Synthesis, Spectral and Solid State Characterization of a New Bioactive Hydrazine Bridged Cyclic Diphosphonium Compound

Abstract
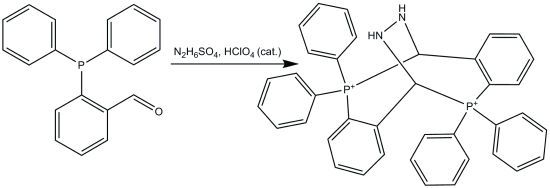
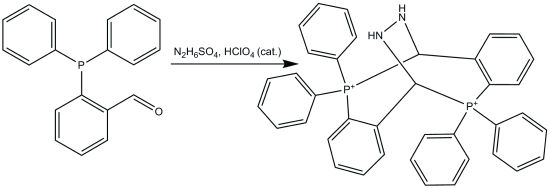
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
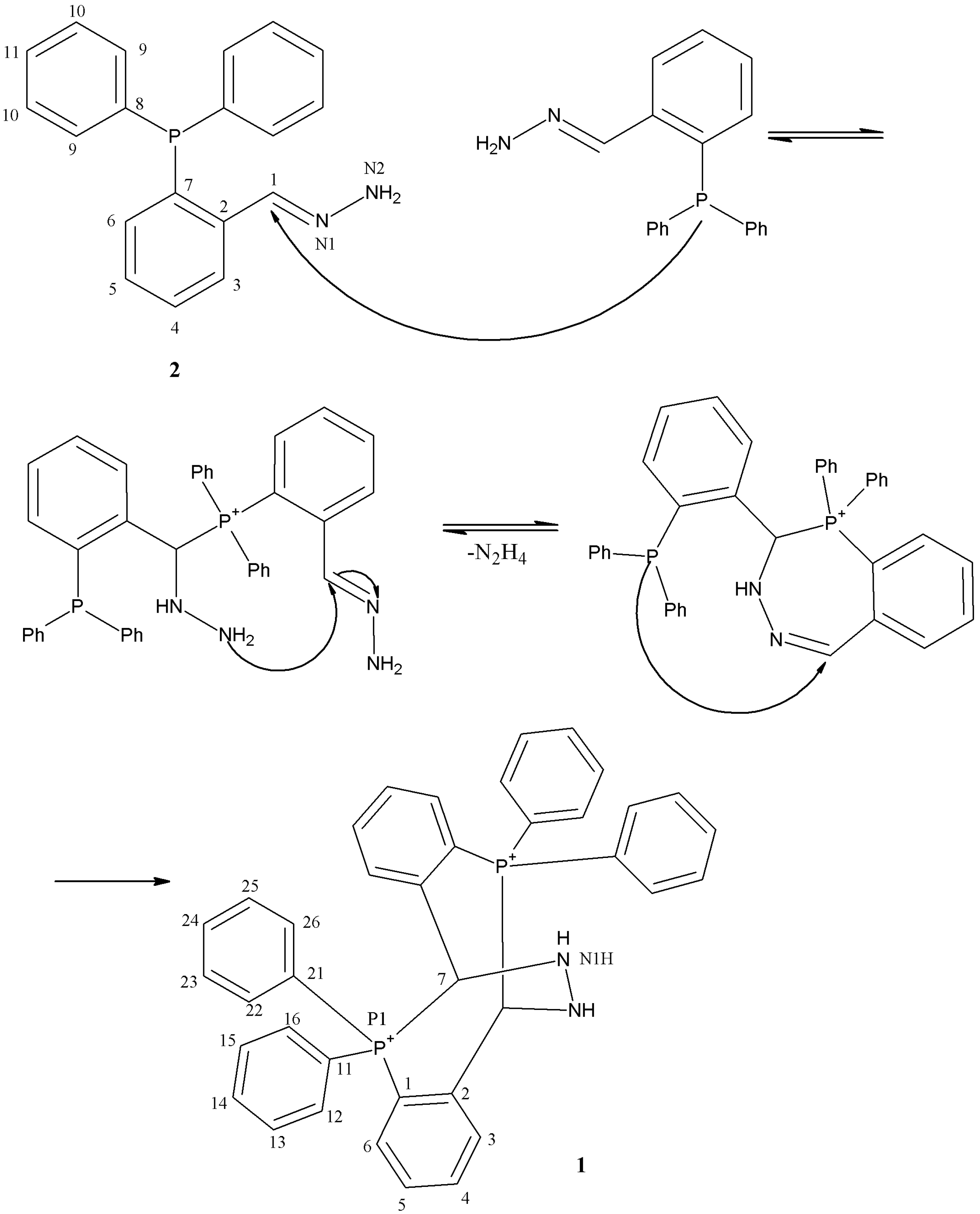{kind=link}
1. Introduction
2. Results and Discussion
2.1. Synthesis

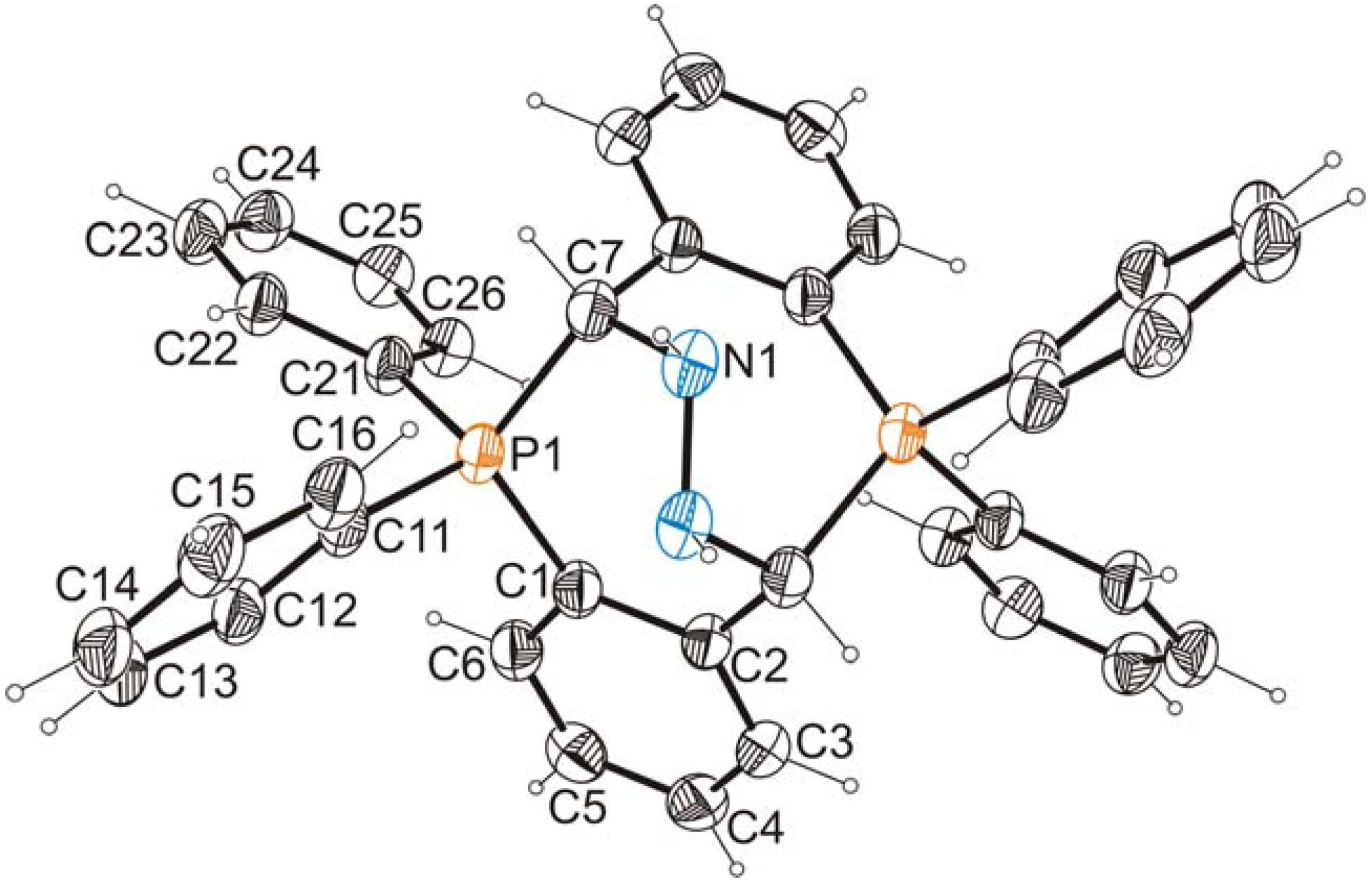
2.2. X-ray Crystallographic Analysis

2.3. NMR Spectra

2.4. Reaction Mechanism
2.5. Biological Activity

3. Experimental
3.1. General
3.2. Synthesis of 5,5,11,11-Tetraphenyl-5,6,11,12-tetrahydro-6,12-biiminodibenzo[b,f][1,5]diphosphociniumDiperchlorate (1) from Malonic Acid Dihydrazide and 2-(Diphenylphosphino)benzaldehyde
3.3. Synthesis of 5,5,11,11-Tetraphenyl-5,6,11,12-tetrahydro-6,12-biiminodibenzo[b,f][1,5]diphosphocinium Diperchlorate (1) from Malonic Acid Dihydrazide and 2-(Diphenylphosphino)benzaldehyde in the Presence of Cd(ClO4)2·6H2O
3.4. Synthesis of 5,5,11,11-Tetraphenyl-5,6,11,12-tetrahydro-6,12-biiminodibenzo[b,f][1,5]diphosphocinium Diperchlorate (1) from Malonic Acid Dihydrazide and 2-(Diphenylphosphino)benzaldehyde in the Presence of Co(ClO4)2·6 H2O
