3.1. Chemistry
Melting points are not corrected and were recorded on a Buchi apparatus. IR spectra (KBr pellets, 400–4000 cm−1) were recorded on a Bruker VECTOR 22 FTIR spectrophotometer. 1H-NMR spectra and NOE spectra were recorded on a Bruker AM 400 instrument at 400 MHz (chemical shifts are expressed as δ values relative to TMS as internal standard). Mass spectra (MS), ESI (positive) were recorded on an Esquire-LC-00075 spectrometer.
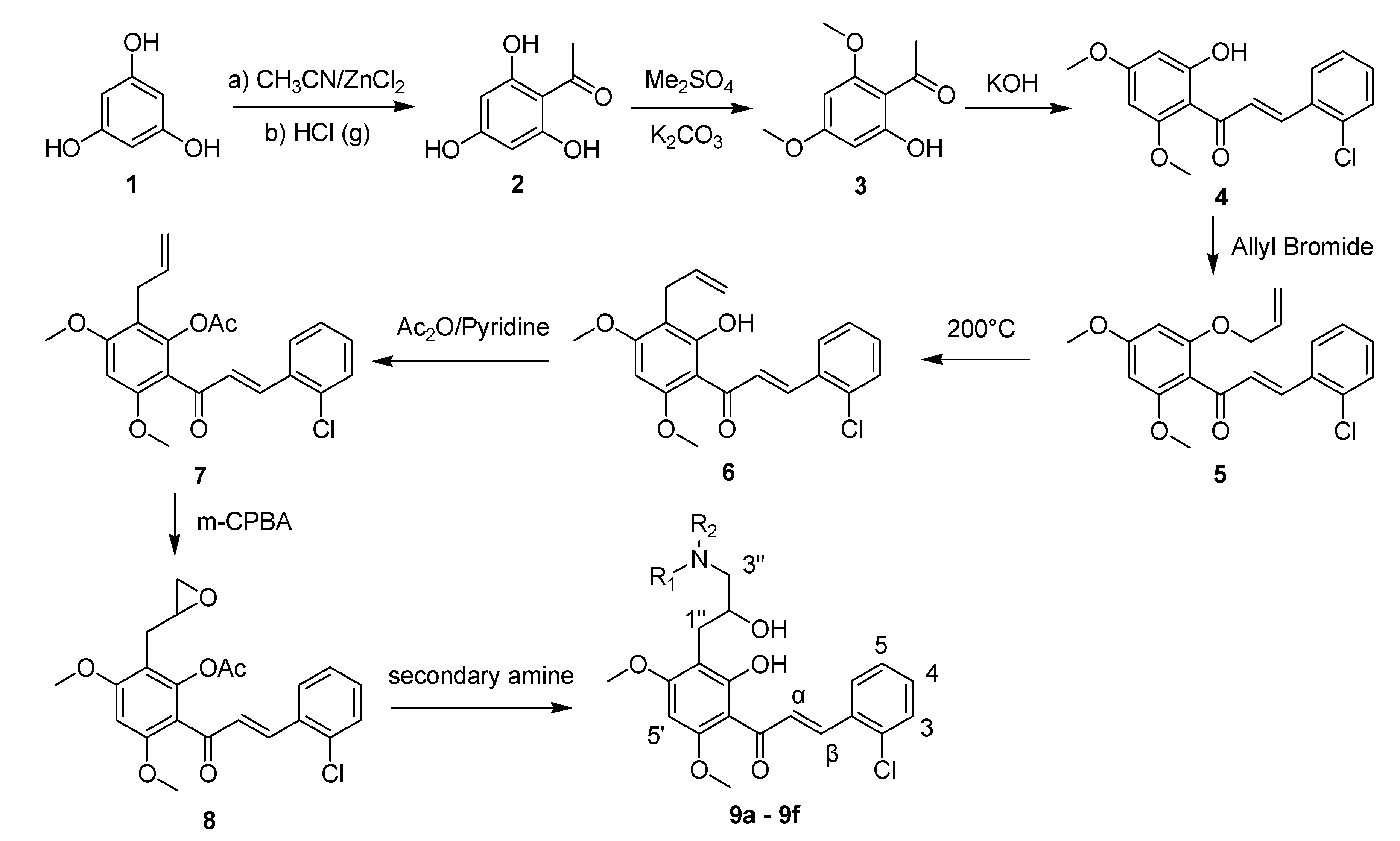
3.1.1. 3-(2-Chlorophenyl)-1-(2-hydroxy-4,6-dimethoxyphenyl)prop-2-en-1-one (4)
To a stirred solution of
3 (0.39 g, 2 mmol) and 2-chlorobenzaldehyde (0.27 g, 3 mmol) in 80% EtOH (10 mL), KOH (1.0 g, 18 mmol) was added at 0 °C. The reaction mixture was stirred at room temperature overnight and then neutralized with 2N HCl and extracted with CH
2Cl
2 (2 × 30 mL). The combined organic fraction was washed with brine, dried over anhydrous Na
2SO
4, and concentrated under reduced pressure. The residue was recrystallized from EtOH to give 0.57 g (89%) of the title compound
4 as a yellow solid. m.p. 135–137 °C (Ref. [
22]: m.p. 136–137 °C); IR (KBr): 3454 (OH), 2953, 2870 (CH
3), 1631 (C=O), 1557 (C=C), 1274 (Ar-O), 1064 (C-O), 750 cm
−1;
1H-NMR (CDCl
3) δ: 14.20 (s, 1H, OH-2'), 8.14 (d, 1H,
J = 15.6 Hz, H-β), 7.87 (d, 1H,
J = 15.6 Hz, H-α), 7.69 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.11 (s, 1H, H-5'), 5.96 (s, 1H, H-3'), 3.90 (s, 3H, OCH
3), 3.84 (s, 3H, OCH
3). ESI-MS:
m/z 319 [M+1]
+.
3.1.2. 1-(2-Allyloxy-4,6-dimethoxyphenyl)-3-(2-chlorophenyl)prop-2-en-1-one (5)
