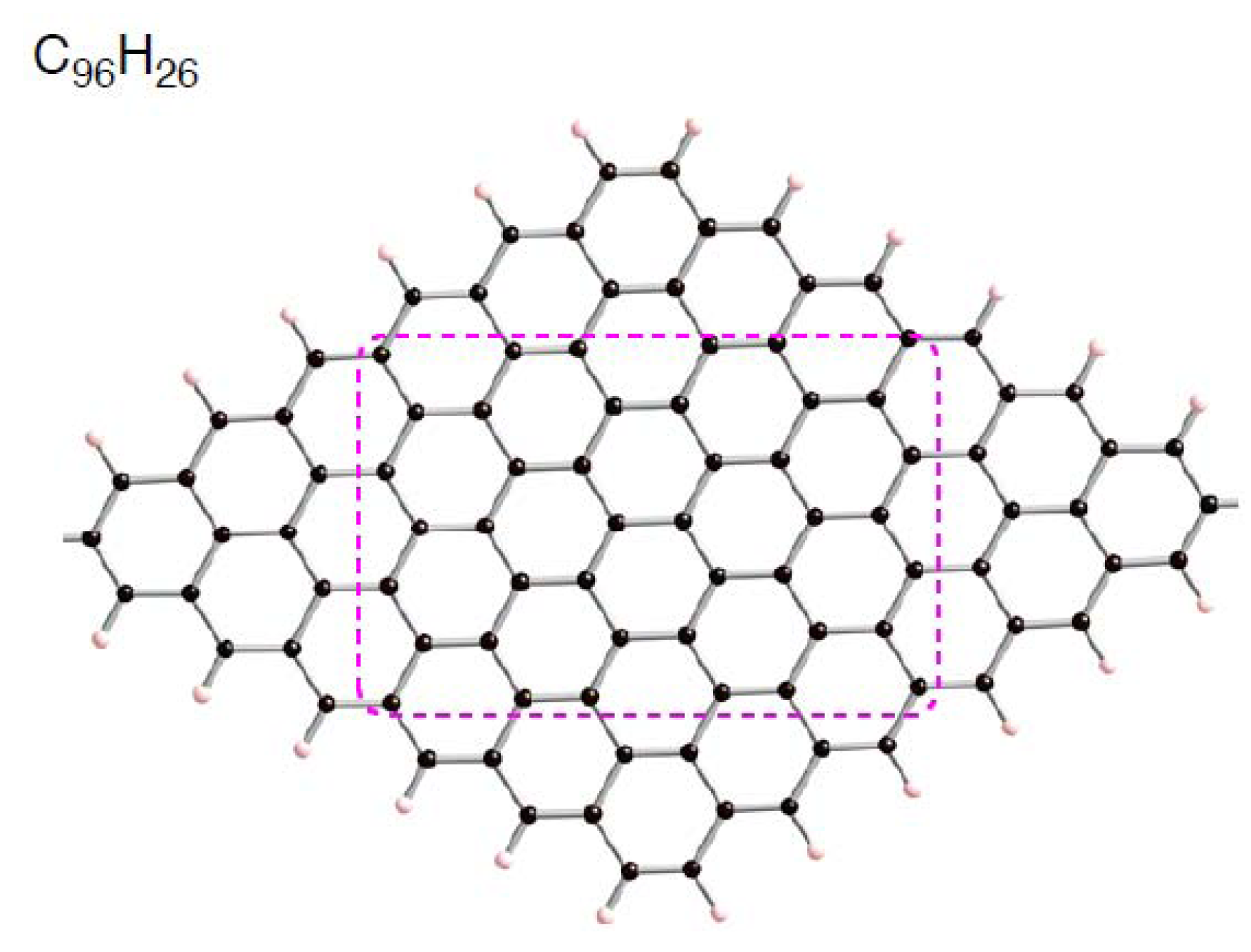
2.3.1. Singlet State
Despite the preferences of the triplet state of C
96–nH
26 over the singlet state, let us first use the singlet state to increase our understanding of how a Pt
k cluster interacts with a defective site on the sp
2 surface. Following the previous study on interactions between a Pt
6 cluster and pristine C
96H
26, we have a special interest on how the presence of a vacancy-type defect of the patch affects the interactions with a Pt
6 cluster. In addition, we will discuss dependences of the interaction energies on size of clusters whose number of contained Pt atoms (
k) being smaller than 6.
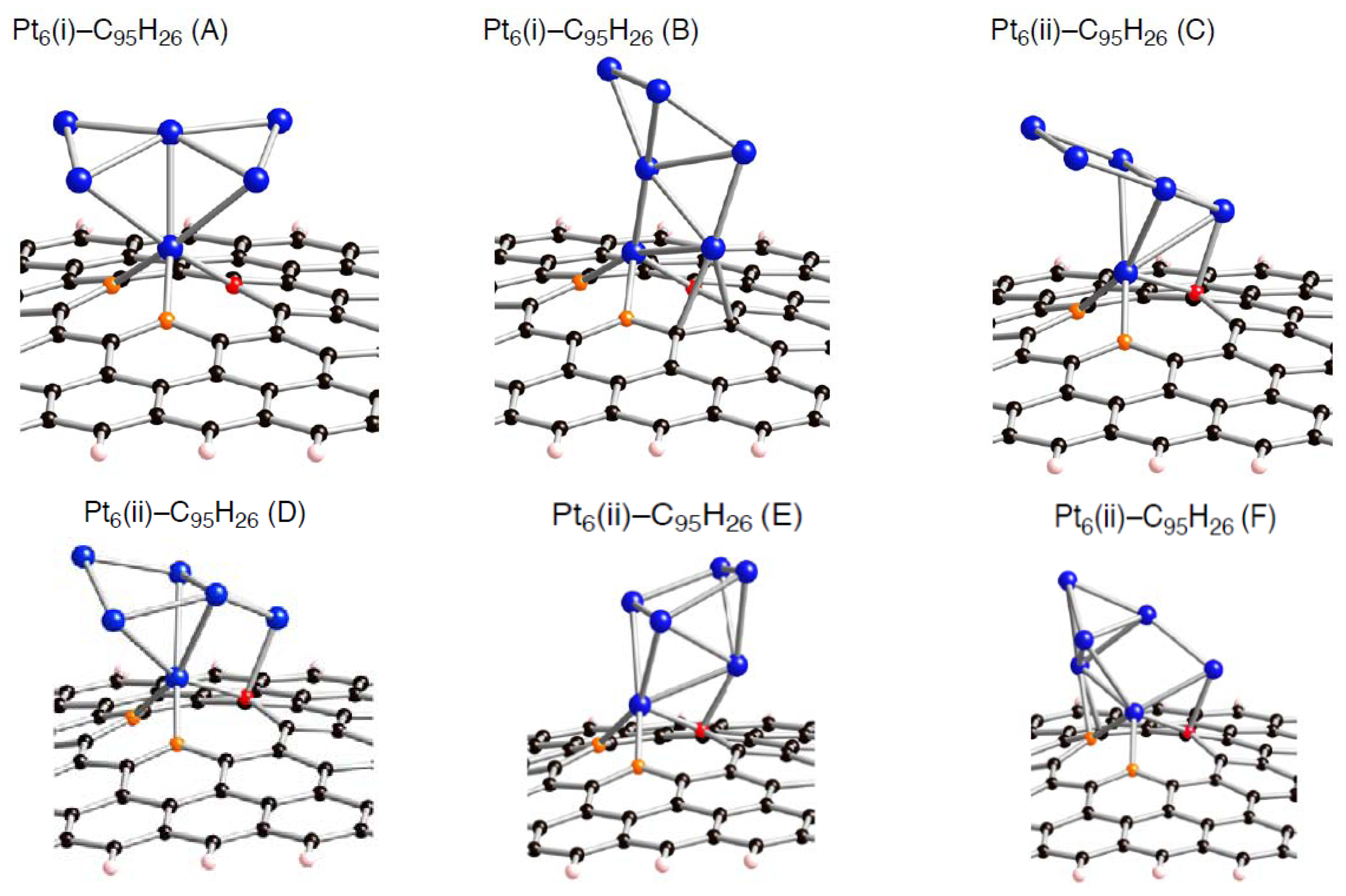
Figure 5–
Figure 7 show optimized structures for a Pt
6 cluster adsorbed on the mono-, di-, and tri-vacancy-type defects, respectively.
Several modes for the cluster bindings were considered. For example, we obtained six optimized geometries for a Pt
6 cluster binding into the mono-vacancy-type defect in
Figure 5. The four Pt
6-C
95H
26 structures displayed in
Figure 5 are relatively stable in energy. In general, stable Pt
k-C
95H
26 structures have a Pt
k-1 moiety contained in stable Pt
k-1-C
95H
26 structures. Of course, there are other possibilities for Pt
k binding modes. However, our computational resource is limited, reluctantly we did not obtain other optimized geometries. We evaluated the binding energy in each configuration defined as [
Ebind =
Etotal(Pt
k-C
96–nH
26) –
Etotal(C
96–nH
26) –
Etotal(Pt
k)], where
k ranges from 1 to 6 (
Table 3–
Table 8).
Figure 5.
Optimized geometries for Pt
6 cluster on the mono-vacancy-type defect in the singlet state (Pt
6-C
95H
26). Optimized bond lengths are given in
Table 3.
Figure 5.
Optimized geometries for Pt
6 cluster on the mono-vacancy-type defect in the singlet state (Pt
6-C
95H
26). Optimized bond lengths are given in
Table 3.
Table 3.
