Microwave Synthesis Under Solvent-Free Conditions and Spectral Studies of Some Mesoporphyrinic Complexes

Abstract
:1. Introduction

2. Results and Discussion
2.1. Infrared Spectra

{kind=link}
{kind=link}
{kind=link}
| Assignments | Zn(II)TMAP | Cu(II)TMAP | Zn(II)TMAPOHP | Cu(II)TMAPOHP |
|---|---|---|---|---|
| νO-H | - | - | 3422 m | 3420 m |
| νC-H | 2922 m | 2923 m | 2922 m | 2924 m |
| νC-H from -O-CH3 | 2853 m | 2852 m | 2852 m | 2853 m |
| νC=O | 1762 m | 1763 m | 1764 m | 1765 m |
| νC-N | 1598 m | 1600 m | 1597 m | 1598 m |
| νC=N | 1505 m | 1501 m | 1506 m | 1503 m |
| νC-Hpyrrole | 1462 m | 1461 m | 1462 m | 1462 m |
| νC-O | 1194 s | 1196 s | 1195 s | 1196 s |
| δC-H | 998 m | 1001 m | 999 m | 1000 m |
| γC-C | 860 w | 862 w | 868 w | 858 w |
| γC-Npyrrole | 797 m | 800 m | 798 m | 788 m |
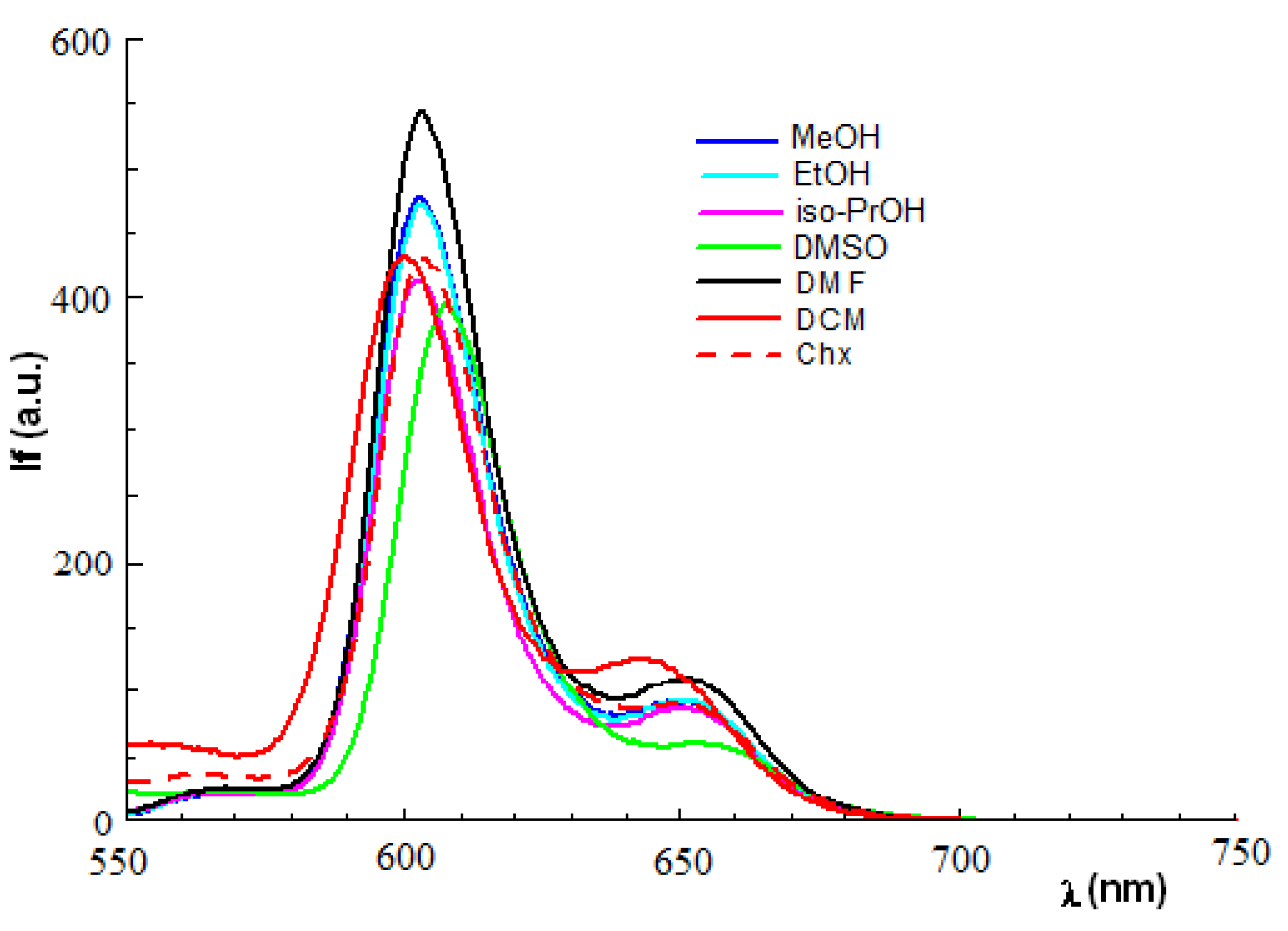
2.2. Absorption and Fluorescence Spectra

| Solvent | λmax (nm) [lgε (L mol−1 cm−1)] | ||||
|---|---|---|---|---|---|
| Soret band B(0,0) | Q bands Qy(0,0) Qx(1,0) | ||||
| Zn(II)-5,10,15,20-meso-tetrakis-(4-acetoxy-3-methoxyphenyl) porphyrin | |||||
| MeOH | 422.5 [5.750] | 556.8 [4.342] | 595.8 [4.017] | ||
| EtOH | 423.9 [5.769] | 557.1 [4.318] | 597.0 [3.924] | ||
| iso-PrOH | 424.4 [5.750] | 557.4 [4.292] | 597.0 [3.857] | ||
| CH2Cl2 | 420.7 [5.718] | 548.2 [4.342] | 585.9 [3.681] | ||
| DMF | 427.1 [5.744] | 559.2 [4.326] | 599.7 [3.833] | ||
| DMSO | 429.6 [5.756] | 561.0 [4.334] | 601.2 [4.049] | ||
| Chx | 418.0 [5.733] | 549.4 [4.292] | 601.8 [3.964] | ||
| Cu(II)- 5, 10, 15, 20-meso-tetrakis-(4-acetoxy-3-methoxyphenyl) porphyrin | |||||
| MeOH | 412.6 [5.595] | 537.4 [4.292] | |||
| EtOH | 413.1 [5.639] | 537.3 [4.107] | |||
| iso-PrOH | 413.3 [5,623] | 537.3 [4.265] | |||
| CH2Cl2 | 416.2 [5.528] | 539.4 [4.182] | |||
| DMF | 417.3 [5.584] | 539.9 [4.301] | |||
| DMSO | 419.6 [5.525] | 541.7 [4.255] | |||
| Chx | 414.9 [5.674] | 538.7 [4.318] | |||
| Zn(II)-5-(4-hydroxyphenyl)-10,15,20–tris-(4-acetoxy-3-methoxyphenyl) porphyrin | |||||
| MeOH | 423.0 [5.706] | 556.1 [4.236] | 597.2 [3.806] | ||
| EtOH | 424.5 [5.725] | 557.4 [4.283] | 597.5 [3.880] | ||
| iso-PrOH | 424.8 [5.735] | 557.6 [4.274] | 597.5 [3.833] | ||
| CH2Cl2 | 421.4 [5.678] | 548.5 [3.301] | 587.4 [3.602] | ||