3.5. Synthesis of 5,5,11,11-Tetraphenyl-5,6,11,12-tetrahydro-6,12-biiminodibenzo[b,f][1,5]diphosphocinium Diperchlorate (1) from Hydrazine Sulfate and 2-(Diphenylphosphino)Benzaldehyde
3.6. X-ray Single Crystal Measurements
3.7. The Brine Shrimp Test
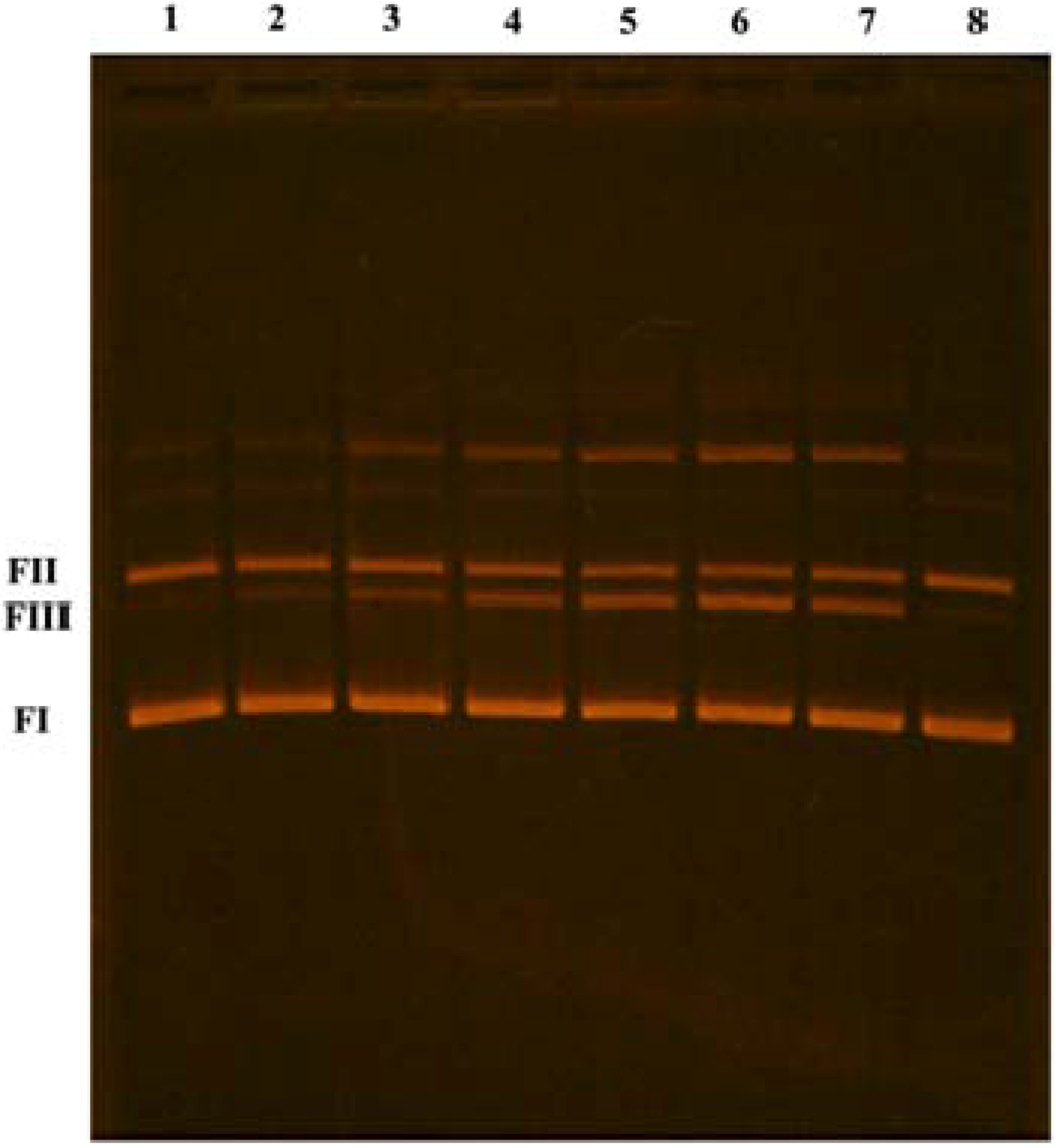
3.8. DNA Cleavage Experiment
4. Conclusions
Supplementary Materials
Acknowledgment
- Sample Availability: Sample of the compound 1 is available from the authors.
References and Notes
- Anđelković, K.; Sladić, D.; Bacchi, A.; Pelizzi, G.; Filipović, N.; Rajković, M. Complexes of iron(II), iron(III) and zinc(II) with condensation derivatives of 2-acetylpyridine and oxalic or malonic dihydrazide. Crystal structure of tris[(1-(2-pyridyl)ethylidene)hydrazine]iron(II) perchlorate. Transition Met. Chem. 2005, 30, 243–250. [Google Scholar] [CrossRef]
- Filipović, N.R.; Bacchi, A.; Lazić, M.; Pelizzi, G.; Radulović, S.; Sladić, D.M.; Todorović, T.R.; Anđelković, K.K. Synthesis, structure and cytotoxic activity evaluation of a dinuclear complex of Cd(II) with N’,N’2-bis[(1E)-1-(2-pyridyl)ethylidene]propanedihydrazide. Inorg. Chem. Commun. 2008, 11, 47–50. [Google Scholar] [CrossRef]
