Selective Oxidation Reactions of Natural Compounds with Hydrogen Peroxide Mediated by Methyltrioxorhenium

 ,
,  ,
, 
Abstract
:1. Introduction
2. Results and Discussion


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Time (h) | Conv. (%) | Product selectivities (%) | ||
|---|---|---|---|---|---|
| 1 | 0.5 | 88 |  |  |  |
| 8 (53) | 9 (28) | 10 (19) | |||
| 2 | 0.5 | 95 |  |  |  |
| 11 (32) | 12 (26) | 13 (42) | |||
| 3 | 18 | 100 |  |  | |
| 14 cis (86) | 15 trans (14) | ||||
| 4 | 20 | 75 |  | ||
| 16 (100) | |||||
| 5 | 8 | 99 |  |  |  |
| 17 (22) | 18 (20) | 19 (58) | |||
| 6 b | 8 | 98 |  |  |  |
| 20 (18) | 21 (17) | 22 (18) | |||
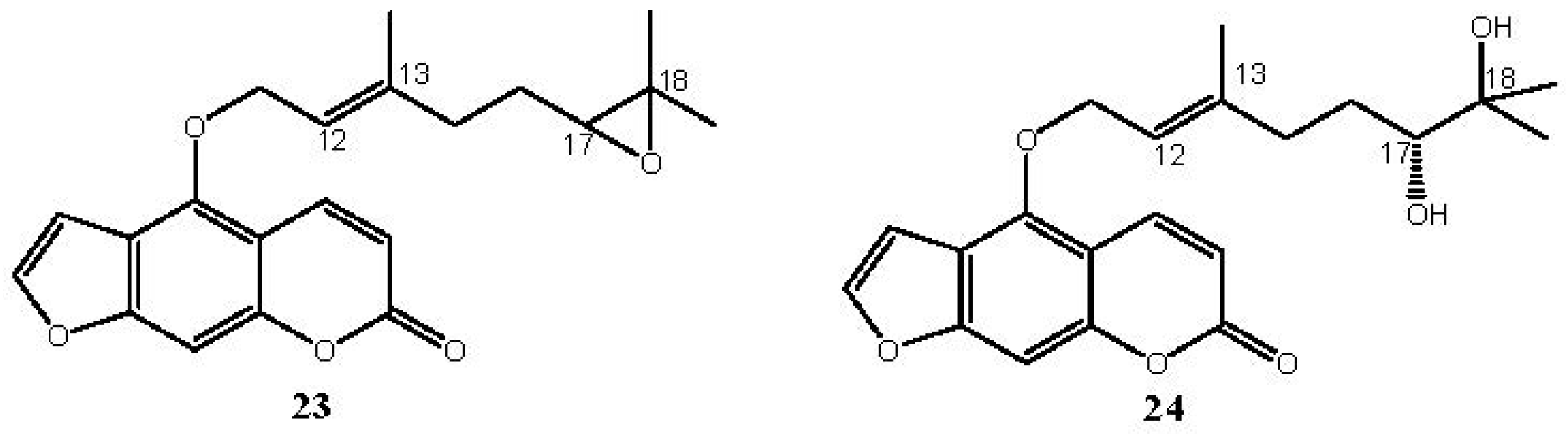
| 7 | 4 | 62 |  | ||
| 23 (100) |
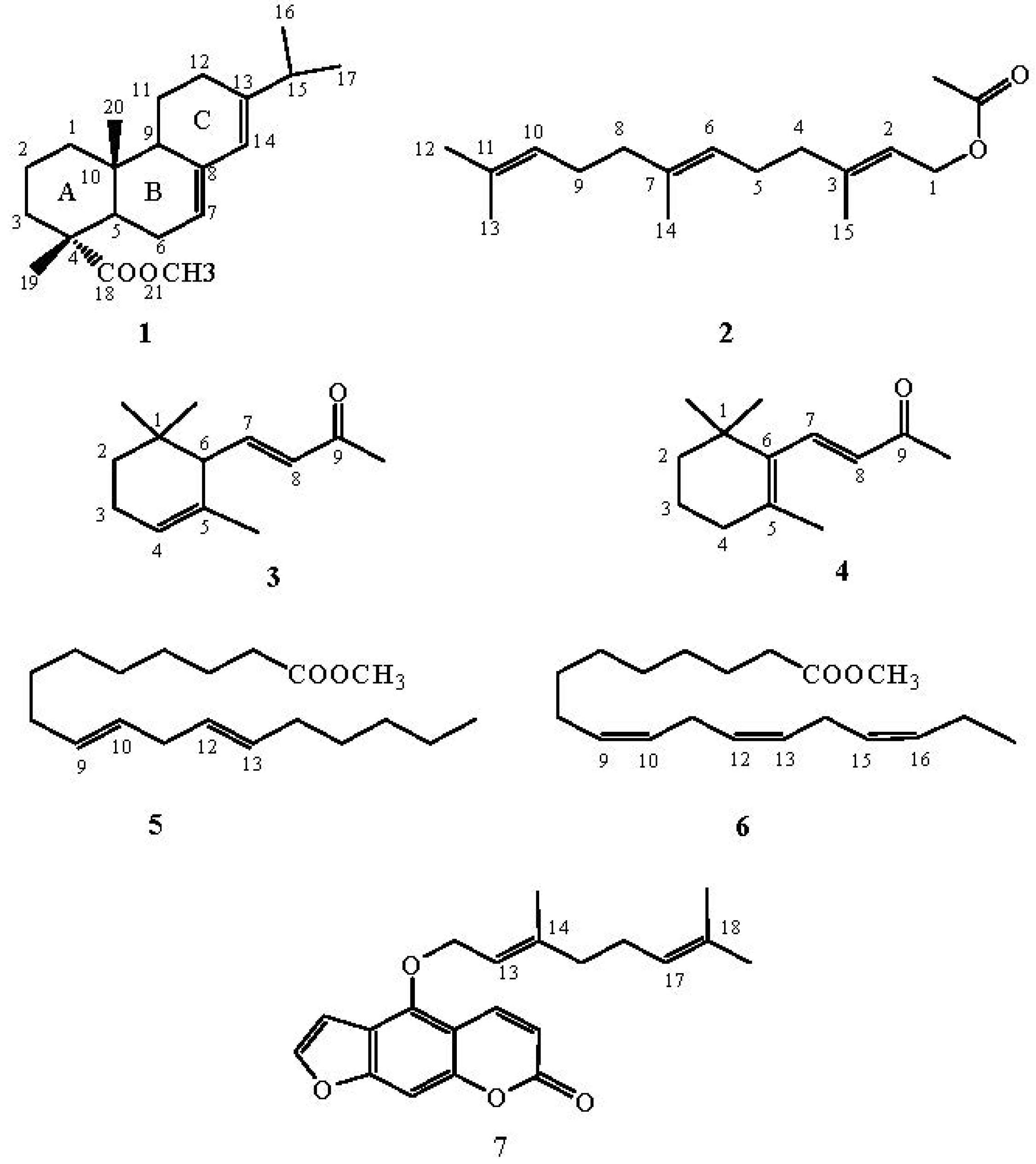
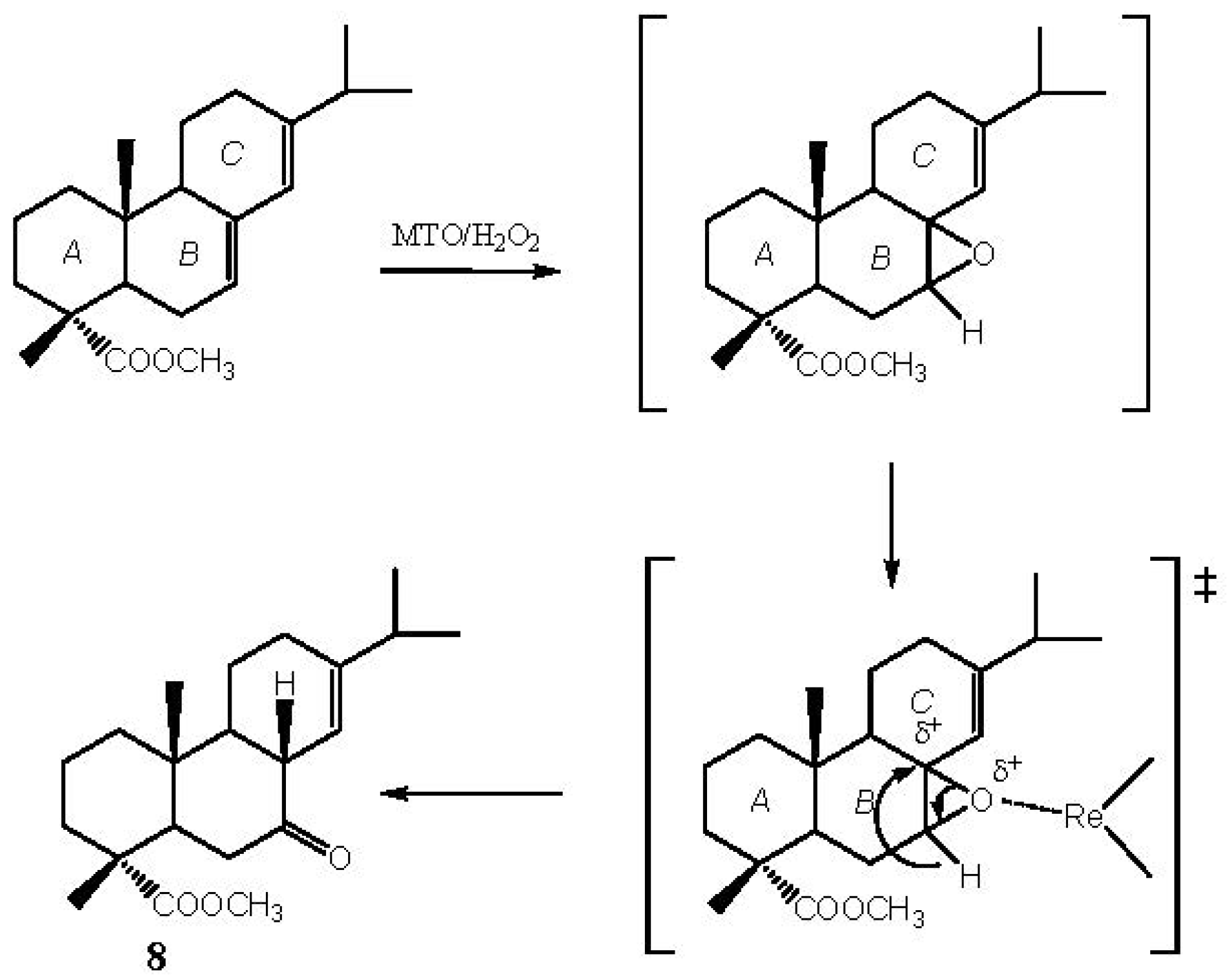
2.1. Methyl Abietate (1)


2.2. All-Trans Farnesyl Acetate (2)
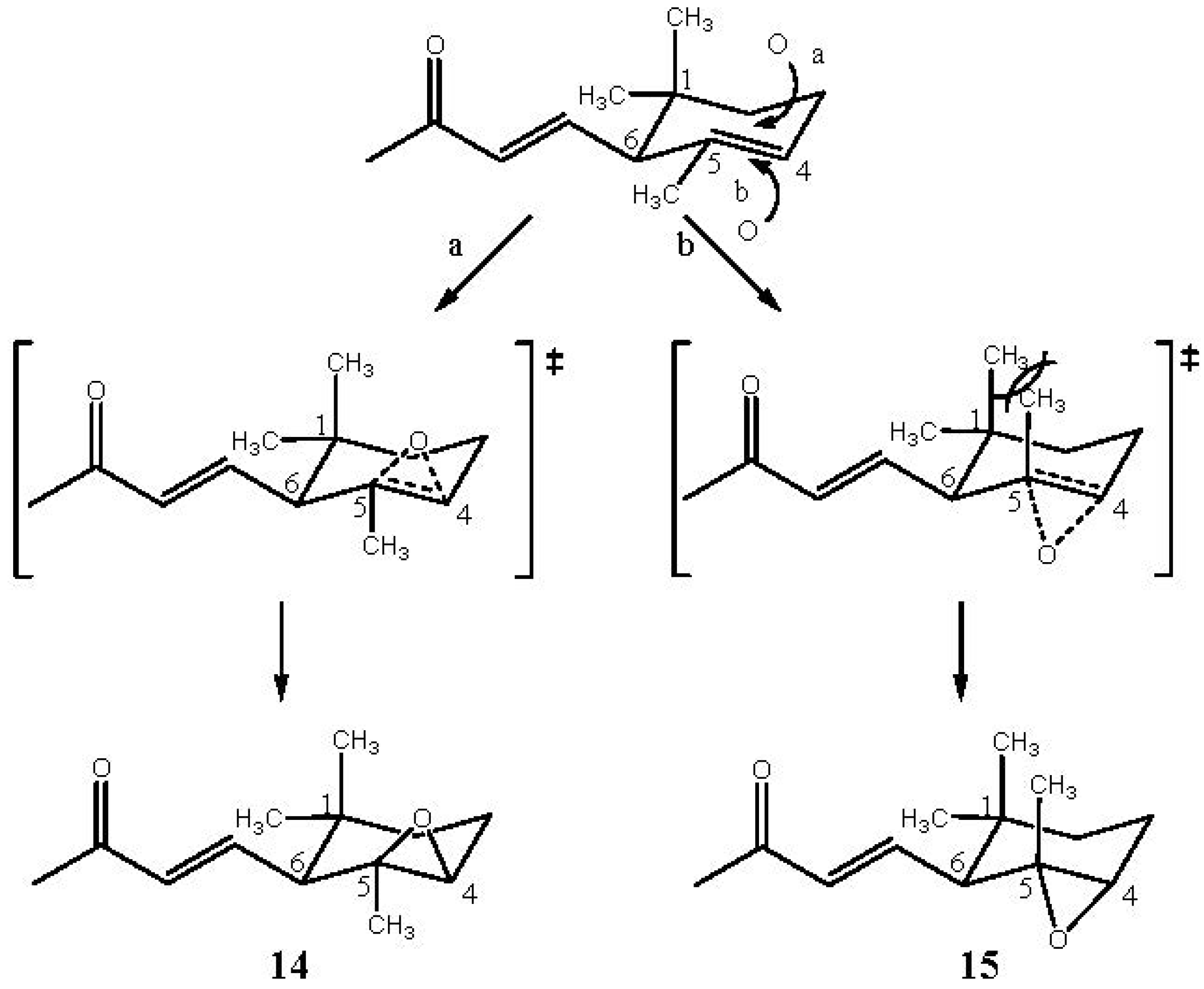
2.3. α-Ionone (3) and β-Ionone (4)


2.4. Methyl Linolelaidate (5)
2.5. Methyl Linolenate (6)
2.6. Bergamottin (7)

3. Experimental
3.1. General Methods
3.2. General Oxidation Procedure
3.2.1. Oxidation of Methyl Abietate (1)
3.2.2. Oxidation of Farnesyl Acetate (2)
3.2.3. Oxidation of α-Ionone (3)
3.2.4. Oxidation of β-Ionone (4)
3.2.5. Oxidation of Methyl Linolelaidate (5)
3.2.6. Oxidation of Methyl Linolenate (6)
3.2.7. Oxidation of Bergamottin (7)
3.2.8. Representative Procedures for the HKR of Terminal Epoxides
4. Conclusions
Supplementary Materials
Acknowledgments
Conflicts of Interest
References
- Bauer, K.; Garbe, D.; Surburg, H. Common Fragance and Flavor Materials. Preparation, Properties and Uses; Wiley: New York, NY, USA, 1997. [Google Scholar]