To a suspension of 4 (3.20 g, 10 mmol) and anhydrous K2CO3 in dry acetone, allyl bromide (9.1 mL, 10 mmol) was added with stirring. The resulting suspension was refluxed for 6 h, then the mixture was filtered, washed with acetone, and the filtrate was evaporated to dryness. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (6:1) as eluent to afford pure 5 (3.40 g, 95%) as a buff-colored liquid. IR (KBr): 3007 (=CH), 2944, 2849 (CH3, CH2), 1620 (C=O), 1535 (C=C), 1272 (Ar-O), 1081 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.79 (d, 1H, J = 16.0 Hz, H-β), 7.66 (m, 1H, H-6), 7.39 (m, 1H, H-3), 7.28 (m, 2H, H-4 and 5), 6.93 (d, 1H, J = 16.0 Hz, H-α), 6.16 (s, 1H, H-5'), 6.14 (s, 1H, H-3'), 5.95 (m, 1H, H-2"), 5.33 (dd, 1H, J1 = 16.8 Hz, J2 = 1.2 Hz, H-3"), 5.50 (m, 2H, H-1"), 3.84 (s, 3H, OCH3), 3.78 (s, 3H, OCH3). ESI-MS: m/z 359 [M+1]+.
3.1.3. 1-(3-Allyl-2-hydroxy-4,6-dimethoxyphenyl)-3-(2-chlorophenyl)prop-2-en-1-one (6)
A solution of 5 (4.00 g, 11.2 mmol) in N,N-dimethylaniline (60 mL) under N2 was heated to reflux for 4 h. The solvent was removed in vacuo, and the residue was recrystallized from EtOH and gave 3.05 g (75%) of title compound 6 as a yellow solid. m.p. 167–168 °C; IR (KBr): 3448 (OH), 2979, 2945 (CH3, CH2), 1630 (C=O), 1581 (C=C), 1274 (Ar-O), 1135 (C-O) cm−1; 1H-NMR (CDCl3) δ: 13.93 (s, 1H, OH-2'), 8.06 (d, 1H, J = 15.6 Hz, H-β), 7.79 (d, 1H, J = 15.6 Hz, H-α), 7.65 (m, 1H, H-6), 7.39 (m, 1H, H-3), 7.26 (m, 2H, H-4 and 5), 5.97 (s, 1H, H-5'), 5.94 (m, 1H, H-2"), 4.95 (m, 2H, H-3"), 3.91 (s, 3H, OCH3), 3.87 (s, 3H, OCH3), 3.31 (m, 2H, H-1"). ESI-MS: m/z 359 [M+1]+.
