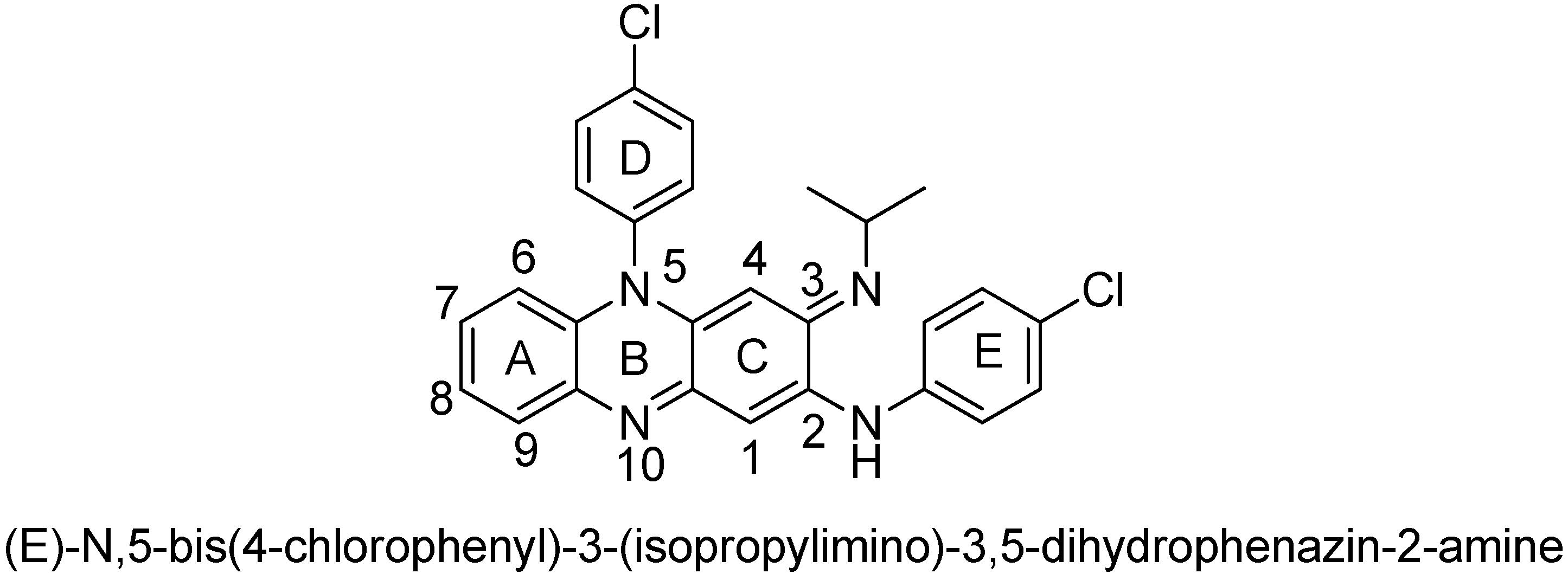
3.2. Chemistry
3.2.1. Synthesis of 2-(4-chloroanilino)-5-fluoronitrobenzene (1)
4-Chloroaniline (12.76 g, 100 mmol), 2,5-difluoronitrobenzene (8.0 g, 50 mmol), anhydrous K2CO3 (3.5 g, 25 mmol) and anhydrous KF (2.9 g, 50 mmol) were mixed and heated at 170 °C for 14 h. After cooling to room temperature, water was added and the solid formed was filtered and washed with water. The crude product was purified via recrystallization from 95% ethanol to give 1 as an orange solid (11.8 g, 89%). 1H-NMR (300 MHz, CDCl3) δ 9.25 (s, 1H), 7.94–7.90 (m, 1H), 7.39 (d, J = 8.7 Hz, 2H), 7.21–7.17 (m, 4H). ESI/MS (m/z): 267 [M+H]+.
3.2.2. Synthesis of 1-(4-chlorophenyl)-6-fluoroquinoxaline-2,3(1H,4H)-dione (3)
Compound 1 above (6.45 g, 24 mmol) was suspended in anhydrous methanol (100 mL). The mixture was hydrogenated with 10% Pd/C (1.3 g) at room temperature at 1 atmosphere pressure of hydrogen for 8 h. After removal of catalyst, the solvent was concentrated under reduced pressure. The residue was dissolved in toluene (150 mL) and ethyl oxalyl chloride (10 mL, 89 mmol) was added. The mixture was refluxed for 1 h and then cooled to room temperature Approximately one half of the solvent was removed under reduced pressure and the resulting solid was filtered and recrystallized from methanol to give 3 as a white solid (4.97 g, 71%). 1H-NMR (300 MHz, DMSO-d6) δ 12.18 (s, 1H), 7.69 (d, J = 8.7 Hz, 2H), 7.43 (d, J = 8.7 Hz, 2H), 6.99 (dd, J = 9.0, 2.7 Hz, 1H), 6.87–9.80 (m, 1H), 6.34 (dd, J = 9.3, 5.1 Hz, 1H). ESI/MS (m/z): 291 [M+H]+.