Key parameters of Pt
k on mono-vacancy defect (C
95H
26) (
k is 1 or 6) in
Figure 5. Separations of a Pt atom from orange atoms (Pt-C(orange)) and those from the red atom (Pt-C(red)). Separations of carbon atoms from a Pt atom except for the nearest Pt atom (other Pt-C), and those between the two orange atoms (C-C). Bond lengths are in Å. The
Ebind and Δ
E(Pt
k) values are given in kcal/mol. Their definition was given in the text.
Table 3.
Key parameters of Ptk on mono-vacancy defect (C95H26) (k is 1 or 6) in Figure 5. Separations of a Pt atom from orange atoms (Pt-C(orange)) and those from the red atom (Pt-C(red)). Separations of carbon atoms from a Pt atom except for the nearest Pt atom (other Pt-C), and those between the two orange atoms (C-C). Bond lengths are in Å. The Ebind and ΔE(Ptk) values are given in kcal/mol. Their definition was given in the text.
| | Ebind | Pt-C(orange) | Pt-C(red) | other Pt-C | C-C Bond | ΔE(Ptk) |
|---|
| Pt1 | –145.7 | 1.953, 1.954 | 1.942 | –– | 2.764 | –– |
| Pt6(i)-(A) | –152.0 | 1.986, 1.988 | 1.968 | –– | 2.745 | 4.2 |
| Pt6(i)-(B) | –146.6 | 1.947, 1.968 | 1.958 | 2.215 | 2.768 | 16.8 |
| Pt6(ii)-(C) | –151.6 | 1.980, 1.981 | 1.991 | 2.074 | 2.728 | 17.4 |
| Pt6(ii)-(D) | –157.1 | 1.983, 1.983 | 2.010 | 2.114 | 2.729 | 8.1 |
| Pt6(ii)-(E) | –136.7 | 1.978, 1.976 | 1.991 | 2.200, 2.083, 2.252 | 2.803 | 15.3 |
| Pt6(ii)-(F) | –143.4 | 1.986, 1.977 | 1.982 | 2.113 | 2.740 | 6.3 |
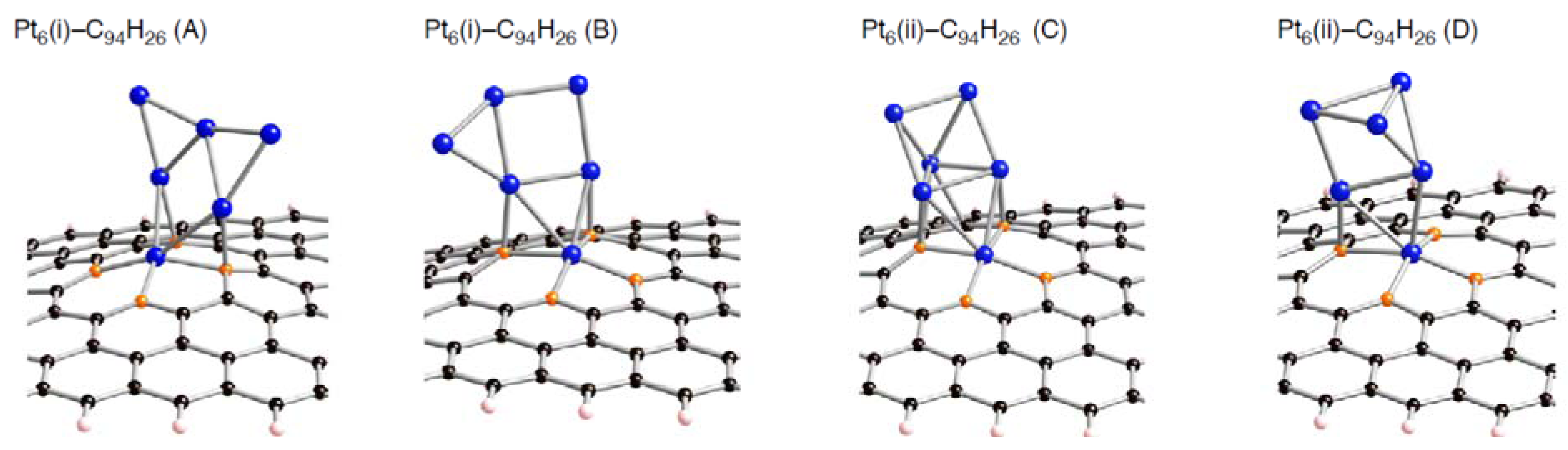
Figure 6.
Optimized geometries for Pt
6 cluster on the di-vacancy-type defect in the singlet state (Pt
6-C
94H
26). Optimized bond lengths are given in
Table 4.
Figure 6.
Optimized geometries for Pt
6 cluster on the di-vacancy-type defect in the singlet state (Pt
6-C
94H
26). Optimized bond lengths are given in
Table 4.
Table 4.
Key parameters of Pt
k on di-vacancy defect (C
94H
26) (
k is 1 or 6) in
Figure 6. Separations of a Pt atom from orange atoms (Pt-C(orange)), those of carbon atoms from a Pt atom except for the nearest Pt atom (other Pt-C), and those between the two orange atoms (C-C). Bond lengths are in Å. The
Ebind and Δ
E(Pt
k) values are given in kcal/mol.
Table 4.
Key parameters of Ptk on di-vacancy defect (C94H26) (k is 1 or 6) in Figure 6. Separations of a Pt atom from orange atoms (Pt-C(orange)), those of carbon atoms from a Pt atom except for the nearest Pt atom (other Pt-C), and those between the two orange atoms (C-C). Bond lengths are in Å. The Ebind and ΔE(Ptk) values are given in kcal/mol.
| | Ebind | Pt-C(orange) | other Pt-C | C-C Bond | ΔE(Ptk) |
|---|
| Pt1 | –106.1 | 1.999, 1.985, 1.999, 1.985 | –– | 2.846, 2.846 | –– |
| Pt6(i)-(A) | –106.7 | 2.010, 2.001, 2.091, 2.117 | 2.088,2.033 | 2.810, 2.940 | 12.7 |
| Pt6(i)-(B) | –106.3 | 2.014, 2.101, 2.109, 2.008 | 2.087,2.108 | 2.918, 2.921 | 14.8 |
| Pt6(ii)-(C) | –94.5 | 2.014, 2.119, 2.088, 2.005 | 2.206,2.314, 2.084, 2.119 | 2.928, 2.921 | 17.3 |
| Pt6(ii)-(D) | –85.1 | 1.994, 2.021, 2.036, 2.114 | 2.040 | 2.836, 2.941 | 17.9 |
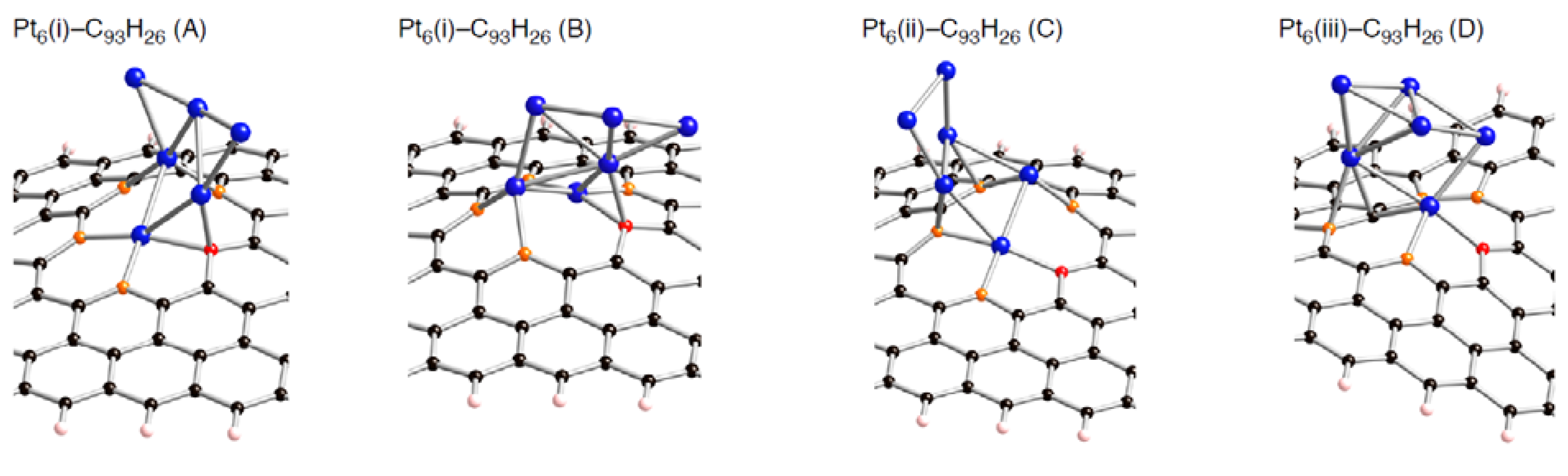
Figure 7.
Optimized geometries for Pt
6 cluster on the tri-vacancy-type defect in the triplet state (Pt
6-C
93H
26). Optimized bond lengths are given in
Table 5.
Figure 7.
Optimized geometries for Pt
6 cluster on the tri-vacancy-type defect in the triplet state (Pt
6-C
93H
26). Optimized bond lengths are given in
Table 5.
Table 5.
Key parameters of Pt
k on tri-vacancy defect (C
93H
26) (
k is 1 or 6) in
Figure 7. Separations of a Pt atom from orange atoms (Pt-C(orange)) and those from the red atom (Pt-C(red)). Separations of carbon atoms from a Pt atom except for the nearest Pt atom (other Pt-C), and those between the two orange atoms (C-C). Bond lengths are in Å. The
Ebind and Δ
E(Pt
k) values are given in kcal/mol.
Table 5.