| DMF | 427.7 [5.693] | 559.8 [4.292] | 600.3 [3.982] | ||
| DMSO | 430.2 [5.648] | 561.4 [4.225] | 602.3 [3.924] | ||
| Chx | 418.4 [5.674] | 546.6 [4.334] | 601.0 [3.84] | ||
| Cu(II)-5-(4-hydroxyphenyl)-10,15,20–tris-(4-acetoxy-3-methoxyphenyl) porphyrin | |||||
| MeOH | 413.3 [5.491] | 538.0 [4.204] | |||
| EtOH | 413.6 [5.515] | 537.6 [4.182] | |||
| iso-PrOH | 414.0 [5,505] | 537.9 [4.170] | |||
| CH2Cl2 | 416.4 [5.483] | 539.5 [4.134] | |||
| DMF | 418.1 [5.465] | 540.3 [4.215] | |||
| DMSO | 420.8 [5.389] | 542.5 [4.167] | |||
| Chx | 415.2 [5.534] | 542.5 [4.158] | |||

| Solvent | λmax (nm) [If] (a.u.) | |
|---|---|---|
| Q(0,0) | Q(0,1) | |
| Zn(II)-5,10,15,20-meso-tetrakis-(4-acetoxy-3-methoxyphenyl)porphyrin | ||
| MeOH | 602.1 [495.5] | 650.0 [49.8] |
| EtOH | 602.3 [534.2] | 650.5 [112.7] |
| iso-PrOH | 602.6 [537.8] | 651.0 [116.2] |
| CH2Cl2 | 600.3 [476.7] | 642.4 [139.7] |
| DMF | 605.2 [533.1] | 653.6 [89.2] |
| DMSO | 607.0 [399.2] | 656.5 [63.4] |
| Chx | 605.8 [307.6] | 653.5 [196.0] |
| Zn(II)-5-(4-hydroxyphenyl)-10,15,20–tris-(4-acetoxy-3-methoxyphenyl)porphyrin | ||
| MeOH | 602.3 [480.2] | 650.0 [93.9] |
| EtOH | 602.5 [472.0] | 650.3 [93.9] |
| iso-PrOH | 602.7 [413.3] | 650.8 [86.9] |
| CH2Cl2 | 600.6 [432.5] | 643.5 [128.0] |
| DMF | 603.1 [544.8] | 651.8 [112.7] |
| DMSO | 607.6 [394.5] | 654.7 [62.2] |
| Chx | 603.9 [430.9] | 651.0 [89.2] |
3. Experimental
3.1. General
3.2. Synthesis of Zinc Mesoporphyrinic Complexes
3.3. Synthesis of Copper Mesoporphyrinic Complexes
4. Conclusions
Acknowledgments
References and Notes
- Detty, M.R.; Gibson, S.L.; Wagner, S.J. Current clinical and preclinical photosensitizers for use in photodynamic therapy. J. Med. Chem. 2004, 47, 3897–3195. [Google Scholar] [CrossRef]
- Kamuhabwa, A.; Agostinis, P.; Ahmed, B.; Landuyt, W.; van Cleynenbreugel, B.; van Poppel, H.; de Witte, P. Hypericin as a potential phototherapeutic agent in superficial transitional cell carcinoma of the bladder. Photochem. Photobiol. Sci. 2004, 3, 772–780. [Google Scholar] [CrossRef]
- Hilderbrand, S.; Weissleder, R. Near-infrared fluorescence: Application to in vivo molecular imaging. Curr. Opin. Chem. Biol. 2010, 14, 71–79. [Google Scholar] [CrossRef]