3.2.3. Synthesis of 1-(4-chlorophenyl)-6-fluoro-7-nitroquinoxaline-2,3(1H,4H)-dione (4)
Compound 3 above (2.9 g, 10 mmol) was dissolved in concentrated sulfuric acid (30 mL) and cooled to −5 °C. KNO3 (1.1 g, 8 mmol) was added into the solution portion-wise. After addition, the mixture was stirred at 0 °C for 1 h and then at room temperature for 1 h. The reaction mixture was slowly poured into ice water, and the solid formed was filtered and washed with water. The product was air-dried to give a light yellow solid 3.4 g (quantitative yield). 1H-NMR (300 MHz, DMSO-d6) δ 12.62 (s, 1H), 7.75 (d, J = 8.7 Hz, 2H), 7.45 (d, J = 8.7 Hz, 2H), 7.19 (d, J = 11.7 Hz, 1H), 6.96 (d, J = 6.9 Hz, 1H). ESI/MS (m/z): 336 [M+H]+.
3.2.4. Synthesis of 1-(4-chlorophenyl)-6-(4-chloroanilino)-7-nitroquinoxaline-2,3(1H,4H)-dione (5)
Compound 4 above (0.34 g, 1.0 mmol), DMSO (3 mL), anhydrous KF (0.12 g, 2.0 mmol), and 4-chloroaniline (1.27 g, 10 mmol) were heated at 140 °C for 25 h. After cooling to room temperature, 5 M HCl was added and the resulting solid was collected by filtration and washed with water. The crude product was purified by column chromatography to give 5 as an orange solid (0.22 g, 50%). 1H-NMR (300 MHz, DMSO-d6) δ 12.09 (s, 1H), 9.35 (s, 1H), 7.76 (d, J = 8.4 Hz, 2H), 7.52–7.46 (m, 4H), 7.35 (d, J = 8.4 Hz, 2H), 7.01 (s, 1H), 6.99 (s, 1H). ESI/MS (m/z): 443 [M+H]+.
3.2.5. Synthesis of 1-(4-chlorophenyl)-6-(4-chloroanilino)-7-(isopropylamino)-quinoxaline-2,3-(1H,4H)-dione (7)
Zinc powder (0.13 g) was added portionwise to a vigorously stirred mixture of 5 (0.08 g, 0.18 mmol) in glacial acetic acid (2 mL) and anhydrous methanol (5 mL). After the reaction was complete, as confirmed by TLC, the reaction mixture was filtered. The filtrate was concentrated under reduced pressure. Water was then added to the residue. After filtration, the yellow solid was dissolved in a mixture of glacial acetic acid (4 mL) and acetone (0.3 mL) in anhydrous methanol (2 mL). The resulting mixture was stirred at room temperature for 30 min., and then sodium borohydride (76 mg, 2.0 mmol) was added and stirred for 30 min. The reaction mixture was concentrated under reduced pressure and water was added to the residue. The resulting solid was collected by filtration, washed with water, and dried. The crude product was purified by column chromatography to give 7 as a solid (30 mg, 37%). 1H-NMR (300 MHz, acetone-d6) δ 7.70 (d, J = 8.7 Hz, 2H), 7.51 (d, J = 8.7 Hz, 2H), 7.15 (d, J = 8.7 Hz, 2H), 7.12 (s, 1H), 6.75 (d, J = 8.7 Hz, 2H), 5.76 (s, 1H), 3.18 (m, 1H), 0.99 (d, J = 6.3 Hz, 6H). ESI/MS (m/z): 445 [M+H]+.
3.2.6. Synthesis of 1-(4-chlorophenyl)-6-(4-chloroanilino)-7-isopropylimino-1,7-dihydroquinoxaline (8)
Lithium aluminum hydride (8 mg, 0.21 mmol) was added into a mixture of 7 (30 mg, 0.07 mmol) in THF (5 mL). The mixture was heated at 50 °C for 1 h under nitrogen atmosphere. After cooling to room temperature, 3 drops of ethyl acetate were added. The mixture was filtered and washed with ethyl acetate. The filtrate was washed with brine. The organic layer was separated and applied to preparative TLC. The TLC plate was heated in an oven (100 °C) for 30 min. before being developed with CHCl3/MeOH (20:1). The red band (Rf: 0.34) was collected and washed with anhydrous methanol. The washing was concentrated, and the residue was further purified by column chromatography (HL20, eluted with MeOH) to give 6 mg of 8 as a red solid (21%). 1H-NMR (300 MHz, CD3OD) δ 8.62 (d, J = 3.6 Hz, 1H) 8.28 (d, J = 3.6 Hz, 1H) 7.78 (d, J = 8.7 Hz, 2H), 7.70 (d, J = 9.0 Hz, 2H), 7.49 (s, 1H), 7.42 (d, J = 8.7 Hz, 2H), 7.31 (d, J = 9.0 Hz, 2H), 6.22 (s, 1H), 3.65–3.61 (m, 1H), 1.23 (d, J = 6.3 Hz, 6H). 13C-NMR (150 MHz, CD3OD) δ 149.3, 146.9, 142.6, 140.1, 139.5, 138.9, 134.3, 132.1, 131.2, 131.0, 129.0, 125.1, 108.6, 92.1, 46.9, 21.4. HRMS (ESI-TOF+): m/z [M+H]+ calcd. for C23H21Cl2N4: 423.1137; found: 423.1141.