Key parameters of Ptk on tri-vacancy defect (C93H26) (k is 1 or 6) in Figure 7. Separations of a Pt atom from orange atoms (Pt-C(orange)) and those from the red atom (Pt-C(red)). Separations of carbon atoms from a Pt atom except for the nearest Pt atom (other Pt-C), and those between the two orange atoms (C-C). Bond lengths are in Å. The Ebind and ΔE(Ptk) values are given in kcal/mol.
| | Ebind | Pt-C(orange) | Pt-C(red) | other Pt-C | C-C Bond | ΔE (Ptk) |
|---|
| Pt1 | –162.6 | 2.114, 2.333, 2.097, 2.641 | 2.059 | –– | 2.637, 2.641 | –– |
| Pt6(i)-(A) | –198.2 | 1.992, 1.959, 1.971, 1.969 | 2.105 | 2.054 | 2.753, 2.830 | 12.2 |
| Pt6(i)-(B) | –193.4 | 2.006, 1.988, 1.974, 1.938 | 2.144 | 2.130 | 2.825, 2.746 | 5.9 |
| Pt6(ii)-(C) | –188.4 | 1.971, 2.081, 1.970, 1.969 | 2.046 | 2.378, 2.217, 2.107 | 2.924, 2.922 | 20.0 |
| Pt6(ii)-(D) | –142.0 | 2.006, 2.015 | 2.021 | 2.130, 2.231 | 2.710 | 17.9 |
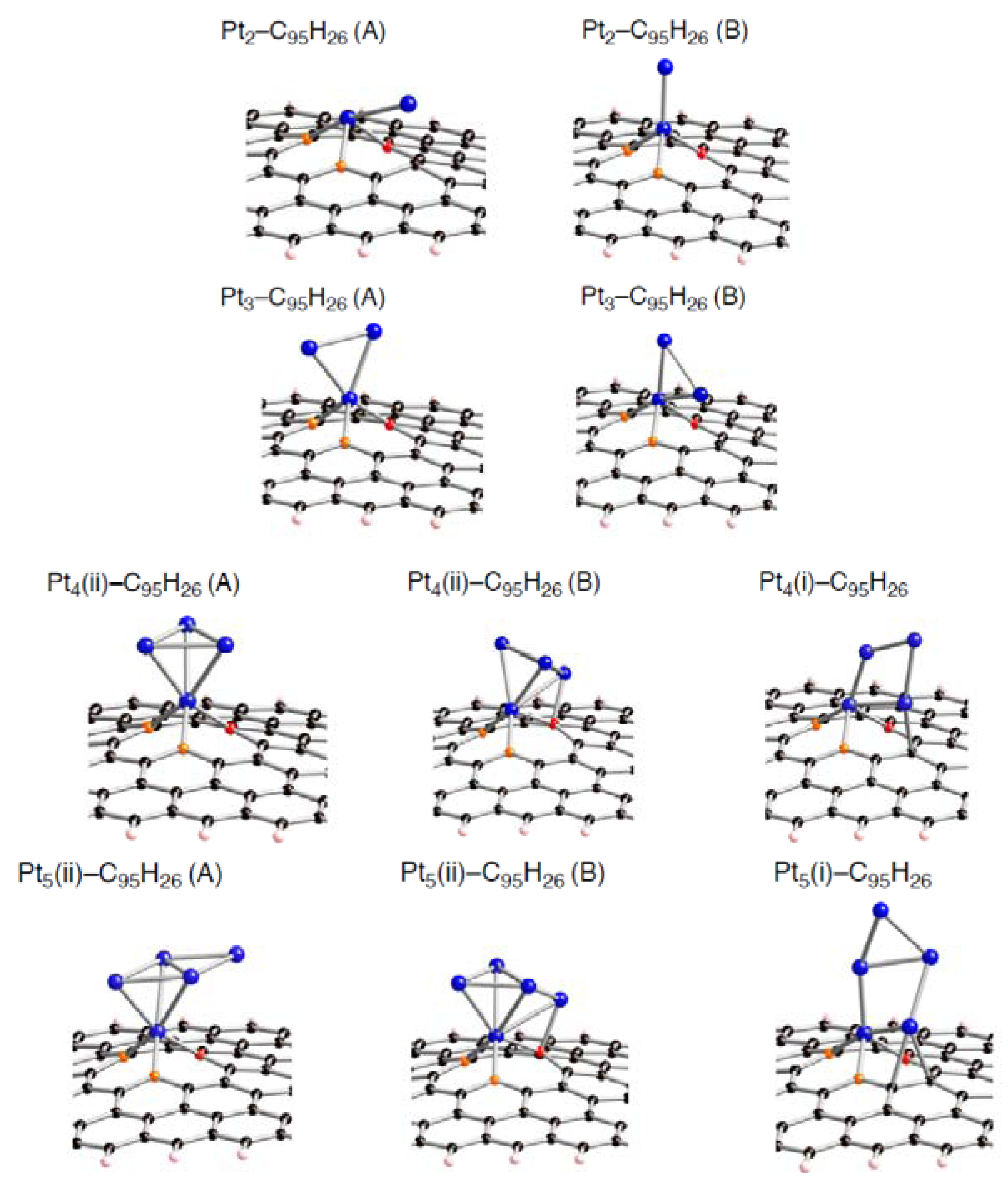
Corresponding optimized structures for the smaller Pt
k clusters adsorbed (
k = 2, 3, 4, and 5) are also displayed in
Figure 8–
Figure 10.
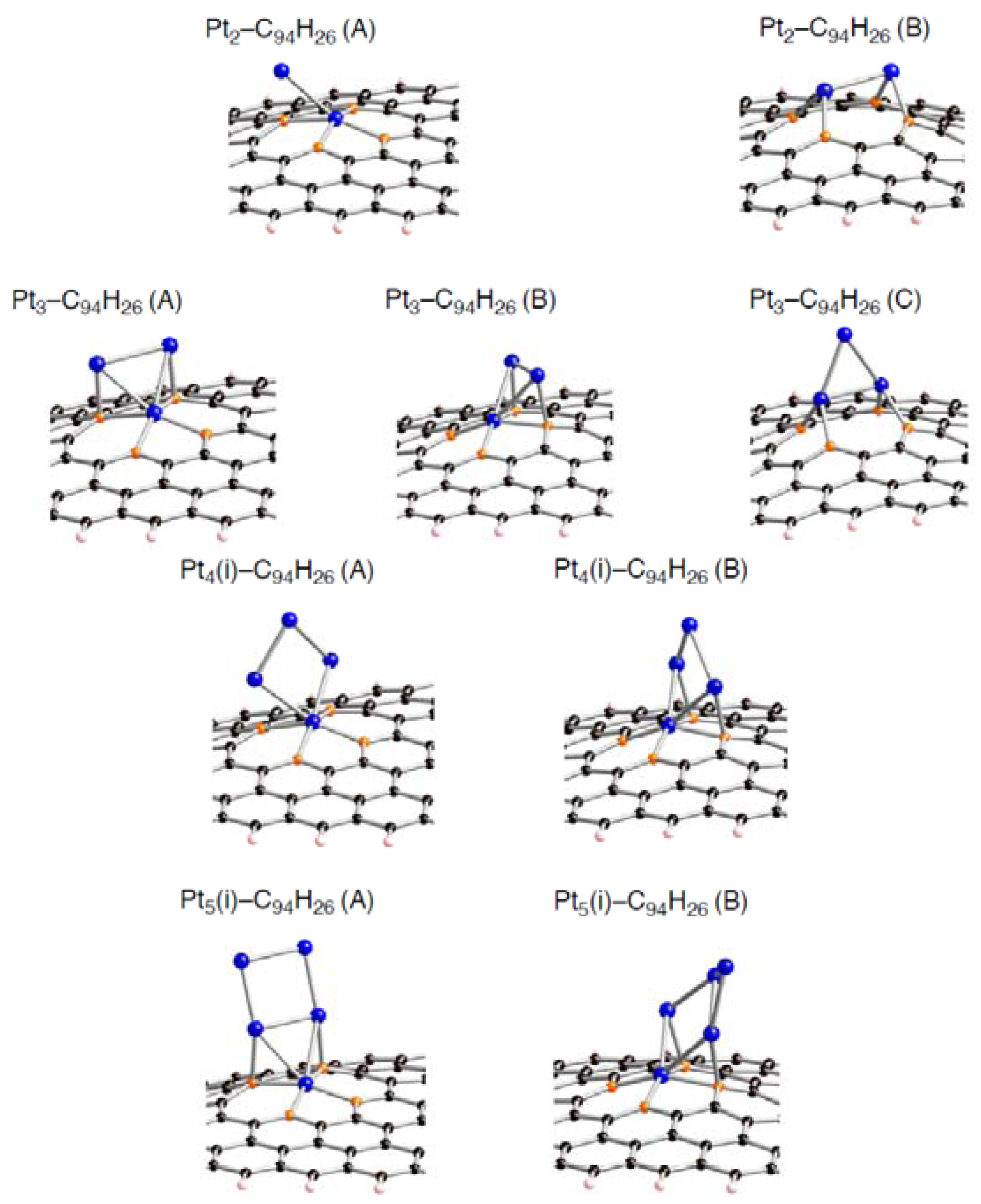
Figure 8.
Optimized geometries for Pt
k cluster (
k = 2~5) on the mono-vacancy-type defect in the singlet state (Pt
k-C
95H
26). Optimized bond lengths are given in
Table 6.
Figure 8.
Optimized geometries for Pt
k cluster (
k = 2~5) on the mono-vacancy-type defect in the singlet state (Pt
k-C
95H
26). Optimized bond lengths are given in
Table 6.
Table 6.
Key parameters of Pt
k on mono-vacancy defect (C
95H
26) (
k = 2 ~ 5) in
Figure 8.
Table 6.