- Chatterjee, D.K.; Fong, L.S.; Zhang, Y. Nanoparticles in photodynamic therapy: An emerging paradigm. Adv. Drug Deliv. Rev. 2008, 60, 1627–1637. [Google Scholar] [CrossRef]
- Bonneau, S.; Bizet, C.V.; Mojzisova, H.; Brault, D. Tetrapyrrole-photosensitizers vectorization and plasma LDL: A physic-chemical approach. Int. J. Pharm. 2007, 344, 78–87. [Google Scholar] [CrossRef]
- Chin, W.W.L.; Lau, O.W.K.; Bhuvaneswari, R.; Heng, P.W.S.; Olivo, M. Chlorin e6-polyvinylpyrrolidone as a fluorescent marker for fluorescence diagnosis of humanbladder cancer implanted on the chick chorioallantoic membrane model. Cancer Lett. 2007, 245, 127–133. [Google Scholar] [CrossRef]
- Stockert, J.C.; Cañete, M.; Juarranz, A.; Villanueva, A.; Horobin, R.W.; Borrell, J.I.; Teixidó, J.; Nonell, S. Porphycenes: Facts and prospects in photodynamic therapy of cancer. Curr. Med. Chem. 2007, 14, 997–1026. [Google Scholar] [CrossRef]
- Banfi, S.; Caruso, E.; Caprioli, S.; Mazzagatti, L.; Canti, G.; Ravizza, R.; Gariboldia, M.; Montia, E. Photodynamic effects of porphyrin and chlorin photosensitizers in human colon adenocarcinoma cells. Bioorg. Med. Chem. 2004, 12, 4853–4860. [Google Scholar] [CrossRef]
- Grosseweiner, L.I. The Science of Phototherapy; CRC Press: London, UK, 1994; pp. 139–155, Chapter 8. [Google Scholar]
- Schweiter, C.; Schmidt, R. Physical mechanisms of generation and deactivation of singlet oxygen. Chem. Rev. 2003, 103, 1685–1758. [Google Scholar]
- Berg, K.; Selbo, P.K.; Weyergang, A.; Dietze, A.; Prasmickaite, L.; Bonsted, A. Porphyrin related photosensitizers for cancer imaging and therapeutic applications. J. Microsc. 2005, 218, 133–147. [Google Scholar] [CrossRef]
- Rosenkranz, A.A.; Jans, D.A.; Sobolev, A.S. Targeted intracellular delivery of photosensitizers to enhance photodynamic efficiency. Immunol. Cell Biol. 2002, 78, 452–464. [Google Scholar]
- Osterloh, J.; Vicente, M.G.H. Mechanisms of porphyrinoid localization in tumors. J. Porph. Phthal. 2002, 5, 305–325. [Google Scholar] [CrossRef]
- Milgrom, L.; MacRobert, S. Light years ahead. Chem. Br. 1998, 34, 45–50. [Google Scholar]
- Sharma, R.K.; Ahuja, G.; Sidhwani, I.T. A new one pot and solvent-free synthesis of nickel porphyrin complex. Green Chem. Lett. Rev. 2009, 2, 101–105. [Google Scholar] [CrossRef]