3.2.7. Synthesis of 2-methylamino-nitrobenzene (9a)
2-Fluoronitrobenzene (2.82 g, 20 mmol) was dissolved in anhydrous ethanol (20 mL), and 33% aqueous methylamine solution (4.6 mL) was added. The mixture was heated to reflux until complete consumption of 2-fluoronitrobenzene. After cooling to room temperature, the solid formed was filtered and washed with 95% ethanol and dried to give the title compound as a red oil (quantitative yield). 1H-NMR (400 MHz, acetone-d6) δ 8.10 (d, J = 8.0 Hz, 1H), 7.54 (t, J = 7.2, 8.4 Hz, 1H), 7.01 (d, J = 8.4 Hz, 1H), 6.68 (t, J = 7.2, 8.0 Hz, 1H), 3.07 (d, J = 5.2 Hz, 3H). ESI/MS (m/z): 153 [M+H]+.
3.2.8. Synthesis of N-(4-chlorophenyl)-2-nitroaniline (9b)
2-Fluoronitrobenzene (14.1 g, 100 mmol), 4-chloroaniline (19.1 g, 150 mmol), anhydrous KF (5.8 g, 100 mmol) and anhydrous K2CO3 (13.8 g, 100 mmol) were heated at 160 °C for 10 h. The reaction mixture was cooled and water and ethyl acetate were added. The aqueous layer was extracted with ethyl acetate. The organic layer was combined and washed with 2 N HCl and dried over anhydrous Na2SO4. The solvent was concentrated under reduced pressure and the residue was recrystallized with anhydrous ethanol to give the title compound as red solid (20 g, 80%). 1H-NMR (300 MHz, CDCl3) δ 9.40 (s, 1H), 8.23–8.19 (d, J = 8.7 Hz, 1H), 7.41–7.37 (m, 3H), 7.23–7.16 (m, 3H), 6.78 (t, J = 8.4, 8.1 Hz, 1H).
3.2.9. Synthesis of 1-methylamino-2-(5-fluoro-2,4-dinitroanillino)-benzene (11a)
A suspension of 9a (1.1 g, 5.0 mmol) in anhydrous methanol (20 mL) was hydrogenated with 10% Pd/C (0.06g) under atmospheric pressure until the reaction mixture turned colourless. After filtration, the filtrate was treated with DFDNB (1.03 g, 5.0 mmol) and Et3N (0.7 mL, 5.0 mmol) and stirred at room temperature for 1 h. The solid formed was filtered and washed with methanol to give yellow solid (78%). 1H-NMR (300 MHz, acetone-d6) δ 9.74 (s, 1H), 9.04–9.02 (d, J = 7.8 Hz, 1H), 7.34–7.29 (t, J = 7.5 Hz, 1H), 7.20–7.17 (d, J = 7.5 Hz, 1H), 6.81–6.76 (d, J = 7.5 Hz, 1H), 6.74–6.71 (t, J = 7.8 Hz, 1H), 6.09 (d, J = 14.1 Hz, 1H), 5.13 (d, J = 3.9 Hz, 1H), 2.80–2.78 (d, J = 5.1 Hz, 3H). ESI/MS (m/z): 307 [M+H]+.
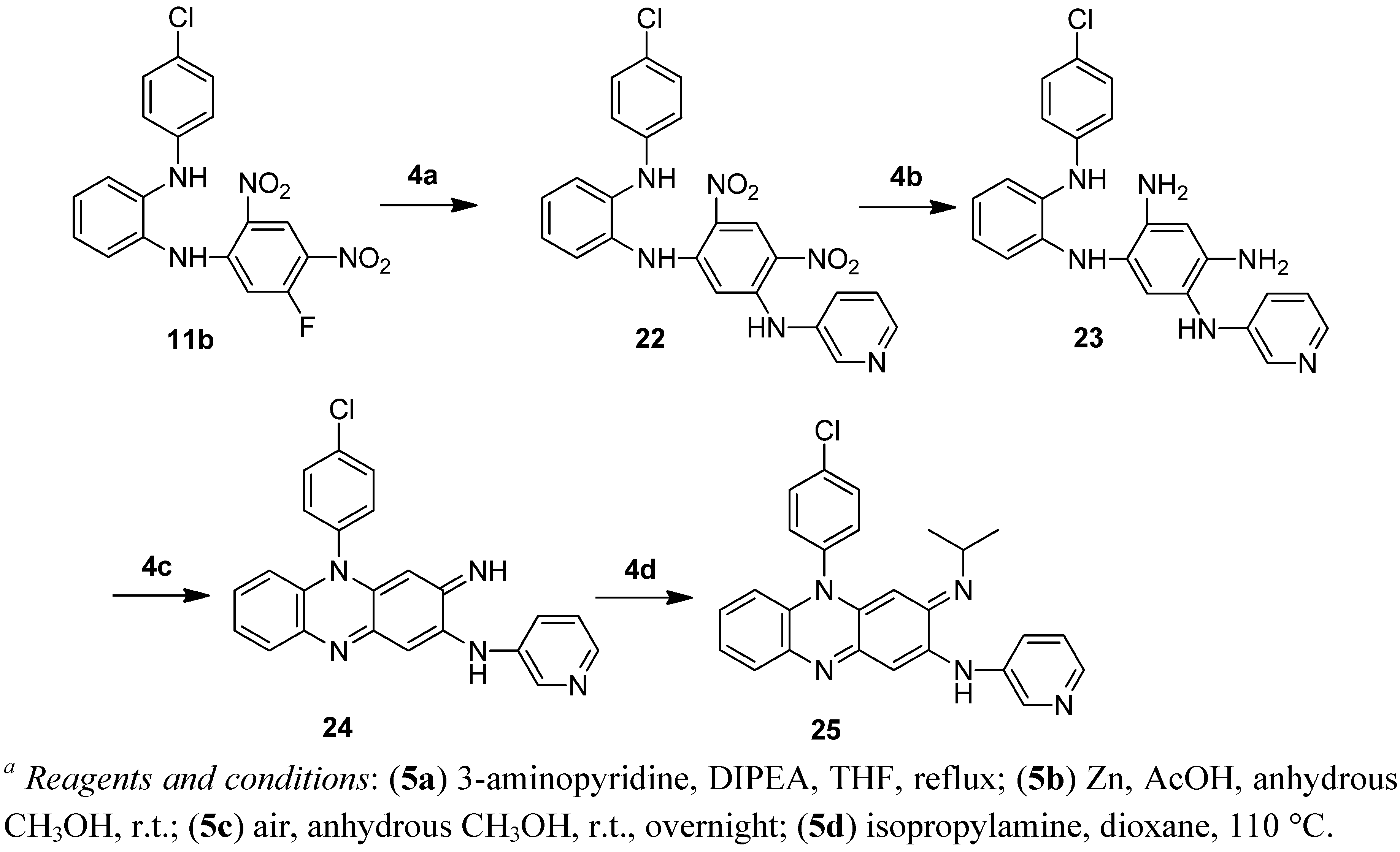
3.2.10. Synthesis of 1-(4-chloroanilino)-2-(5-fluoro-2,4-dinitroanilino)benzene (11b)