3.1.4. 2-Allyl-6-[3-(2-chlorophenyl)acryloyl]-3,5-dimethoxyphenyl acetate (7)
Acetic anhydride (1.27 mL, 13.5 mmol) was added to a solution of 6 (1.60 g, 5 mmol) in pyridine (20 mL). The reaction mixture was stirred at room temperature overnight, then neutralized with 2N HCl, and extracted with CH2Cl2 (3 × 40 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (5:1) as eluent to give pure 7 (1.70 g, 85%) as a yellow solid. m.p. 106–109 °C; IR (KBr): 2979, 2948, 2846 (CH3, CH2), 1762, 1660 (C=O), 1612 (C=C), 1205 (Ar-O), 1091 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.89 (d, 1H, J = 15.6 Hz, H-β), 7.65 (m, 1H, H-6), 7.40 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 7.06 (d, 1H, J = 15.6 Hz, H-α), 5.97 (s, 1H, H-5'), 5.90 (m, 1H, H-2"), 4.96 (m, 2H, H-3"), 3.91 (s, 3H, OCH3), 3.83 (s, 3H, OCH3), 3.32 (m, 2H, H-1"), 2.32 (s, 3H, COCH3). ESI-MS: m/z 401 [M+1]+.
3.1.5. 2-[3-(2-Chlorophenyl)acryloyl]-3,5-dimethoxy-6-oxiranylmethylphenyl acetate (8)
To a stirred solution of 7 (0.60 g, 1.5 mmol) in CH2Cl2 (20 mL) was added a solution of m-chloroperoxybenzoic acid (0.42 g, 2.5 mmol) in CH2Cl2 (40 mL) dropwise during 0.5 h, with the temperature being maintained 0–5 °C. The mixture was stirred at room temperature for 24 h, and then the CH2Cl2 layer was washed successively with aqueous sodium bicarbonate and brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (6:1) as eluent to give pure 8 (0.42 g, 65%) as a yellow solid. m.p.106–109 °C; IR (KBr): 2943, 2842 (CH3, CH2), 1762, 1660 (C=O), 1612 (C=C), 1207 (Ar-O), 1090 (C-O), 768 cm−1; 1H-NMR (CDCl3) δ: 7.89 (d, 1H, J = 15.6 Hz, H-β), 7.65 (m, 1H, H-6), 7.41 (m, 1H, H-3), 7.33 (m, 2H, H-4 and 5), 7.01 (d, 1H, J = 15.6 Hz, H-α), 6.43 (s, 1H, H-5'), 3.92 (s, 3H, OCH3), 3.86 (s, 3H, OCH3), 3.07 (m, 1H, H-2"), 2.70 (dd, 1H, J1 = 16.0 Hz, J2 = 4.0 Hz, H-1"α), 2.67 (dd, 1H, J1 = 15.6 Hz, J2 = 4.4 Hz, H-1"β), 2.59 (t, 1H, J = 7.6 Hz, H-3"α), 2.51 (dd, 1H, J1 = 5.6 Hz, J2 = 2.4 Hz, H-3"β), 2.25 (s, 3H, COCH3). ESI-MS: m/z 417 [M+1]+.