Key parameters of Ptk on mono-vacancy defect (C95H26) (k = 2 ~ 5) in Figure 8.
| | Ebind | Pt-C(orange) | Pt-C(red) | other Pt-C | C-C Bond | ΔE(Ptk) |
|---|
| Pt2-(A) | –131.7 | 1.964, 2.006 | 1.969 | –– | 2.755 | 29.6 |
| Pt2-(B) | –146.1 | 1.965, 1.965 | 1.942 | –– | 2.743 | 9.0 |
| Pt3-(A) | –132.4 | 1.971, 1.972 | 1.971 | –– | 2.715 | 0.9 |
| Pt3-(B) | –126.9 | 1.952, 1.976 | 1.962 | –– | 2.790 | 2.5 |
| Pt4(ii)-(A) | –139.7 | 1.983, 1.984 | 1.967 | –– | 2.759 | 7.4 |
| Pt4(ii)-(B) | –140.0 | 1.981, 1.987 | 1.987 | 2.054 | 2.743 | 7.4 |
| Pt4(i) | –128.3 | 1.953, 1.966 | 1.960 | 2.227 | 2.757 | 19.8 |
| Pt5(ii)-(A) | –140.5 | 1.976, 1.976 | 1.980 | –– | 2.753 | 4.3 |
| Pt5(ii)-(B) | –147.8 | 1.982, 1.982 | 2.002 | 2.105 | 2.741 | 5.4 |
| Pt5(i) | –132.4 | 1.947, 1.966 | 1.972 | 2.211, 2.275 | 2.790 | 4.6 |
Figure 9.
Optimized geometries for Pt
k cluster (
k = 2~5) on the di-vacancy-type defect in the singlet state (Pt
k-C
94H
26). Optimized bond lengths are given in
Table 7.
Figure 9.
Optimized geometries for Pt
k cluster (
k = 2~5) on the di-vacancy-type defect in the singlet state (Pt
k-C
94H
26). Optimized bond lengths are given in
Table 7.
Table 7.
Key parameters of Pt
k on di-vacancy defect (C
94H
26) (
k = 2 ~ 5) in
Figure 9.
Table 7.
Key parameters of Ptk on di-vacancy defect (C94H26) (k = 2 ~ 5) in Figure 9.
| | Ebind | Pt-C(orange) | other Pt-C | C-C Bond | ΔE(Ptk) |
|---|
| Pt2-(A) | –91.2 | 1.999, 2.028, 2.038, 2.066 | –– | 2.831, 2.904 | 10.6 |
| Pt2-(B) | –59.2 | 1.934, 1.935, 1.997, 1.997 | –– | 2.786, 2.740 | 7.1 |
| Pt3-(A) | –70.9 | 2.025, 2.118, 2.025, 2.119 | 2.045, 2.046 | 2.893, 2.893 | 9.6 |
| Pt3-(B) | –63.2 | 1.990, 2.013, 2.125, 2.199 | 2.081, 2.082 | 2.791, 2.976 | 7.0 |
| Pt3-(C) | –57.6 | 1.972, 1.987, 1.971, 1.986 | –– | 2.690, 2.690 | 1.1 |
| Pt4(i)-(A) | –84.8 | 2.034, 2.035, 2.101, 2.100 | 2.053, 2.054 | 2.852, 2.853 | 6.4 |
| Pt4(i)-(B) | –75.8 | 2.004, 2.005, 2.064, 2.134 | 2.067, 2.066 | 2.786, 2.896 | 4.7 |
| Pt5(i)-(A) | –93.4 | 2.005, 2.006, 2.085, 2.099 | 2.122, 2.138 | 2.917, 2.923 | 12.3 |
| Pt5(i)-(B) | –81.3 | 1.991, 2.001, 2.113, 2.123 | 2.053, 2.082 | 2.806, 2.932 | 13.4 |
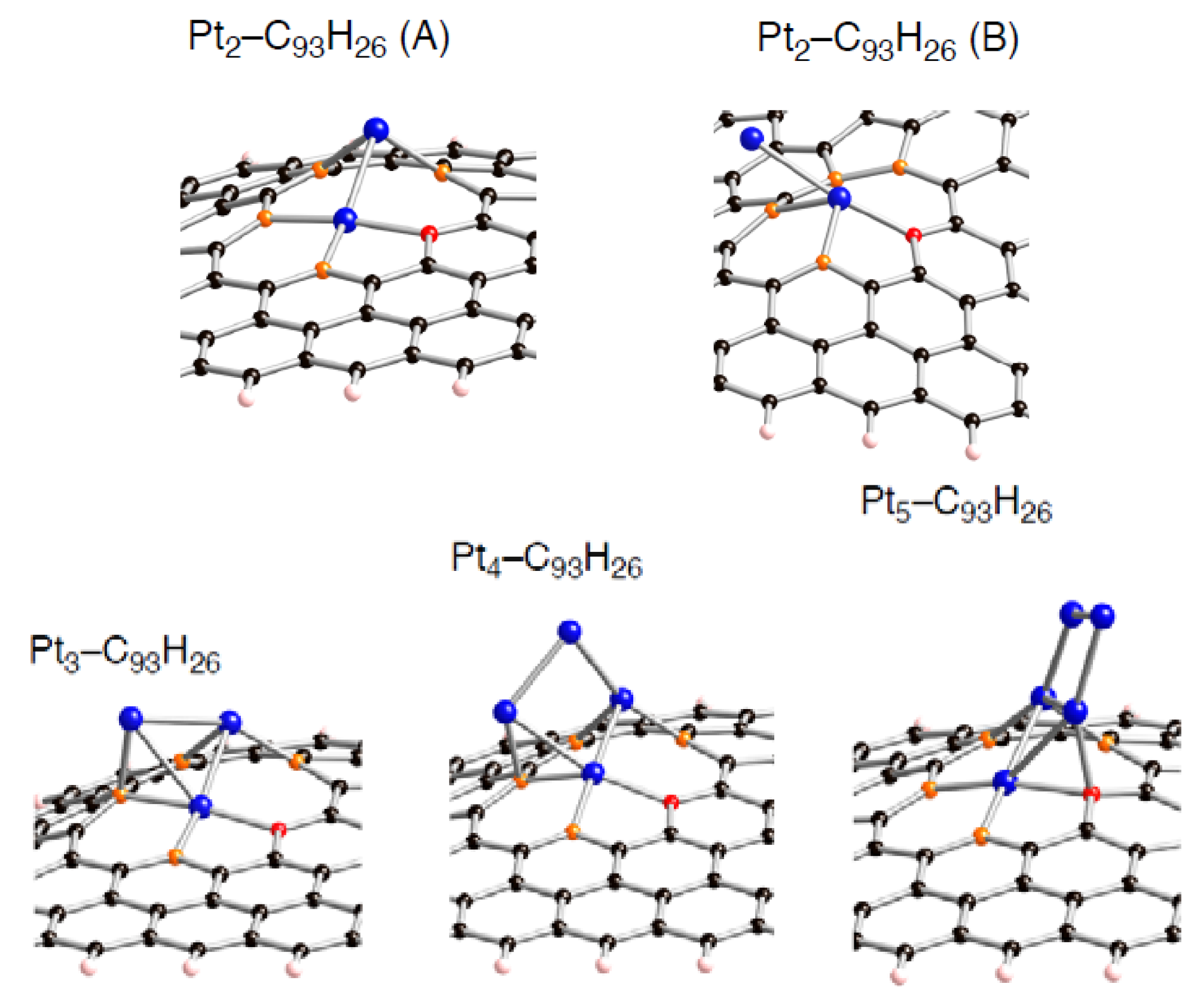
Figure 10.
Optimized geometries for Pt
k cluster (
k = 2~5) on the tri-vacancy-type defect in the singlet state (Pt
k-C
93H
26). Optimized bond lengths are given in
Table 8.
Figure 10.
Optimized geometries for Pt
k cluster (
k = 2~5) on the tri-vacancy-type defect in the singlet state (Pt
k-C
93H
26). Optimized bond lengths are given in
Table 8.
Table 8.
Key parameters of Pt
k on tri-vacancy defect (C
93H
26) (
k = 2 ~ 5) in
Figure 10. Separations of a Pt atom from orange atoms (Pt-C(orange)) and those from the red atom (Pt-C(red)). Separations of carbon atoms from a Pt atom except for the nearest Pt atom (other Pt-C), and those between the two orange atoms (C-C). Bond lengths are in Å. The
Ebind and Δ
E(Pt
k) values are given in kcal/mol. Their definition was given in the text.
Table 8.
Key parameters of Ptk on tri-vacancy defect (C93H26) (k = 2 ~ 5) in Figure 10. Separations of a Pt atom from orange atoms (Pt-C(orange)) and those from the red atom (Pt-C(red)). Separations of carbon atoms from a Pt atom except for the nearest Pt atom (other Pt-C), and those between the two orange atoms (C-C). Bond lengths are in Å. The Ebind and ΔE(Ptk) values are given in kcal/mol. Their definition was given in the text.
| | Ebind | Pt-C(orange) | Pt-C(red) | other Pt-C | C--C Bond | ΔE(Ptk) |
|---|
| Pt2-(A) | –172.3 | 1.944, 1.968, 1.988, 1.950 | 2.038 | –– | 2.839, 2.852 | 22.0 |
| Pt2-(B) | –132.0 | 2.002, 1.985 | 2.009 | 2.021 | 2.874, 1.630 | 8.3 |
| Pt3 | –167.5 | 1.948, 1.964, 1.989, 2.124 | 2.021 | 2.067 | 2.999, 2.832 | 9.8 |
| Pt4 | –180.3 | 1.978, 2.068, 1.973, 1.981 | 2.046 | 2.050 | 2.897, 2.704 | 3.6 |
| Pt5 | –173.0 | 1.950, 1.976, 1.934, 1.997 | 2.099 | –– | 2.840, 2.750 | 13.9 |
Here
Etotal(Pt
k-C
96–nH
26) is the total energy of an optimized C
96–nH
26 geometry,
Etotal(C
96–nH
26) is that of the optimized C
96–nH
26 geometry, and
Etotal(Pt
k) is that of the optimized Pt
k cluster. For the DFT calculations of the binding energies, a counterpoise correction for basis set superposition error (BSSE) was included [
55]. When an
Ebind value has a negative sign, the binding of a Pt cluster or the Pt atom into C
96–nH
26 is energetically preferable. As shown in
Table 3–
Table 5, the calculated
Ebind values in the single Pt addition are similar to those reported in [
35]. These similarities verify the reliability of our DFT results.