Zinc powder (9 g) was added portionwise into a mixture of 9b (2.5 g, 10 mmol) in CH2Cl2 (50 mL) and glacial acetic acid (3 mL) and stirred at room temperature for 4 h. After filtration, the filtrate was concentrated, and the residue was treated with DFDNB (2.04 g, 10 mmol) and Et3N (1.01 g, 10 mmol) in anhydrous methanol (100 mL). The mixture was refluxed for 5 h. and then cooled to room temperature. The solid formed was filtered, washed with ethanol and dried to give the title compound as a red solid (2.60 g, 65%). M.p. 216–217 °C. 1H-NMR (300 MHz, DMSO-d6) δ 9.96 (s, 1H), 8.87 (d, J = 8.1 Hz, 1H), 7.87 (s, 1H), 7.35–7.30 (m, 3H), 7.22–7.19 (d, J = 9.0 Hz, 2H), 7.07–7.02 (m, 1H), 6.97–6.94 (d, J = 9.0 Hz, 2H), 6.46 (d, J = 14.4 Hz, 1H). ESI/MS (m/z): 403 [M+H]+.
3.2.11. Synthesis of 1-[5-(4-chloroanilino)-2,4-dinitroanilino]-2-methylaminobenzene (12a)
A stirred suspension of 11a (1.34 g, 3.6 mmol), 4-chloroanilline (0.92 g, 7.2 mmol), and anhydrous KF (0.21 g, 3.6 mmol) in anhydrous ethanol (30 mL) was refluxed for 4 h. After being cooled to room temperature, the solid formed was filtered and washed with anhydrous ethanol to give the title compound as an orange solid (1.52 g, 88%). 1H-NMR (300 MHz, DMSO-d6) δ 9.48 (brs, 2H), 9.00 (s, 1H), 7.30 (d, J = 8.7 Hz, 2H), 7.19 (d, J = 9.0 Hz, 2H), 7.08 (t, J = 7.2 Hz, 1H), 6.99 (dd, J = 7.5, 1.5 Hz, 1H), 6.70 (d, J = 7.8 Hz, 1H), 6.56 (t, J = 7.5 Hz, 1H), 6.00 (s, 1H), 4.85 (d, J = 8.7 Hz, 1H), 3.23–3.20 (m, 1H), 1.80–1.76 (m, 2H), 1.68–1.56 (m, 3H), 1.36–1.24 (m, 2H), 1.12–1.01 (m, 3H).
3.2.12. Synthesis of 1-[5-methylamino-2,4-dinitroanilino]-2-(4-chlorophenyl)benzene (12b)
A mixture of 11b (1.05 g, 2.6 mmol), 30% methylamine in ethanol (5 mL, 48.4 mmol) in anhydrous ethanol (100 mL) was refluxed for 4 h. The reaction mixture was cooled to room temperature and filtered. The solid obtained was washed with ethanol and dried to give the title compound as a red solid (1.0 g, 93%). It was used directly in the next step without further purification. M.p. 239–240 °C. 1H-NMR (300 MHz, DMSO-d6) δ 9.56 (s, 1H), 8.96 (s, 1H), 8.39 (d, J = 4.8 Hz, 1H), 7.87(s, 1H), 7.41 (d, J = 6.9 Hz, 1H), 7.34–7.24 (m, 2H), 7.19 (d, J = 9.0 Hz, 2H), 7.08–7.02 (m, 1H), 6.95 (d, J = 9.0 Hz, 2H), 5.71 (s, 1H), 2.65 (d, J = 5.1 Hz, 3H). ESI/MS (m/z): 436 [M+Na]+.