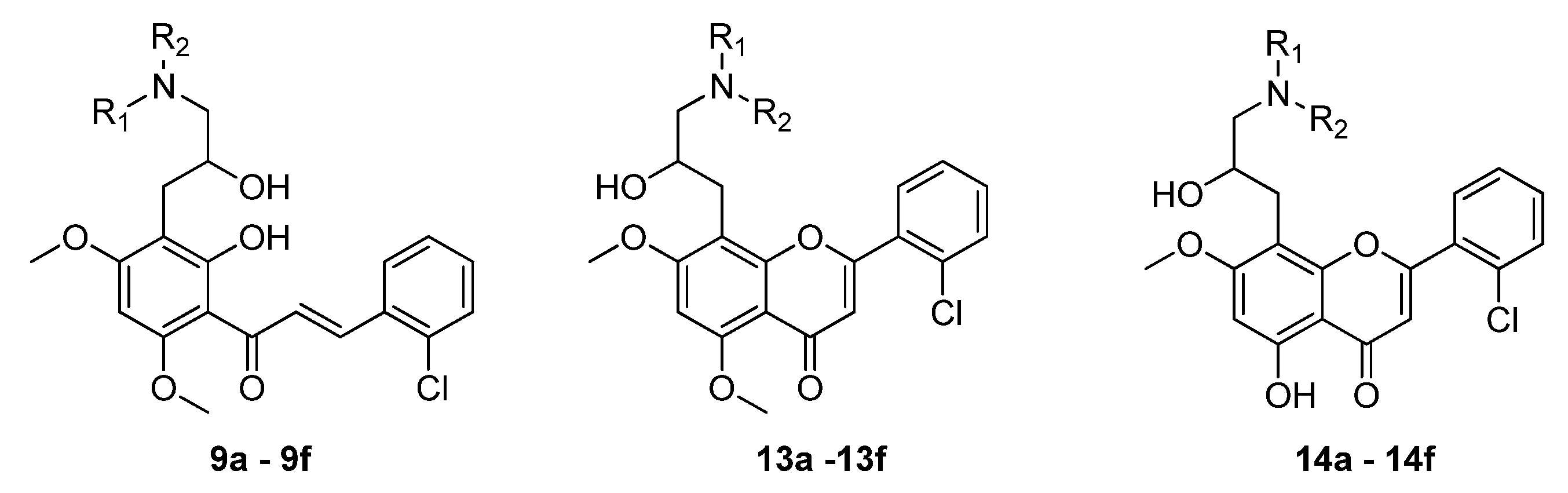
3.1.6. General Procedure to Obtain Target Chalcones 9a–f
To a stirred solution of 8 (0.15 g, 0.36 mmol) and a suitable secondary amine (3.6 mmol) in CH3CN (2 mL), a catalytic amount of triethylamine (0.1 mL) was added. The resulting mixture was stirred at room temperature for 24 h. Then acetonitrile was removed in vacuo, extracted with CH2Cl2, the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc/Et3N (100:100:2.5) as eluent to give pure target chalcones 9a–f. In this manner, the following target chalcones were obtained.
3-(2-Chlorophenyl)-1-{2-hydroxy-3-[2-hydroxy-3-(morpholino-4-yl)propyl]-4,6-dimethoxyphenyl}prop-2-en-1-one (9a). Yield: 72%, m.p. 141–143 °C; IR (KBr): 3458 (OH), 2920, 2851 (CH3, CH2), 1626 (C=O), 1566 (C=C), 1267 (Ar-O), 1172 (C-O), 758 cm−1; 1H-NMR (CDCl3) δ: 13.93 (s, 1H, OH-2'), 8.11 (d, 1H, J = 15.6 Hz, H-β), 7.81(d, 1H, J = 15.6 Hz, H-α), 7.69 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.01 (s, 1H, H-5'), 4.00 (s, 3H, OCH3), 3.95 (s, 3H, OCH3), 3.81 (m, 1H, H-2"), 3.70 (m, 4H, CH2O, CH2 × 2), 3.10 (dd, 1H, J1 = 13.6Hz, J2 = 7.2 Hz, H-1"α), 2.91 (dd, 1H, J1 = 13.2 Hz, J2 = 7.2 Hz, H-1"β), 2.49 (m, 2H, H-3"), 2.24 (m, 4H, NCH2, CH2 × 2). ESI-MS: m/z 462 [M+1]+.