- Strukul, G.; Scarso, A. Environmentally Benign Oxidants; John Wiley & Sons: New York, NY, USA, 2013. [Google Scholar]
- Herrmann, W.A.; Fischer, R.W. Methyltrioxorhenium as catalyst for olefin oxidation. Angew. Chem. Ed. Engl. 1991, 30, 1638–1641. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Fischer, R.; Scherer, M.U.W. Methyltrioxorhenium(VII) als epoxidationskatalysator: Struktur der aktiven spezies und katalysemechanismus. Angew. Chem. 1993, 105, 1209–1212. [Google Scholar] [CrossRef]
- Al-Ajlouni, A.M.; Espenson, J.H. Epoxidation of styrenes by hydrogen peroxide as catalyzed by methylrhenium trioxide. J. Am. Chem. Soc. 1995, 117, 9243–9250. [Google Scholar] [CrossRef]
- Adam, W.; Saha-Moller, C.R.; Weichold, O. Epoxidation of trans-cyclooctene by methyltrioxorhenium/H2O2: Reaction of trans-epoxide with the monoperoxo complex. J. Org. Chem. 2000, 65, 5001–5004. [Google Scholar] [CrossRef]
- Murray, R.W.; Iyanar, K. Oxidation of [60] fullerene by the methyltrioxorhenium-hydrogen peroxide system. Tetrahedron Lett. 1997, 38, 335–338. [Google Scholar] [CrossRef]
- Murray, R.W.; Iyanar, K.; Chen, J.; Wearing, J.T. Synthesis of nitrones using the methyltrioxorhenium/hydrogen peroxide system. J. Org. Chem. 1996, 61, 8099–8102. [Google Scholar] [CrossRef]
- Goti, A.; Nannelli, L. Synthesis of nitrones by methyltrioxorhenium catalyzed direct oxidation of secondary amines. Tetrahedron Lett. 1996, 37, 6025–6028. [Google Scholar] [CrossRef]
- Zhu, Z.; Espenson, J.H. Oxidation of alkynes by hydrogen peroxide catalyzed by methylrhenium trioxide. J. Org. Chem. 1995, 60, 7728–7732. [Google Scholar] [CrossRef]