3.2.13. Synthesis of 2-(4-chloroanilino)-5-methyl-3-isopropylimino-3,5-dihydrophenazine (15a)
Zinc powder (1.96 g) was added portionwise to a suspension of 12a (0.48 g, 1.0 mmol) in glacial acetic acid (10 mL) and heated at 50 °C for 30 min. After filtration, the filtrate was stirred in contact with air overnight. The reaction mixture was concentrated under reduced pressure and adjusted to alkaline with ammonia, and then the solid formed was filtered and washed with water to give a black solid 0.34 g. The black solid was taken up in dioxane (5 mL) and isopropylamine (2.2 mL, 25.7 mmol) was added. The mixture was heated in a sealed bomb at 110 °C for 7 h. After being cooled to room temperature, water was added to the mixture and the solid formed was filtered and purified by column chromatography (eluted with P.E./ethyl acetate: 4/1 to 2/1 to 100% ethyl acetate) to give 0.13 g of the title compound (29%). M.p. 98–102 °C. 1H-NMR (300 MHz, CDCl3) δ 7.65 (dd, J = 7.8, 1.5 Hz, 2H), 7.44 (d, J = 8.4 Hz, 1H), 7.32–7.31 (m, 1H), 7.30 (s, 4 H), 7.19–7.14 (m, 1H), 6.78 (s, 1H), 6.17 (s, 1H), 4.39 (m, 1H), 3.92–3.88 (m, 1H), 2.58–2.52 (m, 2H), 2.07–1.85 (m, 5 H), 1.56–1.51 (m, 2H), 1.35–1.34 (m, 1H), 1.31 (d, J = 6.3 Hz, 6H). 13C-NMR (100 MHz, CDCl3) δ 152.3, 150.6, 143.1, 138.8, 137.0, 133.7, 131.2, 129.2, 128.6, 127.8, 126.9, 122.5, 122.4, 114.2, 99.7, 90.5, 61.2, 49.7, 28.6, 26.5, 25.7, 23.9. HRMS (ESI-TOF+): m/z [M+H]+ calcd. for C27H30ClN4: 445.2159; found: 445.2161.
3.2.14. Synthesis of 2-methylamino-5-(4-chlorophenyl)-3-imino-3,5-dihydrophenazine (14b)
Zinc powder (4.0 g, 61.5 mmol) was added portionwise to a suspension of 12b (0.8 g, 1.9 mmol) in glacial acetic acid (80 Ml) and stirred for 4 h. The reaction mixture was filtered and washed with glacial acetic acid. The filtrate was stirred in contact with air overnight. The solvent was concentrated and the residue was treated with water and adjusted to alkaline with ammonia. The solid formed was filtered, washed with water and dried. The crude solid was purified by neutral aluminum oxide (100–200 mesh) column chromatography eluted with ethyl acetate/methanol (10:1 to 5:1) to give the title compound as a dark red solid (0.28 g, 43%). M.p. 218–220 °C. 1H-NMR (300 MHz, DMSO-d6) δ 9.20 (s, 1H), 7.83 (d, J = 8.7 Hz, 2H), 7.65 (d, J = 5.1 Hz, 1H), 7.55 (d, J = 8.4 Hz, 2H), 7.24–7.21 (m, 2H), 6.95 (brs, 1H), 6.48 (d, J = 8.4 Hz, 1H), 6.10 (s, 1H), 5.31 (s, 1H), 2.88 (d, J = 2.7 Hz, 3H). ESI/MS (m/z): 335 [M+H]+.
3.2.15. Synthesis of 2-methylamino-3-isopropylimino-5-(4-chlorophenyl)-3,5-dihydro-phenazine (15b)
A mixture of 14b (0.15 g, 0.45 mmol), isopropylamine (1 mL, 12.0 mmol) and dioxane (20 mL) was heated in a sealed tube at 120 °C for 10 h. After being cooled to room temperature, the reaction mixture was concentrated under reduced pressure. The residue was purified by neutral aluminum oxide (100–200 mesh) column chromatography eluted with hexane/ethyl acetate (2:1) to give 150 mg of 15b as a red solid (88%). M.p. 235–237 °C. 1H-NMR (300MHz, acetone-d6) δ 7.85–7.82 (d, J = 8.7 Hz, 2H), 7.61–7.58 (d, J = 9.3 Hz, 1H), 7.54–7.51 (d, J = 8.4 Hz, 2H), 7.18–7.10 (m, 2H), 6.95 (brs, 1H), 6.48–6.45 (d, J = 9.0 Hz, 1H), 5.99 (s, 1H), 5.24 (s, 1H), 3.44–3.36 (m, 1H), 2.96 (d, J = 2.7 Hz, 3H), 0.98 (d, J = 6.0 Hz, 6H). 13C-NMR (125 MHz, CDCl3) δ 151.1, 150.8, 149.2, 136.4, 135.7, 135.5, 134.7, 131.5, 131.1, 130.5, 127.7, 126.5, 122.7, 113.7, 95.2, 88.9, 49.2, 29.2, 23.5. HRMS (ESI-TOF+): m/z [M+H]+ calcd. for C22H22ClN4: 377.1527; found: 377.1510.