- Vujčić, M.; Lazić, M.; Milenković, M.; Sladić, D.; Radulović, S.; Filipović, N.; Anđelković, K. A comparative study of DNA binding and cell cycle phase perturbation by the dinuclear complex of Cd(II) with the condensation product of 2-acetylpyridine and malonic acid dihydrazide N’,N’2-bis[(1E)-1-(2-pyridyl)ethylidene]propanedihydrazide. J. Biochem. Mol. Toxicol. 2011, 25, 175–182. [Google Scholar] [CrossRef]
- Eshkourfu, R.; Čobeljić, B.; Vujčić, M.; Turel, I.; Pevec, A.; Sepčić, K.; Zec, M.; Radulović, S.; Srdić-Radić, T.; Mitić, D.; Anđelković, K.; Sladić, D. Synthesis, characterization, cytotoxic activity and DNA binding properties of the novel dinuclear cobalt(III) complex with the condensation product of 2-acetylpyridine and malonic acid dihydrazide. J. Inorg. Biochem. 2011, 105, 1196–1203. [Google Scholar] [CrossRef]
- Todorović, T.R.; Rychlewska, U.; Warżajtis, B.; Radanović, D.D.; Filipović, N.R.; Pajić, I.A.; Sladić, D.M.; Anđelković, K.K. Synthesis, characterization and antimicrobial activity of Ni(II) and Zn(II) complexes with N’,N’2-bis[(1E)-1-(2-pyridyl)ethylidene]propanedihydrazide. Crystal structures of two highly solvated bimetallic complexes of Ni(II). Polyhedron 2009, 28, 2397–2402. [Google Scholar]