- Wang, Y.; Espenson, J.H. Oxidation of symmetric disulfides with hydrogen peroxide catalyzed by methyltrioxorhenium(VII). J. Org. Chem. 2000, 65, 104–107. [Google Scholar] [CrossRef]
- Adam, W.; Herrmann, W.A.; Lin, J.; Saha-Moller, C.R.; Fischer, R.W.; Correia, J.D.J. Homogeneous-catalytic oxidation of arenes and a new synthesis of vitamin K3. Angew. Chem. Ed. Engl. 1994, 33, 2475–2477. [Google Scholar]
- Zhu, Z.; Espenson, J.H. Kinetics and mechanism of oxidation of anilines by hydrogen peroxide as catalyzed by methylrhenium trioxide. J. Org. Chem. 1995, 60, 1326–1332. [Google Scholar] [CrossRef]
- Adam, W.; Mitchell, C.M.; Saha-Moller, C. Chemoselective methyltrioxorhenium(VII)-catalyzed sulfoxidations with hydrogen peroxide. Tetrahedron 1994, 50, 13121–13124. [Google Scholar] [CrossRef]
- Vassel, K.A.; Espenson, J.H. Oxidation of organic sulfides by electrophilically activated hydrogen peroxide: The catalytic ability of methylrhenium trioxide. Inorg. Chem. 1994, 33, 5491–5498. [Google Scholar] [CrossRef]
- Adam, W.; Mitchell, C.M.; Saha-Moller, C.R. Regio- and diastereoselective catalytic epoxidation of acyclic allylic alcohols with methyltrioxorhenium: A mechanisti comparison with metal (peroxy and peroxo complexes) and nonmetal (peracids and dioxirane) oxidants. J. Org. Chem. 1999, 64, 3699–3707. [Google Scholar] [CrossRef]
- Adam, W.; Mitchell, C.M.; Saha-Moller, C. Steric and electronic effects in the diastereoselective catalytic epoxidation of cyclic allylic alcohols with methyltrioxorhenium (MTO). Eur. J. Org. Chem. 1999, 1999, 785–790. [Google Scholar]
- Lane, B.S.; Burgess, K. Metal-catalyzed epoxidations of alkenes with hydrogen peroxide. Chem. Rev. 2003, 103, 2457–2473. [Google Scholar] [CrossRef]