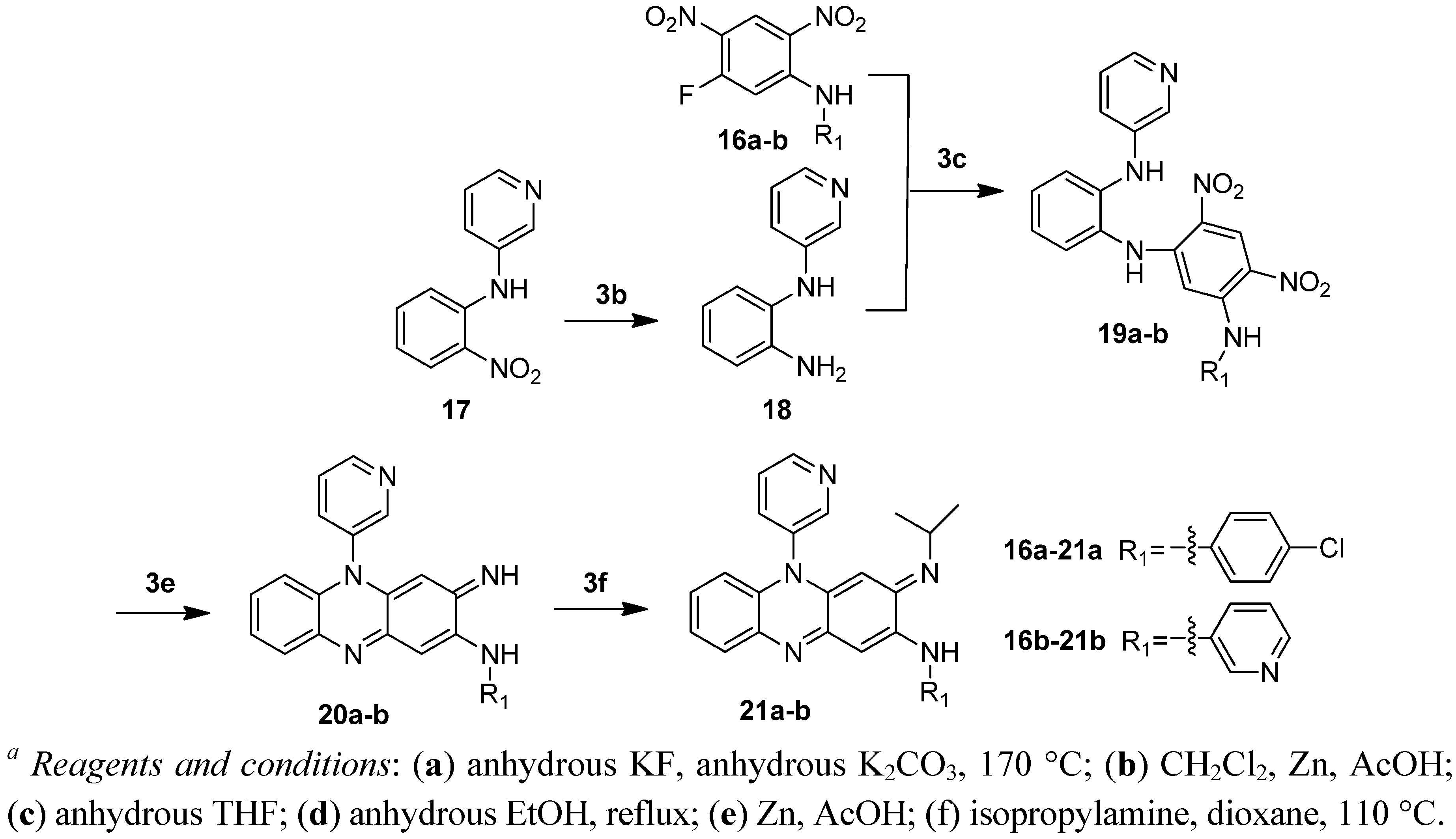
3.2.16. N1-(5-((4-chlorophenyl)amino)-2,4-dinitrophenyl)-N2-(pyridin-3-yl)benzene-1,2-diamine (19a)
Compound 17 (0.215g, 1.0 mmol) was suspended in anhydrous methanol (10 mL). Pd/C (10%, 40 mg) was added and the mixture was hydrogenated under atmospheric pressure until the reaction mixture turned colourless. The Pd/C was filtered off. The filtrate was concentrated in vacuo to give a light yellow oil. The oil was dissolved in THF (10 mL) and to this solution 16a (0.312 g 1.0 mmol) and diisopropylethylamine (0.17 mL, 1.0 mmol) were added. The mixture was refluxed for 20 h and then the reaction mixture was cooled to room temperature and concentrated in vacuo. The residue was added with water and filtered. The crude product was purified via chromatography (PE/EA: 2/1 to 1/1) to give the title compound as a yellow oil 0.243 g, yield 51%. 1H-NMR (300 MHz, DMSO-d6): 9.67 (brs, 1H), 9.52 (brs, 1H), 8.98 (s, 1H), 8.14 (s, 1H), 8.03–8.01 (m, 1H), 7.88 (s, 1H), 7.38 (d, J = 8.7 Hz, 2H), 7.28–7.13 (m, 7H), 7.01–6.96 (m, 1H), 6.02 (s, 1H).