Looking at the
Ebind values, the bindings of a Pt
k cluster into the sp
2 surface are strongly facilitated by introducing vacancy-type defects. In fact, their stabilizing energies (–
Ebind) in stable Pt
k-C
95H
26 structures are around 150 kcal/mol. These values are much larger than the pristine cases (about 50 kcal/mol [
22]). Similar enhancement in the stabilization energies was also found in the Pt
k-C
94H
26 and Pt
k-C
93H
26 structures. Judging from the E
bind values, reactivity of vacancy-type defects toward Pt clusters declines in the order: tri-vacancy > mono-vacancy > di-vacancy. These results suggest that the tri-vacancy defect is more suitable for binding of Pt clusters into carbon surface rather than the mono- and di-vacancy defects.
From
Table 3–
Table 8, we see different behaviors between the three types of defect in terms of dependences of the
Ebind values on Pt cluster size. Most stable Pt
k-C
95H
26 structures except for Pt
3-C
95H
26 have E
bind values similar to that in Pt
1-C
95H
26. In contrast, the
Ebind values in Pt
k-C
94H
26 and Pt
k-C
93H
26 are deviated from those in Pt
1-C
94H
26 and Pt
1-C
93H
26. When a Pt
k cluster binds into the di-vacancy-defect, their E
bind values are smaller than the Pt
1-C
94H
26 value. However, these absolute values increase gradually with an increase of the cluster size, and seem to converge to the Pt
1-C
94H
26 value at
k = 6. In the Pt
k-C
93H
26 cases, the E
bind values, being around 180 kcal/mol, are always larger than the Pt
1-C
93H
26 value.
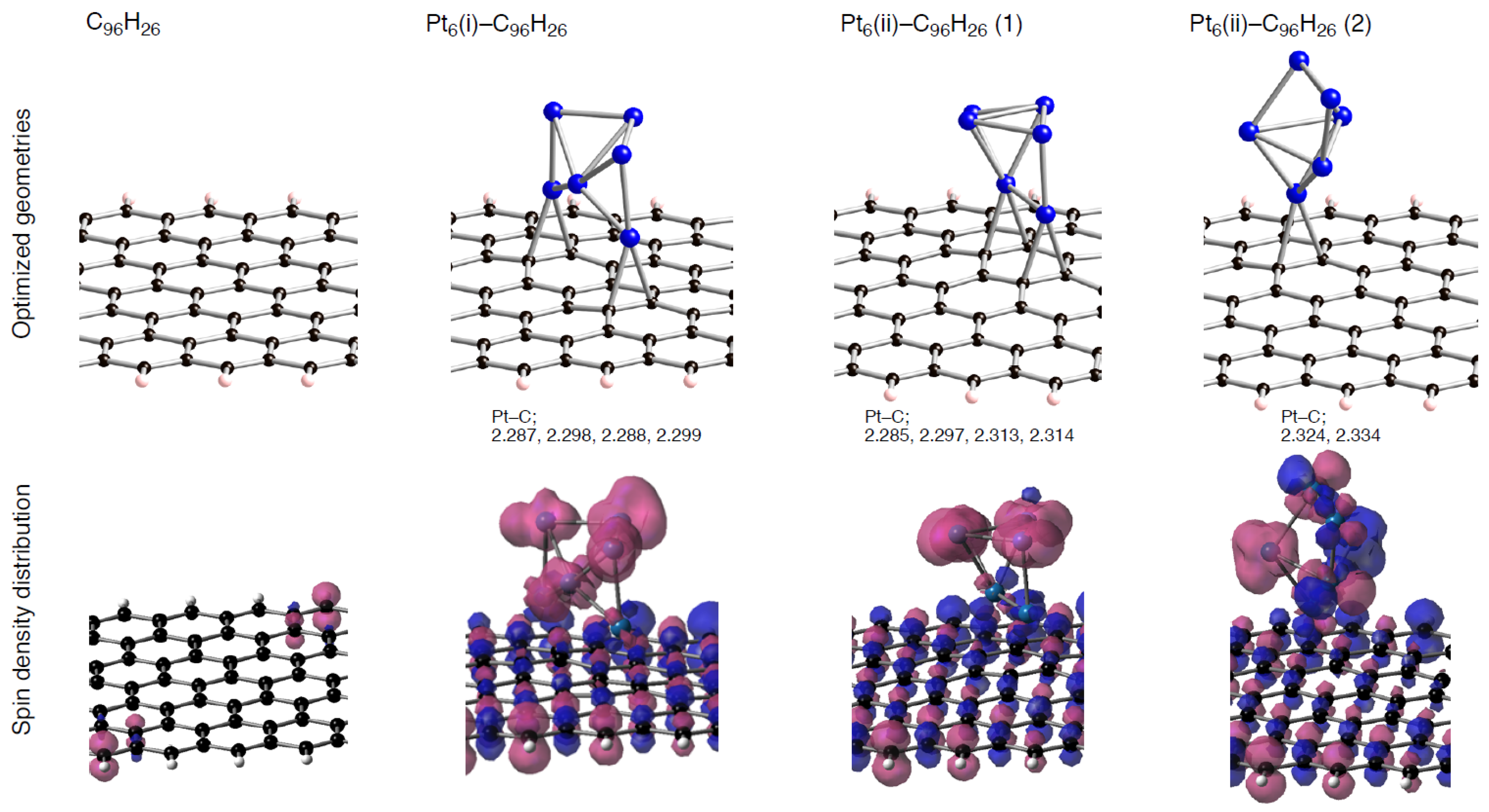
To understand the energetics in the optimized Pt
k-C
96–nH
26 structures (
Table 3–
Table 8), let us first look at in detail geometrical features of Pt
1-C
96–nH
26. These key geometrical parameters in the Pt
1-C
96-nH
26 configurations (lengths of newly formed Pt-C bonds and of lengthening CC bonds) are listed in
Table 3–
Table 5.
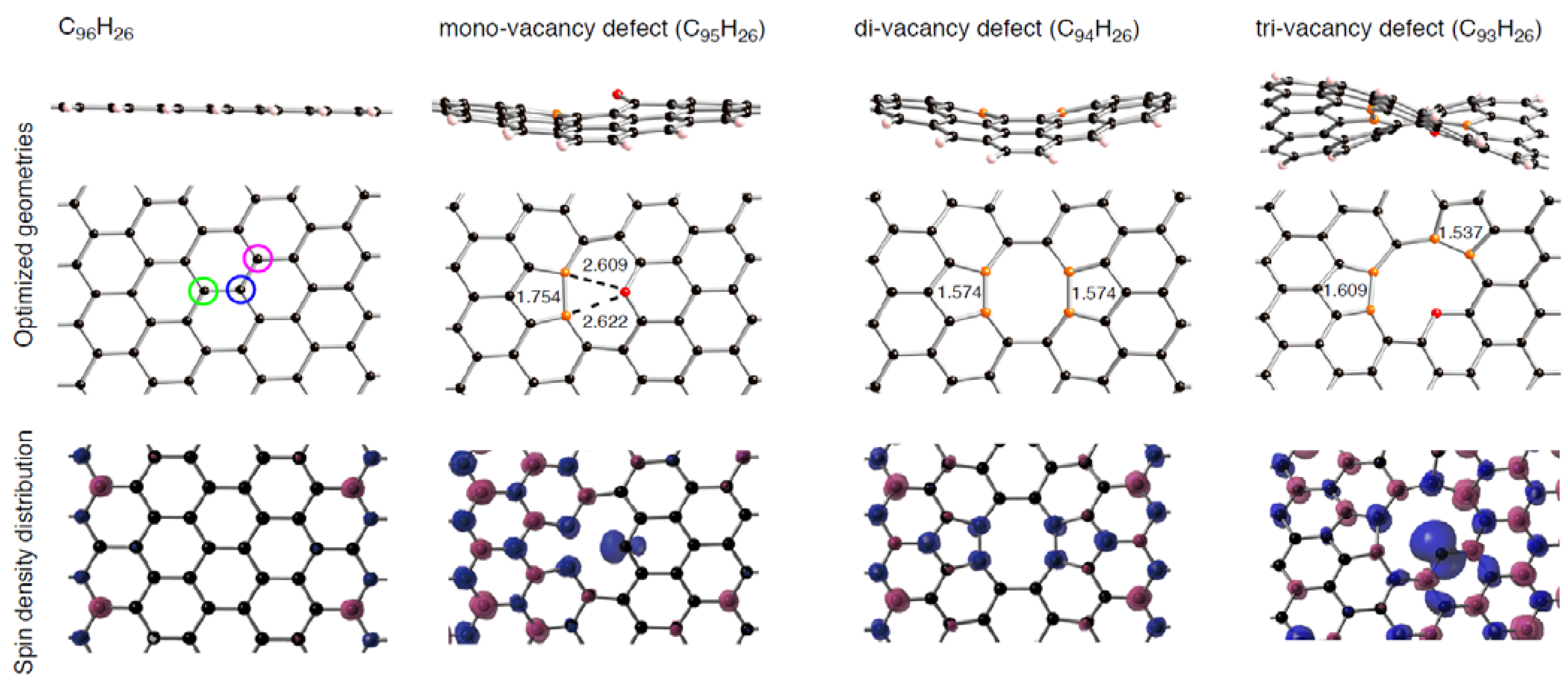
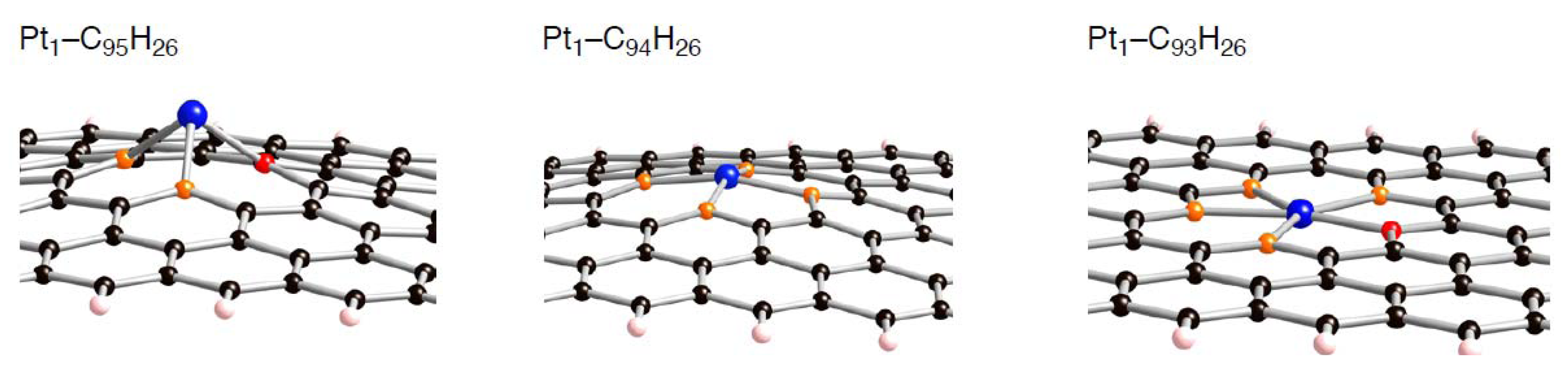
In these tables, we can distinguish two types of the formed Pt-C bond, by whether a Pt atom binds into orange or red atoms. When the single Pt atom binds into the mono-vacancy defect, it inserts between the orange atoms in the five-membered ring, and then two P-C(orange) bonds are formed newly. As a result of the Pt addition, the separation between the orange atoms lengthens from 1.754 to 2.764 Å. At the same time, the Pt atom also coordinates to the unsaturated red atom. The binding Pt atom lifts from the sp2 surface, because the hole is not large enough to accommodate the Pt atom. Similar Pt lifting can be seen in the Pt1-C94H26 configuration, where the Pt atom inserts between orange atoms in both five-membered rings, and it breaks the connections. The degree of Pt lifting in Pt1-C94H26 is less significant than that in Pt1-C95H26, due to relatively larger hole in C94H26.