- Yamazaki, S. An effective procedure for the synthesis of acid-sensitive epoxides: Use of 1-methylimidazole as the additive on methyltrioxorhenium-catalyzed epoxidation of alkenes with hydrogen peroxide. Org. Biomol. Chem. 2010, 8, 2377–2385. [Google Scholar] [CrossRef]
- Yamazaki, S. Methyltrioxorhenium-catalyzed epoxidation of homoallylic alcohols with hydrogen peroxide. J. Org. Chem. 2012, 77, 9884–9888. [Google Scholar]
- Crucianelli, M.; Saladino, R.; de Angelis, F. Methyltrioxorhenium catalysis in nonconventional solvents: A great catalyst in a safe reaction medium. ChemSusChem 2010, 3, 524–540. [Google Scholar] [CrossRef]
- Ballistreri, F.P.; Chillemi, R.; Sciuto, S.; Tomaselli, G.A.; Toscano, R.M. Regio and stereoselective oxidations of usaturated steroidal compounds with H2O2 mediated by CH3ReO3. Steroids 2006, 71, 565–577. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Fischer, R.A.; Scherer, W.; Rauch, M.U. Methyltrioxorhenium(VII) as catalyst for epoxidations: Structure of the active species and mechanism of catalysis. Angew. Chem. Int. Ed. 1993, 32, 1157–1160. [Google Scholar] [CrossRef]
- Al-Ajlouni, A.M.; Espenson, J.H. Kinetics and mechanism of the epoxidation of alkyl-substituted alkenes by hydrogen peroxide, Catalyzed by methylrhenium trioxide. J. Org. Chem. 1996, 61, 3969–3976. [Google Scholar] [CrossRef]
- Copéret, C.; Adolfsson, H.; Sharpless, K.B. A simple and efficient method for epoxidation of terminal alkenes. Chem. Commun. 1997. [Google Scholar] [CrossRef]
- Herrmann, W.A.; Ding, H.; Kratzer, R.M.; Kühn, F.E.; Haider, J.J.; Fischer, R.W.J. Multiple bonds between transition metals and main-group elements Part 168. I Methyltrioxorhenium/Lewisbase catalysts in olefin Epoxidation. Organomet. Chem. 1997, 549, 319–322. [Google Scholar] [CrossRef]