3.2.17. N1-(2,4-dinitro-5-(pyridin-3-ylamino)phenyl)-N2-(pyridin-3-yl)benzene-1,2-diamine (19b)
Compound 17 (0.215 g, 1.0 mmol) was suspended in anhydrous methanol (10 mL). Pd/C (10%, 40 mg) was added to the suspension and the mixture was hydrogenated under atmospheric pressure until the reaction mixture turned colourless. Then the Pd/C was filtered off. The filtrate was concentrated in vacuo to give a light yellow oil. The oil obtained was dissolved in THF (10 mL), and the solution was added 16b (0.278 g, 1.0 mmol) and diisopropylethylamine (0.17 mL, 1.0 mmol). The reaction mixture was refluxed for another 20 h. and then cooled to room temperature The mixture was concentrated in vacuo and the residues were added with distilled water and filtered. The crude product was purified via chromatography (PE/EA: 2/1 to 1/2) to produce the title compound as a yellow solid 0.3 g, yield 68%. 1H-NMR (300 MHz, DMSO-d6): 9.72 (brs, 1H), 9.52 (brs, 1H), 8.99 (s, 1H), 8.41–8.36 (m, 1H), 8.15 (t, J = 1.5 Hz, 1H), 8.02 (t, J = 3.0 Hz, 1H), 7.85 (s, 1H), 7.64–7.60 (m, 1H), 7.35 (dd, J = 8.1 Hz, 4.8 Hz, 1H), 7.23 (d, J = 7.8 Hz, 1H), 7.17–7.14 (m, 4H), 6.94–6.88 (m, 1H), 5.92 (s, 1H).
3.2.18. (E)-N-(4-chlorophenyl)-3-(isopropylimino)-5-(pyridin-3-yl)-3,5-dihydrophenazin-2-amine (21a)
Compound 19a (0.243 g, 0.5 mmol) was suspended in glacial acetic acid (10 mL). The suspension was added with zinc powder (0.66 g) portionwise in an ice bath. After addition, the mixture was stirred at room temperature for 30 min. and then heated at 50 °C for 30 min. After being cooled to room temperature, the reaction mixture was filtered and washed with glacial acetic acid. The filtrate was concentrated in vacuo. The residues were added with concentrated aqueous ammonia until the mixture became basic and then the solid was filtered out, washed with water and dried to give black solid. The black solid was dissolved in anhydrous methanol and the solution was added with solution of ammonia in methanol to adjust the solution to basic. The mixture was stirred in contact with air overnight and then concentrated in vacuo. The residues were mixed with dioxane (5 mL) and isopropylamine (2 mL, 24 mmol). The mixture was heated at 110 °C in a sealed tube for 10 h. After being cooled to room temperature, the reaction mixture was added with water and filtered to give crude product. The crude product was purified via chromatography (PE/EA: 2/1 to EA) to produce 21 mg of the title compound as a red solid, yield 9%. Mp: 237–239 °C. 1H-NMR (300 MHz, DMSO-d6): 8.93 (dd, J = 4.8 Hz, 1.5 Hz, 1H), 8.77 (dd, J = 1.8 Hz, 1H), 8.62 (brs, 1H), 8.15–8.11 (m, 1H), 7.87–7.83 (m, 1H), 7.67–7.63 (m, 1H), 7.48–7.41 (m, 4H), 7.24–7.21 (m, 2H), 6.73 (s, 1H), 6.44–6.41 (m, 1H), 5.12 (s, 1H), 3.39–3.30 (m, 1H), 1.03 (d, J = 6.3 Hz, 6H). 13C-NMR (100 MHz, DMSO-d6): 151.0, 150.1, 143.0, 138.8, 137.5, 135.2, 134.7, 133.9, 131.2, 129.3, 128.2, 127.9, 126.8, 126.0, 122.9, 113.9, 88.5, 48.9, 44.8, 23.3. HRMS (m/z) calcd. for C26H23N5Cl (M+H+): 440.1636, found 440.1635.