In contrast, the hole of C93H26, surrounded by ten carbon atoms, can house the Pt atom, and therefore the binding Pt atom is on the sp2 surface. Then, four Pt-C bonds are formed, accompanying the cleavage of the bonds between orange atoms in the five-membered rings. Moreover, the Pt binding into the unsaturated C atom was also seen. When a Ptk cluster binds into a vacancy-type defect, slightly longer separations of the nearest Pt atom from reactive (orange and red) atoms were found. Despite the stabilization operated between a Ptk cluster and C96–nH26, slightly longer Pt-C bonds imply weakening interactions of the nearest Pt atom from the reactive carbon atoms compared with Pt1-C96–nH26 case.
Compensating the weakening of the interactions, remaining Pt atoms of a clusters are additionally bound to carbon atoms of a defective site to maximize the Pt-C interactions. Then their clusters are more or less deformed from the most stable configuration in the gas-phase [
56,
57,
58]. The degree of cluster deformation was estimated by using Δ
E(Pt
k), defined as [
E(Pt
k on surface) –
E(Pt
k)], where
E(Pt
k on surface) is the total energy of Pt
k cluster taken from an optimized Pt
k-C
96–nH
26 structure and
E(Pt
k) is that of the optimized geometry for the bare Pt
k cluster. Positive Δ
E(Pt
k) values in
Table 3–
Table 8 suggest destabilization from cluster deformation upon the interactions with a vacancy-type defect. Although we cannot find a clear correlation between E
bind and Δ
E(Pt
k) values, the balance between the stabilization from the Pt-C bond formation and the destabilization from the cluster deformation is a key in determining the stability. From
Figure 5–
Figure 7 and
Figure 11, we found clear differences between Pt
k-C
93H
26 and Pt
k-C
95H
26 (Pt
k-C
94H
26) in terms of the number of Pt atoms binding directly into orange atoms in the defective site to cleave connections between adjacent orange atoms.
Figure 11.
Optimized geometries for the singlet Pt atom on the mono-, di-, and tri-vacancy-type defects in the triplet state (Pt
1-C
95H
26, Pt
1-C
94H
26, and Pt
1-C
93H
26, respectively). Optimized bond lengths are given in
Table 3–
Table 5.
Figure 11.
Optimized geometries for the singlet Pt atom on the mono-, di-, and tri-vacancy-type defects in the triplet state (Pt
1-C
95H
26, Pt
1-C
94H
26, and Pt
1-C
93H
26, respectively). Optimized bond lengths are given in
Table 3–
Table 5.
In Pt
k-C
95H
26 (Pt
k-C
94H
26), one Pt atom participates in cleaving the orange connection(s), irrespective of the cluster size. In the tri-vacancy cases containing larger ten-membered ring, two Pt atoms in a cluster bind to the defective site to split two orange connections. The accommodation of two Pt atoms cannot be seen in the Pt
1-C
94H
26 structure, and thus the significant enhanced stabilization in Pt
k-C
93H
26 is understandable. Moreover the acceptability of the ten-membered-ring to trap Pt atoms differentiates C
93H
26 from C
95H
26 and C
94H
26 in terms of their reactivity toward Pt
k clusters. Due to the strong interactions between a Pt
6 cluster and a vacancy-type defect, we can see unique orbital features, which cannot be seen in C
96–nH
26 (
Figure 12).