- Collman, J.P.; Decreau, R.A. Microwave-assisted synthesis of corroles. Tetrahedron Lett. 2003, 44, 1207–1210. [Google Scholar] [CrossRef]
- Kappe, C.O.; Dallinger, D. The impact of microwave synthesis on drug discovery. Nat. Rev. Drug Discov. 2006, 5, 51–63. [Google Scholar]
- Bougrin, K.; Loupy, A.; Soufiaoui, M. Microwave-assisted solvent-free heterocyclic synthesis. J. Photochem. Photobiol. C: Photochem. 2005, 6, 139–167. [Google Scholar] [CrossRef]
- Buchler, J.W. Static Coordination Chemistry of Metalloporphyrins. In Porphyrins and Metalloporphyrins; Smith, K.M., Ed.; Elsevier: Amsterdam, The Netherlands, 1975; pp. 157–224, Chapter 5. [Google Scholar]
- Boscencu, R.; Socoteanu, R.; Oliveira, A.S.; Vieira Ferreira, L.F.; Nacea, V.; Patrinoiu, G. Synthesis and characterization of some unsymmetrically-substituted mesoporphyrinic mono-hydroxyphenyl complexes of Copper(II). Pol. J. Chem. 2008, 82, 509–522. [Google Scholar]
- Boscencu, R.; Socoteanu, R.; Oliveira, A.S.; Ferreira, L.F.V. Studies on Zn(II) monohydroxyphenyl mesoporphyrinic complexes. Synthesis and characterization. J. Serb. Chem. Soc. 2008, 73, 713–726. [Google Scholar] [CrossRef]
- Machado, A.E.; Gomes, W.R.; Araújo, D.M.; Miglio, H.S.; Ueno, L.T.; Paula, R.D.; Cavaleiro, J.A.; Neto, N.M.B. Synthesis and spectroscopic characterization of two tetrasubstituted cationic porphyrin derivatives. Molecules 2011, 16, 5807–5821. [Google Scholar]
- Liu, M.O.; Tai, C.H.; Hu, A.T. Synthesis of metalloporphyrins by microwave irradiation and their fluorescent properties. Mater. Chem. Phys. 2005, 92, 322–326. [Google Scholar] [CrossRef]
- Lindsey, J.S. Synthetic routes to meso-patterned porphyrins. Acc. Chem. Res. 2010, 43, 300–311. [Google Scholar] [CrossRef]
- Senge, M.O. Nucleophilic substitution as a tool for the synthesis of unsymmetrical porphyrins. Acc. Chem. Res. 2005, 38, 733–743. [Google Scholar] [CrossRef]
- Senge, M.O.; Shaker, Y.M.; Pintea, M.; Ryppa, C.; Hatscher, S.S.; Ryan, A.; Sergeeva, Y. Synthesis of meso-substituted ABCD-type porphyrins by functionalization reactions. Eur. J. Org. Chem. 2010, 2, 237–258. [Google Scholar]