3.2.19. (E)-3-(isopropylimino)-N,5-di(pyridin-3-yl)-3,5-dihydrophenazin-2-amine (21b)
Compound 19b (0.3 g, 0.7 mmol) was suspended in glacial acetic acid (5 mL). Zinc powder (1.33 g) was added portionwise to the suspension in an ice bath. After addition, the mixture was heated at 50 °C for 1 h. After being cooled to room temperature, the reaction mixture was filtered and washed with glacial acetic acid. The filtrate was concentrated in vacuo. The residues were added with concentrated aqueous ammonia until the mixture became basic and then the solid was filtered out, washed with water and dried to give a dark brown solid. The solid was dissolved in anhydrous methanol and the solution was added with ammonia in methanol to adjust the solution to basic. The mixture was stirred in contact with air for 7 h. and then concentrated in vacuo. The residues were mixed with dioxane (5 mL) and isopropylamine (0.036 mol, 3 mL). The mixture was heated at 110 °C in a sealed bomb for 10 h. After being cooled to room temperature, the reaction mixture was added with water and filtered to give crude product. The crude product was purified via chromatography (PE/EA: 2/1 to EA) to produce 40 mg of the title compound as a red solid, yield 15%. Mp: 174–175 °C. 1H-NMR (400 MHz, DMSO-d6): 8.92 (dd, J = 4.8 Hz, 1.2 Hz, 1H), 8.76 (d, J = 2.4 Hz, 1H), 8.62 (brs, 1H), 8.60 (d, J = 2.4 Hz, 1H), 8.32 (d, J = 4.0 Hz, 1H), 8.13–8.11 (m, 1H), 7.87–7.83 (m, 1H), 7.65–7.63 (m, 1H), 7.44 (dd, J = 8.0 Hz, 4.8 Hz, 1H), 7.23–7.20 (m, 2H), 6.65 (s, 1H), 6.43–6.41 (m, 1H), 5.12 (s, 1H), 3.36–3.33 (m, 1H), 1.03 (d, J = 6.0 Hz, 6H). 13C-NMR (100 MHz, DMSO-d6): 151.0, 150.1, 150.0, 144.2, 143.9, 143.6, 137.4, 136.7, 135.2, 134.7, 133.9, 131.2, 128.5, 128.1, 127.9, 126.0, 123.9, 123.0, 113.9, 98.8, 88.5, 48.9, 23.3. HRMS (m/z) calcd. for C25H23N6 (M+H+): 407.1978, found 407.1980.
3.2.20. Synthesis of 1-[5-(3-pyridylamino)-2, 4-dinitroanilino]-2-(4-chloroanilino)benzene (22)
A mixture of compound 11b (40.3 g, 100 mmol), 3-aminopyridine (14.1 g, 150 mmol) and triethylamine (14 mL, 100 mmol) in THF (200 mL) was refluxed under nitrogen atmosphere for 28 h. About 150 mL of THF was evaporated at atmospheric pressure. CH2Cl2 was added and the solid formed was filtered and washed with CH2Cl2 to give the title compound 22 (31.2 g, 66%). 1H-NMR (300 MHz, DMSO-d6) δ 9.72 (s, 1H), 9.00 (s, 1H), 8.40–8.37 (m, 2H), 7.77 (s, 1H), 7.63–7.59 (m, 1H), 7.37–7.33 (m, 1H), 7.21–7.09 (m, 5H), 6.92–6.83 (m, 1H), 6.82–6.78 (m, 2H), 5.92 (s, 1H). ESI/MS (m/z): 477 [M+H]+.
3.2.21. Synthesis of 2-(3-pyridylamino)-5-(4-chlorophenyl)-3-imino-3,5-dihydrophenazine (24)
Zinc powder (71 g, 1.1 mol) was added portionwise to a mixture of compound 22 (28.6 g, 60 mmol) in glacial acetic acid (150 mL) cooled in an ice water bath. The mixture was stirred until the colour turned to light green, and then filtered, washed with glacial acetic acid and anhydrous methanol. The filtrate was concentrated and the residue was treated with water and adjusted to alkaline with ammonia. The solid formed was filtered, washed with water and then dissolved in anhydrous methanol. The methanol solution was stirred in contact with air overnight. The solid formed was filtered to produce 23.1 g of crude 24 which was used in the next step without further purification.
3.2.22. Synthesis of 5-(4-chlorophenyl)-3-isopropylimino-2-(3-pyridylamino)-3,5-dihydrophenazine (25)
A mixture of compound 24 (23.1 g), isopropylamine (100 mL, 1.2 mol) and dioxane (120 mL) was heated in a sealed tube at 110 °C for 7 h. After cooling, the reaction mixture was treated with water and the solid formed was filtered off and washed with water. The crude product was purified by column chromatography (eluted with hexane/ethyl acetate: 2:1) to produce the title compound 25 as a red solid (11.2 g, 44%). M.p. 194–196 °C. 1H-NMR (300 MHz, DMSO-d6) δ 8.63 (brs, 1H), 8.61 (d, J = 2.4 Hz, 1H), 8.32 (dd, J = 4.8, 1.2 Hz, 1H), 7.90–7.84 (m, 3H), 7.65–7.58 (m, 3H), 7.25–7.18 (m, 2H), 6.66 (s, 1H), 6.47–6.44 (m, 1H), 5.76 (s, 1H), 3.43–3.35 (m, 1H), 1.06 (d, J = 6.3 Hz, 6H). 13C-NMR (100 MHz, DMSO-d6) δ 150.2, 149.9, 144.1, 143.8, 143.5, 136.8, 135.8, 135.3, 134.4, 131.6, 131.1, 130.9, 128.3, 128.0, 127.8, 123.9, 122.9, 114.1, 98.8, 88.3, 48.9, 23.3. HRMS (ESI-TOF+): [M+H]+ calcd. for C26H23ClN5: 440.1641; found: 440.1643.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