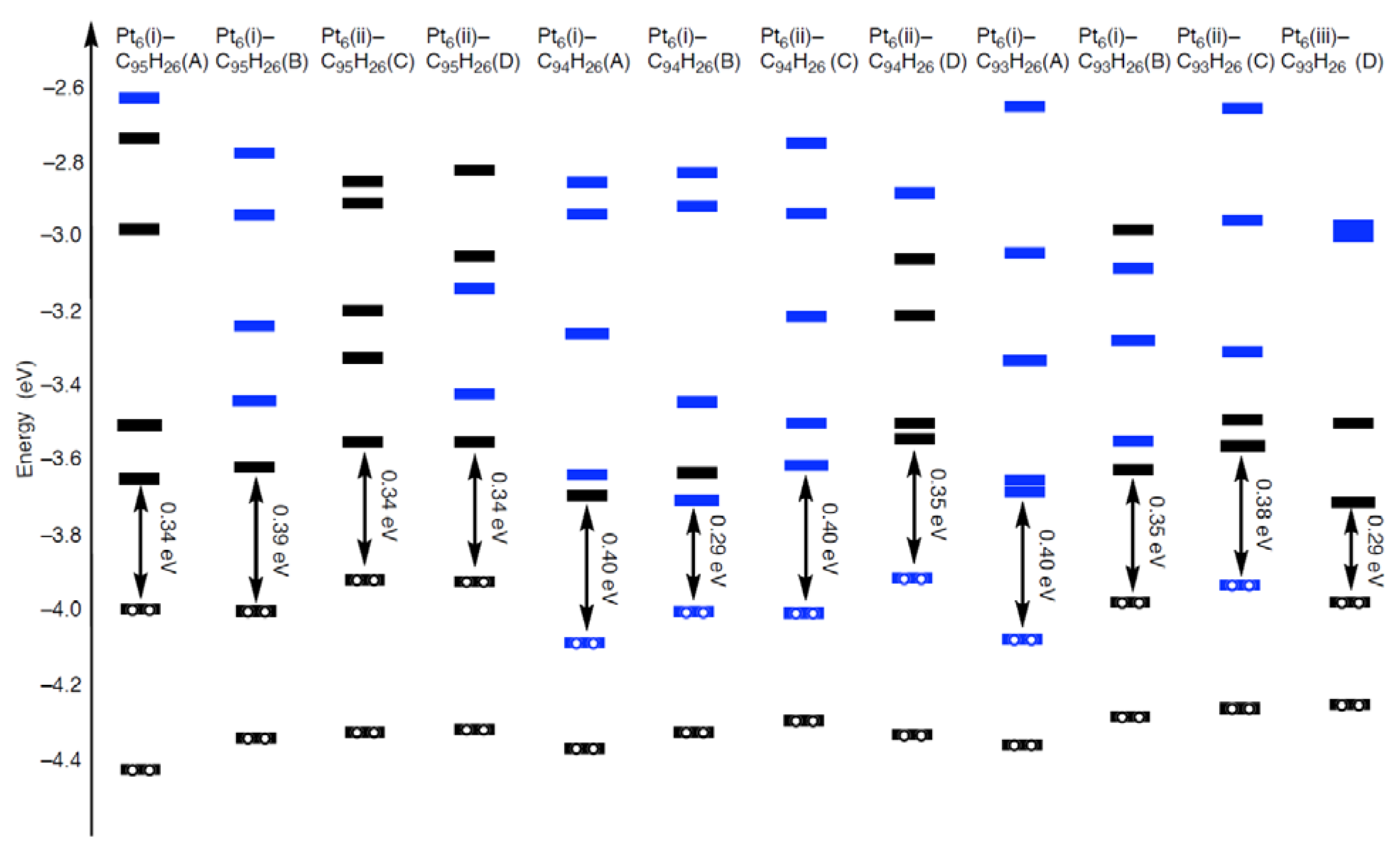
In fact, 5d(Pt)-based orbitals, given by blue bars in
Figure 12, appear in the frontier orbital regions of the Pt
6-C
94H
26 and Pt
6-C
93H
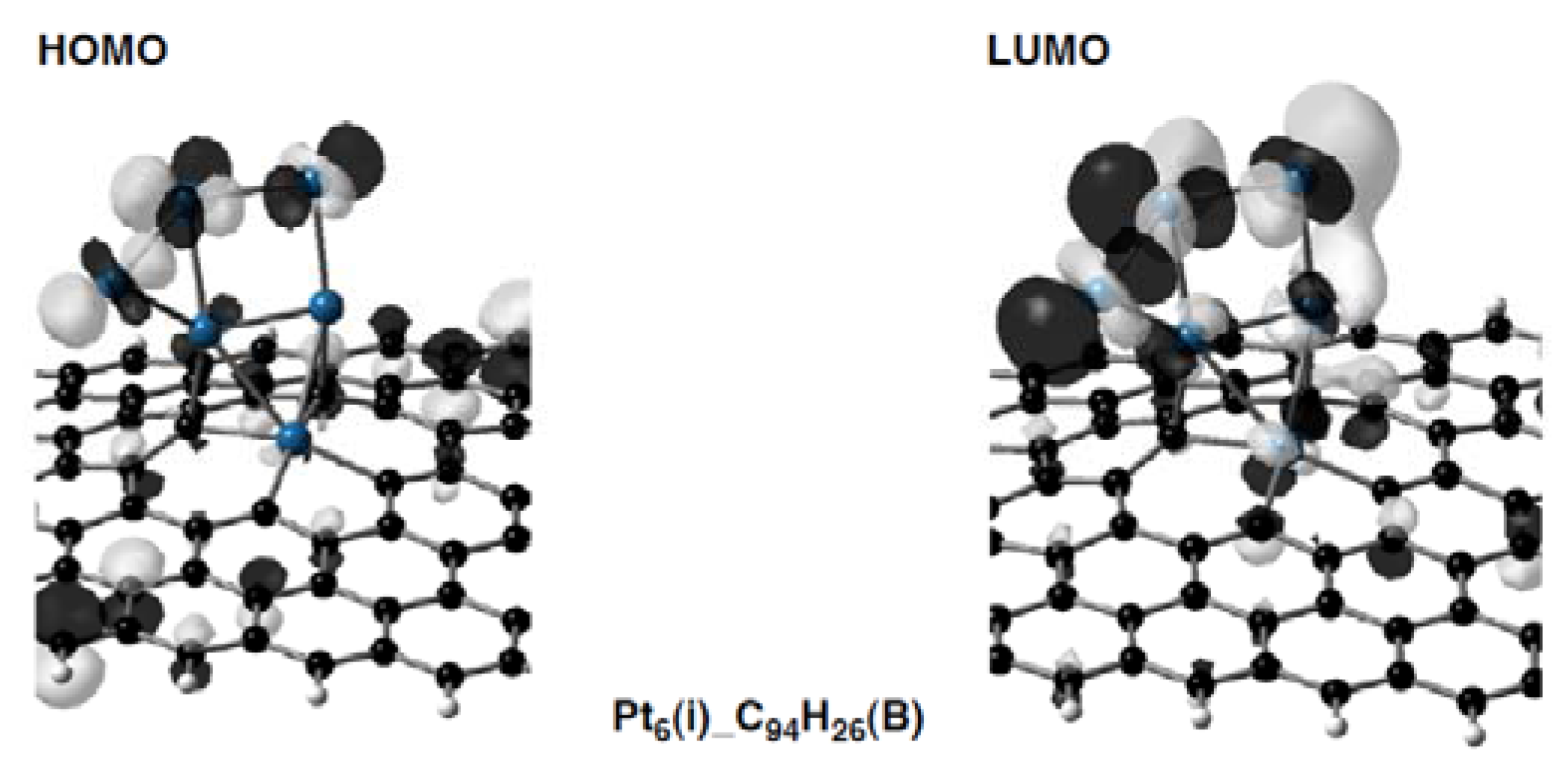
26 configurations. As the most striking case, we can see in
Figure 13 that the Pt
6(i)-C
94H
26(B) configuration has the HOMO and LUMO consisting of 5d(Pt) orbitals. On the other hand, levels of 5d(Pt)-based orbitals in the Pt
6-C
95H
26 strongly depend on their cluster-shape. In the Pt
6(i)-C
95H
26(A) and Pt
6(ii)-C
95H
26(C) configurations, such 5d(Pt)-based orbital lies larger than 1.3 eV above the LUMO, whereas the LUMO+1 consists of 5d(Pt)-based orbitals in the other Pt
6-C
95H
26 configurations.
2.3.2. Triplet State
As shown in
Figure 12, the all optimized geometries in the single state have relatively small HOMO-LUMO gaps (0.29 ~ 0.40 eV). Thus, higher spin states can be energetically stable relative to the singlet states. Along the assumption, we obtained their triplet states, and estimated the energy difference between the two spin states (ΔE
state(Pt
6-C
96–nH
26)), as tabulated in
Table 9.
Figure 12.
Orbital energies (eV) in the frontier orbital region of the optimized Pt
6-C
95H
26, Pt
6-C
94H
26, and Pt
6-C
93H
26 configurations whose structures are given in
Figure 5–
Figure 7. The HOMO-LUMO gaps are given. Orbitals originated from 5d(Pt) orbitals are denoted by blue bars, and those with no or less 5d(Pt) orbital contribution are denoted by black bars.
Figure 12.
Orbital energies (eV) in the frontier orbital region of the optimized Pt
6-C
95H
26, Pt
6-C
94H
26, and Pt
6-C
93H
26 configurations whose structures are given in
Figure 5–
Figure 7. The HOMO-LUMO gaps are given. Orbitals originated from 5d(Pt) orbitals are denoted by blue bars, and those with no or less 5d(Pt) orbital contribution are denoted by black bars.
Figure 13.
Frontier orbitals (the HOMO and LUMO) in the Pt
6(i)-C
94H
26(B) configuration (
Figure 6) are given as a representative Pt
6-C
96–nH
26 configuration.
Figure 13.
Frontier orbitals (the HOMO and LUMO) in the Pt
6(i)-C
94H
26(B) configuration (
Figure 6) are given as a representative Pt
6-C
96–nH
26 configuration.
Table 9.
Energy difference between the singlet and triplet states in the Pt6-C96–nH26 configurations (ΔEstate in kcal/mol) a.
Table 9.
Energy difference between the singlet and triplet states in the Pt6-C96–nH26 configurations (ΔEstate in kcal/mol) a.
| | Pt6(i)-(A) | Pt6(i)-(B) | Pt6(ii) | Pt6(iii) |
|---|
| Clusters on C95H26 | –14.3 | –17.8 | –14.0 | b |
| Clusters on C94H26 | –23.3 | –16.2 | –23.3 | –38.5 |
| Clusters on C93H26 | –15.0 | –23.7 | –13.7 | b |
Table 9 shows that their triplet spin states are energetically stable relative to the singlet states, as expected. According to DFT calculations, most Pt
6-C
96–nH
26 configurations have radical Pt
6 clusters on defective graphene patches As representative cases, spin density distributions on the Pt
6(i) cluster binding into C
95H
26 or C
94H
26 are shown in
Figure 14.