- Bayly, S.R.; Cowley, A.R.; Dilworth, J.R.; Ward, C.V. Ruthenium complexes with tridentate PNX (X = O, S) donor ligands. Dalton Trans. 2008, 2190–2198. [Google Scholar]
- Liu, P.N.; Su, F.H.; Wen, T.B.; Sung, H.H.-Y.; Williams, I.D.; Jia, G. Selective and Efficient Cycloisomerization of Alkynols Catalyzed by a New Ruthenium Complex with a Tetradentate Nitrogen–Phosphorus Mixed Ligand. Chem. Eur. J. 2010, 16, 7889–7897. [Google Scholar]
- Thibault, M.; Lucier, B.E.G.; Schurko, R.W.; Fontaine, F. Synthesis and solid-state characterization of platinum complexes with hexadentate amino- and iminophosphine ligands. Dalton Trans. 2009, 7701–7716. [Google Scholar]
- Campo, O.; Carbayo, A.; Cuevas, J.V.; García-Herbosa, G.; Muñoz, A. Isomeric Preference in Complexes of Palladium(II) with Chelating P,N-Donor Ligands. Eur. J. Inorg. Chem. 2009, 2009, 2254–2260. [Google Scholar]
- Sui-Seng, C.; Haque, F.N.; Hadzovic, A.; Pütz, A.; Reuss, V.; Meyer, N.; Lough, A.J.; Zimmer-De Iuliis, M.; Morris, R.H. Synthesis and Characterization of Iron(II) Complexes with TetradentateDiiminodiphosphine or DiaminodiphosphineLigands as Precatalysts for the Hydrogenation of Acetophenone. Inorg. Chem. 2009, 48, 735–743. [Google Scholar]