- Wang, W.D.; Espenson, J.H. Effects of pyridine and its derivatives on the equilibria and kinetics pertaining to epoxidation reactions catalyzed by methyltrioxorhenium. J. Am. Chem. Soc. 1998, 120, 11335–11341. [Google Scholar] [CrossRef]
- Tan, H.; Espenson, J.H. Kinetics and mechanism of the dihydroxylation and epoxidation of conjugated dienes with hydrogen peroxide catalyzed by methylrhenium trioxide. Inorg. Chem. 1998, 37, 467–472. [Google Scholar] [CrossRef]
- Espenson, J.H. Atom-transfer reactions catalyzed by methyltrioxorhenium(VII)—mechanisms and applications. Chem. Commun. 1999, 6, 479–488. [Google Scholar] [CrossRef]
- Presser, A.; Hasingerl, E.; Weis, R.; Hüfner, A. Synthetic transformations of abietic acid IV[1]. B- and C-ring oxidation. Monatsh. Chem. 1998, 129, 921–930. [Google Scholar]
- Suda, K.; Kikkawa, T.; Nakajima, S.; Takanami, T. Highly regio- and stereoselective rearrangement of epoxides to aldehydes catalyzed by high-valent metalloporphyrin complex, Cr(TPP)OTf. J. Am. Chem. Soc. 2004, 126, 9554–9555. [Google Scholar] [CrossRef]
- Silva, L.F., Jr. Construction of cyclopentyl units by ring contraction reactions. Tetrahedron 2002, 58, 9137–9161. [Google Scholar] [CrossRef]
- Silko, Y.A.; Raldugin, V.A.; Shmidt, E.N.; Mamatyuk, V.I.; Pentegova, V.A. Oxidation of methyl abietate with peracetic acid. Izv. Sibir. Otd. Akad. Nauk Khim. 1984, 3, 107–112. [Google Scholar]
- Silko, Y.A.; Raldugin, V.A.; Shmidt, E.N.; Mamatyuk, V.I.; Pentegova, V.A. Methyl abietate epoxides and their reaction products. Izv. Sibir. Otd. Akad. Nauk Khim. 1983, 2, 124–128. [Google Scholar]