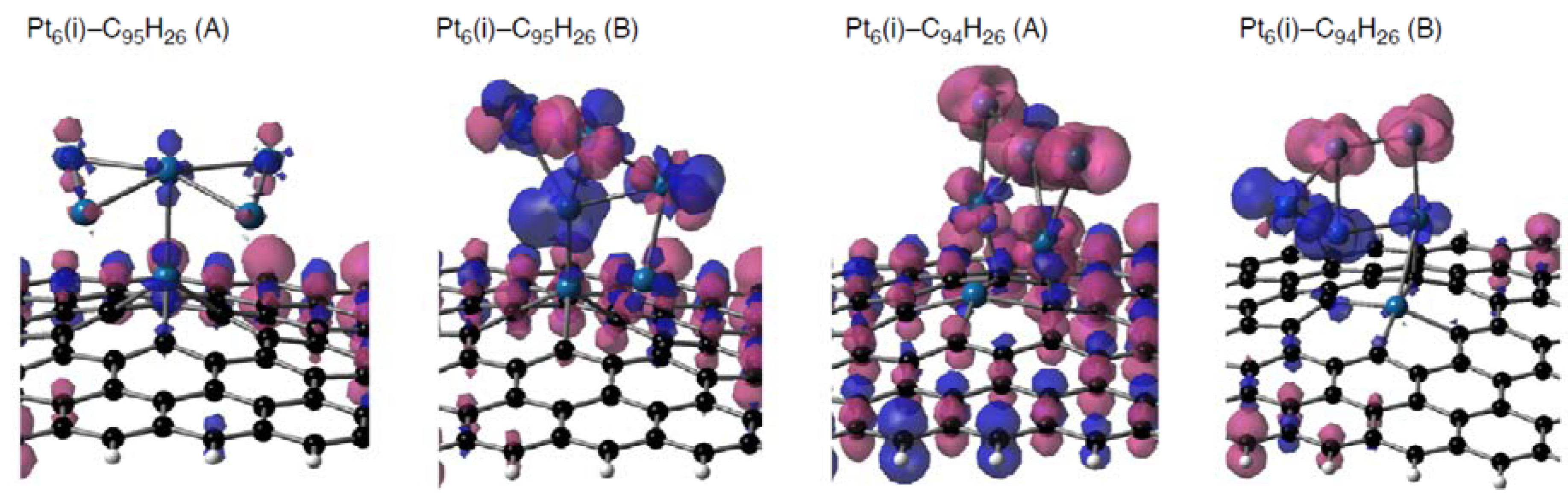
Figure 14 shows that substantial spin densities appear on the adsorbed clusters in the three Pt
6-C
96–nH
26 configurations (Pt
6(i)-C
95H
26(B), and two Pt
6(i)-C
94H
26 configurations), whereas the Pt
6(i)-C
95H
26(A) configuration does not have such radical cluster due to the absence of 5d(Pt)-based frontier orbitals (
Figure 12). In the three configurations with radical clusters, a variety of the spin density distributions was found. In the Pt
6(i)-C
95H
26(B) and Pt
6(i)-C
94H
26(B), the spin density distributions are strongly deviated from those in the bare Pt
6 cluster, whereas the Pt
6(i)-C
94H
26(B) has similar distributions. Furthermore, we found a relationship between spin densities of Pt clusters and those on the defective sp
2 surface. When spin density distributions of the Pt cluster of a Pt
6-C
96-nH
26 configuration are (not) similar to the bare case, spin densities are (not) delocalized over the carbon surface. Similar tendencies were found in spin density distributions of the stable triplet Pt
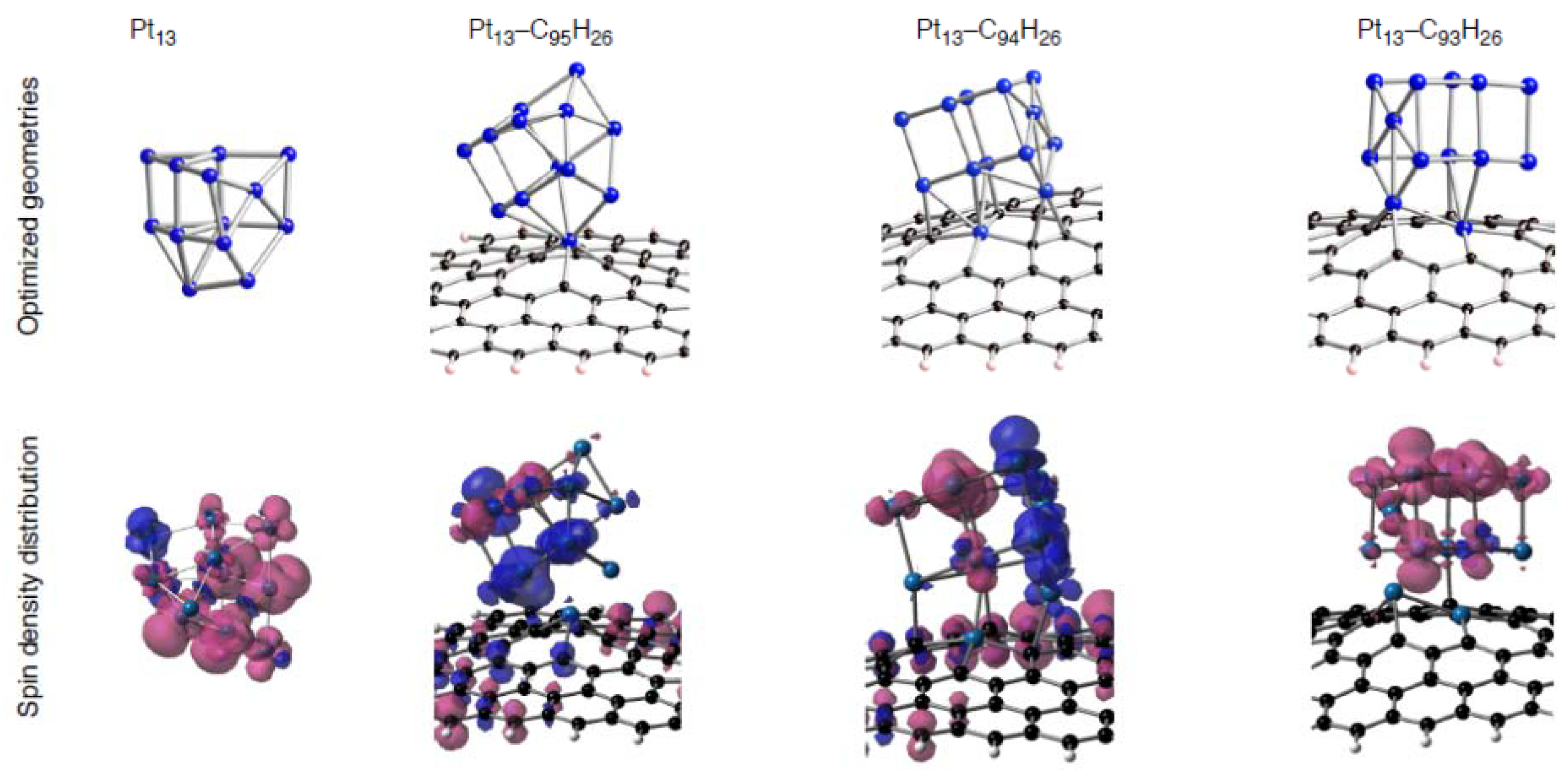
13-C
96–nH
26 configurations [
59], because radical Pt clusters exist on the defective graphene patches as shown in
Figure 15.
Figure 14.
Spin density distributions of representative Pt
6-C
96–nH
26 configurations (Pt
6(i) cluster on C
95H
26 or C
94H
26 in two binding fashions, displayed in
Figure 5 and
Figure 6). Isosurface α- and β-spins are given by pink and blue, respectively.
Figure 14.
Spin density distributions of representative Pt
6-C
96–nH
26 configurations (Pt
6(i) cluster on C
95H
26 or C
94H
26 in two binding fashions, displayed in
Figure 5 and
Figure 6). Isosurface α- and β-spins are given by pink and blue, respectively.
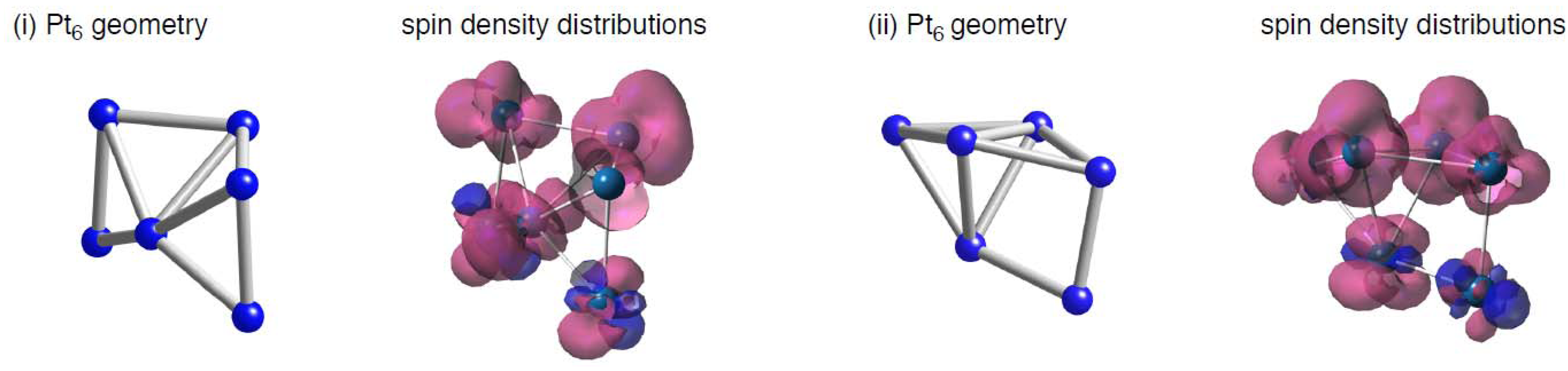
Finally, let us compare the spin density distributions on Pt
6 clusters on defective sp
2 surfaces (
Figure 14) with the pristine case (
Figure 2). As mentioned above, spin density maps on Pt
6 cluster on pristine C
96H
26 are similar to those in the bare cluster. However, such similarity cannot be always found in the defective graphene cases. The different tendencies come from whether underlying carbon atoms have radical characters or not in
Figure 2 and
Figure 4. In the pristine patch, underlying carbon atoms do not have radical characters, and they cannot perturb spin density distributions of the absorbed cluster. In contrast, unpaired electrons exist on underlying carbon atoms in the defective sites, and thus spin density distributions of the adsorbed clusters are modulated by the Pt-C interactions. Therefore, perturbation of the radical sp
2 surface by introduction of vacancy-type defects can change characters of adsorbed Pt clusters.
Figure 15.
Spin density distributions of representative Pt13-C96–nH26 configurations. Isosurface α- and β-spins are given by pink and blue, respectively.
Figure 15.
Spin density distributions of representative Pt13-C96–nH26 configurations. Isosurface α- and β-spins are given by pink and blue, respectively.
The DFT findings are important in the catalytic activity of Pt clusters on sp
2 surface, because the supported clusters can serve as active site for catalytic reactions. For example, if radical Pt clusters exist on carbon surface, it can cleave the H-H bond of hydrogen molecules in a homolytic manner. Otherwise, the H-H bond is activated by the clusters via a non-radical mechanism in [
22]. Thus, the DFT calculations propose that one can change chemical reactivity of Pt clusters on graphene patches by introducing of vacancy-type defects on the surface.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}