- Boscencu, R.; Ilie, M.; Socoteanu, R.; Oliveira, A.S.; Constantin, C.; Neagu, M.; Manda, G.; Vieira Ferreira, L.F. Microwave synthesis, basic spectral and biological evaluation of some Copper (II) mesoporphyrinic complexes. Molecules 2010, 15, 3731–3743. [Google Scholar] [CrossRef]
- Boscencu, R. Unsymmetrical mesoporphyrinic complexes of Copper (II) and Zinc (II). microwave-assisted synthesis, spectral characterization and cytotoxicity evaluation. Molecules 2011, 16, 5604–5617. [Google Scholar] [CrossRef]
- Gouterman, M. Optical Spectra and Electronic Structure of Porphyrins and Related Rings. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; Volume 3, pp. 11–87. [Google Scholar]
- Gouterman, M.; Wagniere, G.H.; Snyder, L.C. Spectra of porphyrins: Part II. Four orbital model. J. Mol. Spectrosc. 1963, 11, 108–127. [Google Scholar] [CrossRef]
- Lin, W.C. Electron Spin Resonance and Electronic Structure of Metalloporphyrins. In The Porphyrins; Dolphin, D., Ed.; Academic Press: New York, NY, USA, 1978; Volume 4, pp. 358–364. [Google Scholar]
- Manoharan, P.T.; Roger, M.T. ESR Study of Copper (II) and Silver (II) Tetraphenylporphyrin. In Electron Spin Resonance of Metal Complexes; Yen, T.F, Ed.; Plenum Press: New York, NY, USA, 1969; pp. 143–173. [Google Scholar]
- Kivelson, D.; Neiman, R.R. ESR studies on the bonding in copper complexes. J. Chem. Phys. 1961, 35, 149–155. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds Zn(II)TMAP, Cu(II)TMAP, Zn(II)TMAPOHP, Cu(II)TMAPOHP are available from the authors.
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boscencu, R. Microwave Synthesis Under Solvent-Free Conditions and Spectral Studies of Some Mesoporphyrinic Complexes. Molecules 2012, 17, 5592-5603. https://doi.org/10.3390/molecules17055592
Boscencu R. Microwave Synthesis Under Solvent-Free Conditions and Spectral Studies of Some Mesoporphyrinic Complexes. Molecules. 2012; 17(5):5592-5603. https://doi.org/10.3390/molecules17055592
Chicago/Turabian StyleBoscencu, Rica. 2012. "Microwave Synthesis Under Solvent-Free Conditions and Spectral Studies of Some Mesoporphyrinic Complexes" Molecules 17, no. 5: 5592-5603. https://doi.org/10.3390/molecules17055592
APA StyleBoscencu, R. (2012). Microwave Synthesis Under Solvent-Free Conditions and Spectral Studies of Some Mesoporphyrinic Complexes. Molecules, 17(5), 5592-5603. https://doi.org/10.3390/molecules17055592