- Tsangarakis, C.; Arkoudis, E.; Raptis, C.; Stratakis, M. Selective monocyclization of epoxy terpenoids promoted by zeolite NaY. A short biomimetic synthesis of elegansidiol and farnesiferols B-D. Org. Lett. 2007, 9, 583–586. [Google Scholar] [CrossRef]
- Van Tamelen, E.E.; McCormickg, J.P. Terpene terminal epoxides. Mechanistic aspects of conversion to the bicyclic level. J. Am. Chem. Soc. 1969, 91, 1847–1848. [Google Scholar] [CrossRef]
- Cane, D.E.; Iyengar, R.; Shiao, M. Cyclonerodiol biosynthesis and the enzymatic conversion of farnesyl to nerolidyl pyrophosphate. J. Am. Chem. Soc. 1981, 103, 914–931. [Google Scholar] [CrossRef]
- Tong, R.; McDonald, F.E. Mimicking biosynthesis: Total synthesis of the triterpene natural product abudinol B from a squalene-like precursor. Angew. Chem. Int. Ed. 2008, 47, 4377–4379. [Google Scholar] [CrossRef]
- Uebelhart, P.; Baumeler, A.; Haag, H.; Prewo, R.; Bieri, J.H.; Eugster, C.H. Optisch aktive 4,5-Epoxy-4,5-dihydro-α-ionone und Synthese der stereoisomeren 4,5:4′,5′-Diepoxy-4,5,4′,5′-tetrahydro-ϵ,ϵ-carotine und der sterische Verlauf ihrer Hydrolyse. Helv. Chim. Acta 1986, 69, 816–834. [Google Scholar] [CrossRef]
- Sakamaki, H.; Itoh, K.; Chai, W.; Hayashida, Y.; Kitanaka, S.; Horiuchi, C.A. Biotransformation of (±)-α-ionone and β-ionone by cultured cells of Caragana chamlagu. J. Mol. Catal. B-Enzym. 2004, 27, 177–181. [Google Scholar] [CrossRef]
- Gunstone, F.D.; Schuler, H.R. Fatty acids, Part 46 PMR spectra of several epoxyoctadecenoic, epoxyoctadecynoic, and diepoxyoctadecanoic esters. Chem. Phys. Lipids 1975, 15, 189–197. [Google Scholar] [CrossRef]
