecules171113087