- Williams, D.B.G.; Pretorius, M. Synthesis and evaluation of phosphine–N ligands in transition metal-catalysed C-C bond forming reactions. J. Mol. Catal. A: Chem. 2008, 284, 77–84. [Google Scholar] [CrossRef]
- Radulović, V.; Bacchi, A.; Pelizzi, G.; Sladić, D.; Brčeski, I.; Anđelković, K. Synthesis, Structure, and Antimicrobial Activity of Complexes of Pt(II), Pd(II), and Ni(II) with the Condensation Product of 2-(Diphenylphosphino)benzaldehyde and Semioxamazide. Monatsh. Chem. 2006, 137, 681–691. [Google Scholar] [CrossRef]
- Malešević, N.; Srdić, T.; Radulović, S.; Sladić, D.; Radulović, V.; Brčeski, I.; Anđelković, K. Synthesis and characterization of a novel Pd(II) complex with the condensation product of 2-(diphenylphosphino) benzaldehyde and ethyl hydrazinoacetate. Cytotoxic activity of the synthesized complex and related Pd(II) and Pt(II) complexes. J. Inorg. Biochem. 2006, 100, 1811–1818. [Google Scholar] [CrossRef]
- International Tables for Crystallography; Wilson, A.J.C. (Ed.) Kluwer Academic Publishers: Dordrecht, The Netherlands, 1995; Volume C, pp. 796–811.