- Du, G.; Tekin, A.; Hammond, E.G.; Woo, L.K. Catalytic epoxidation of methyl linoleate. J. Am. Oil Chem. Soc. 2004, 81, 477–480. [Google Scholar] [CrossRef]
- Cui, P.H.; Duke, R.K.; Tattam, B.N.; Duke, C.C. Monoepoxy octadecadienoates and monoepoxy octadecatrienoates 2: Mass spectral characterization. Chem. Phys. Lipids 2008, 152, 65–70. [Google Scholar] [CrossRef]
- Cui, P.H.; Duke, R.K.; Duke, C.C. Monoepoxy octadecadienoates and monoepoxy octadecatrienoates: 1: NMR spectral characterization. Chem. Phys. Lipids 2008, 152, 122–130. [Google Scholar] [CrossRef]
- Bailey, D.G.; Malcolm, J.; Arnold, O.; Spence, J.D. Grapefruit juice-drug interactions. Br. J. Clin. Pharmacol. 1998, 46, 101–110. [Google Scholar]
- Row, E.C.; Brown, S.A.; Stachulski, A.V.; Lennard, M.S. Design, Synthesis and evaluation of furanocoumarin monomers as inhibitors of CYP3A4. Org. Biomol. Chem. 2006, 4, 1604–1610. [Google Scholar] [CrossRef]
- Ohta, T.; Maruyama, T.; Nagahashi, M.; Miyamoto, Y.; Hosoi, S.; Kiuchi, F.; Yamazoe, Y.; Tsukamoto, S. Paradisin C: A new CYP3A4 inhibitor from grapefruit juice. Tetrahedron 2002, 58, 6631–663. [Google Scholar] [CrossRef]
- Schaus, S.E.; Brandes, B.D.; Larrow, J.F.; Tokunaga, M.; Hansen, K.B.; Gould, A.E.; Furrow, M.E.; Jacobsen, E.N. Highly selective hydrolytic kinetic resolution of terminal epoxides catalyzed by chiral (salen)CoIII complexes. Practical synthesis of enantioenriched terminal epoxides and 1,2-diols. J. Am. Chem. Soc. 2002, 124, 1307–1315. [Google Scholar] [CrossRef]
- Larrow, J.F.; Jacobsen, E.N. Asymmetric processes catalyzed by chiral (salen)metal complexes. Top. J. Organomet. Chem. 2004, 6, 123–152. [Google Scholar]
- Nielsen, L.P.C.; Stevenson, C.P.; Blackmond, D.G.; Jacobsen, E.N. Mechanistic investigation leads to a synthetic improvement in the hydrolytic kinetic resolution of terminal epoxides. J. Am. Chem. Soc. 2004, 126, 1360–1362. [Google Scholar] [CrossRef]
- Abad, A.; Arno, M.; Domingo, L.R.; Zaragoza, R.J. Synthesis of (+)-podocarp-8(14)-en-13-one and methyl-(+)-13-oxo-podocarp-8(14)-en-18-oate from abietic acid. Tetrahedron 1985, 41, 4937–4940. [Google Scholar] [CrossRef]
- Sample Availability: Samples are not available.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Amato, M.E.; Ballistreri, F.P.; Pappalardo, A.; Tomaselli, G.A.; Toscano, R.M.; Sfrazzetto, G.T. Selective Oxidation Reactions of Natural Compounds with Hydrogen Peroxide Mediated by Methyltrioxorhenium. Molecules 2013, 18, 13754-13768. https://doi.org/10.3390/molecules181113754
Amato ME, Ballistreri FP, Pappalardo A, Tomaselli GA, Toscano RM, Sfrazzetto GT. Selective Oxidation Reactions of Natural Compounds with Hydrogen Peroxide Mediated by Methyltrioxorhenium. Molecules. 2013; 18(11):13754-13768. https://doi.org/10.3390/molecules181113754
Chicago/Turabian StyleAmato, Maria E., Francesco P. Ballistreri, Andrea Pappalardo, Gaetano A. Tomaselli, Rosa M. Toscano, and Giuseppe Trusso Sfrazzetto. 2013. "Selective Oxidation Reactions of Natural Compounds with Hydrogen Peroxide Mediated by Methyltrioxorhenium" Molecules 18, no. 11: 13754-13768. https://doi.org/10.3390/molecules181113754
APA StyleAmato, M. E., Ballistreri, F. P., Pappalardo, A., Tomaselli, G. A., Toscano, R. M., & Sfrazzetto, G. T. (2013). Selective Oxidation Reactions of Natural Compounds with Hydrogen Peroxide Mediated by Methyltrioxorhenium. Molecules, 18(11), 13754-13768. https://doi.org/10.3390/molecules181113754