- Dalpozzo, R.; Bartoli, G.; Sambri, L.; Melchiorre, P. Perchloric Acid and Its Salts: Very Powerful Catalysts in Organic Chemistry. Chem. Rev. 2010, 110, 3501–3551. [Google Scholar] [CrossRef]
- Ben-Aroya, B.B.; Portnoy, M. Addition of borane-protected secondary phosphines to imines. A route to protected mono-N-substituted-α-aminophosphines. Tetrahedron Lett. 2000, 41, 6143–6147. [Google Scholar] [CrossRef]
- Bhutani, H.; Singh, S.; Jindal, K.C.; Chakraboti, A.K. Mechanistic explanation to the catalysis by pyrazinamide and ethambutol of reaction between rifampicin and isoniazid in anti TB FDCs. J. Pharm. Biomed. Anal. 2005, 39, 892–899. [Google Scholar] [CrossRef]
- Maerkl, G. Innermolekulare C-Alkylierung von Phosphin-methylenen zu cyclischen Phosphoniumsalzen und Phosphor-Yliden. Z. Naturforsch. 1963, 18b, 84–85. [Google Scholar]
- Meyer, B.N.; Ferrigni, N.R.; Putnam, J.E.; Jacobsen, L.B.; Nichols, D.E.; McLaughlin, J.L. Brine shrimp: A convenient bioassay for active plant constituents. Planta Med. 1982, 45, 31–34. [Google Scholar] [CrossRef]
- Anderson, J.E.; Goetz, C.M.; McLaughlin, J.L.; Suffness, M. A blind comparison of simple bench-top bioassays and human tumour cell cytotoxicities as antitumor prescreens. Phytochem. Anal. 1991, 2, 107–111. [Google Scholar] [CrossRef]
- Oxford Diffraction CrysAlis CCD and CrysAlisRED including ABSPACK, Version 1.171 Oxford Diffraction. Oxfordshire, England, 2000.
- Sheldrick, G.M. Phase annealing in SHELX-90: Direct methods for larger structures. ActaCrystallogr. Sect. A 1990, 46, 467–473. [Google Scholar]
- Sheldrick, G.M. A short history of SHELX. ActaCrystallogr. Sect. A 2008, 64, 112–122. [Google Scholar]
- Spek, A.L. PLATON, A Multipurpose Crystallographic Tool; Utrecht University: Utrecht, The Netherlands, 2010. [Google Scholar]
- XP. Siemens Analytical X-ray Instruments Inc.: Madison, Wisconsin, USA, 1990.
- Dower, W.J.; Miller, J.F.; Ragsdale, C.W. High efficiency transformation of E. coli by high voltage electroporation. Nucleic Acids Res. 1988, 16, 6127–6145. [Google Scholar]
- Birnboim, H.C.; Doly, J. A rapid alkaline extraction procedure for screening recombinant plasmid DNA. Nucleic Acids Res. 1979, 7, 1513–1523. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Milenković, M.; Warżajtis, B.; Rychlewska, U.; Radanović, D.; Anđelković, K.; Božić, T.; Vujčić, M.; Sladić, D. Synthesis, Spectral and Solid State Characterization of a New Bioactive Hydrazine Bridged Cyclic Diphosphonium Compound. Molecules 2012, 17, 2567-2578. https://doi.org/10.3390/molecules17032567
Milenković M, Warżajtis B, Rychlewska U, Radanović D, Anđelković K, Božić T, Vujčić M, Sladić D. Synthesis, Spectral and Solid State Characterization of a New Bioactive Hydrazine Bridged Cyclic Diphosphonium Compound. Molecules. 2012; 17(3):2567-2578. https://doi.org/10.3390/molecules17032567
Chicago/Turabian StyleMilenković, Milica, Beata Warżajtis, Urszula Rychlewska, Dušanka Radanović, Katarina Anđelković, Tatjana Božić, Miroslava Vujčić, and Dušan Sladić. 2012. "Synthesis, Spectral and Solid State Characterization of a New Bioactive Hydrazine Bridged Cyclic Diphosphonium Compound" Molecules 17, no. 3: 2567-2578. https://doi.org/10.3390/molecules17032567
APA StyleMilenković, M., Warżajtis, B., Rychlewska, U., Radanović, D., Anđelković, K., Božić, T., Vujčić, M., & Sladić, D. (2012). Synthesis, Spectral and Solid State Characterization of a New Bioactive Hydrazine Bridged Cyclic Diphosphonium Compound. Molecules, 17(3), 2567-2578. https://doi.org/10.3390/molecules17032567