3-(2-Chlorophenyl)-1-{2-hydroxy-3-[2-hydroxy-3-(piperidin-1-yl)propyl]-4,6-dimethoxyphenyl} prop-2-en-1-one (9b). Yield: 75%, m.p. 135–136 °C; IR (KBr): 3443 (OH), 2936, 2887 (CH3, CH2), 1628 (C=O), 1611, 1576 (C=C), 1272 (Ar-O), 1107 (C-O), 754 cm−1; 1H-NMR (CDCl3) δ: 8.10 (d, 1H, J = 15.6 Hz, H-β), 7.81 (d, 1H, J = 15.6 Hz, H-α), 7.69 (m, H-6), 7.43 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.01 (s, 1H, H-5'), 3.94 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.91 (m, 1H, H-2"), 2.87 (dd, 1H, J1 =13.6 Hz, J2 = 6.4 Hz, H-1"α), 2.73 (dd, 1H, J1 = 13.2 Hz, J2 =6.8 Hz, H-1"β), 2.55 (m, 2H, H-3"), 2.32 (m, 4H, NCH2, CH2 × 2), 1.53 (m, 4H, NCH2CH2, CH2 × 2), 1.41 (m, 2H, CH2CH2CH2); 13C-NMR (DMSO-d6) δ: 192.2, 167.0, 164.1, 162.0, 141.1, 134.6, 133.3, 131.6, 130.8, 129.1, 128.4, 127.3, 108.9, 108.1, 90.2, 71.1, 60.7, 56.8, 56.6, 53.3, 33.2, 23.8, 21.2; ESI-MS: m/z 460 [M+1]+.
3-(2-Chlorophenyl)-1-{2-hydroxy-3-[2-hydroxy-3-(pyrrolidin-1-yl)propyl]-4,6-dimethoxyphenyl} prop-2-en-1-one (9c). Yield: 55%, m.p. 132–135 °C; IR (KBr): 3453 (OH), 2937, 2879 (CH3, CH2), 1628 (C=O), 1601, 1576 (C=C), 1274 (Ar-O), 1110 (C-O), 754 cm−1; 1H-NMR (CDCl3) δ: 13.95 (s, 1H, OH-2'), 8.10 (d, 1H, J = 15.6 Hz, H-β), 7.79 (d, 1H, J = 15.6 Hz, H-α), 7.69 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.31 (m, 2H, H-4 and 5), 6.02 (s, 1H, H-5'), 3.94 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.91 (m, 1H, H-2"), 2.87 (dd, 1H, J1 =13.2 Hz, J2 = 5.6 Hz, H-1"α), 2.79 (dd, 1H, J1 = 13.2 Hz, J2 =5.8 Hz, H-1"β), 2.69 (m, 4H, NCH2, CH2 × 2), 2.52 (m, 2H, H-3"), 1.73 (s, 4H, NCH2CH2, CH2 × 2). ESI-MS: m/z 446 [M+1]+.
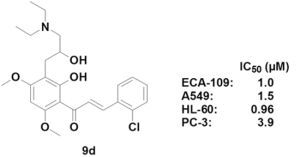
3-(2-Chlorophenyl)-1-[3-(3-diethylamino-2-hydroxypropyl)-2-hydroxy-4,6-dimethoxyphenyl]prop-2-en-1-one (9d). Yield: 63%, m.p. 114–116 °C; IR (KBr): 3450 (OH), 2936, 2859 (CH3, CH2), 1625 (C=O), 1579 (C=C), 1272 (Ar-O), 1123 (C-O), 759 cm−1; 1H-NMR (CDCl3) δ: 13.94 (s, 1H, OH-2'), 8.10 (d, 1H, J = 15.6 Hz, H-β), 7.84 (d, 1H, J = 15.6 Hz, H-α), 7.74 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.31 (m, 2H, H-4 and 5), 5.98 (s, 1H, H-5'), 3.97 (s, 3H, OCH3), 3.90 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 3.03 (dd, 1H, J1 =13.6 Hz, J2 =7.2 Hz, H-1"α), 2.84 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"β), 2.39 (m, 6H, H-3" and NCH2), 0.94 (t, 6H, NCH2CH3, CH3 × 2); 13C-NMR (DMSO-d6) δ: 192.5, 169.6, 164.9, 162.1, 140.9, 134.2, 132.3, 130.3, 129.8, 127.2, 127.1, 126.8, 108.0, 105.1, 90.5, 70.8, 61.0, 55.2, 54.6, 47.7, 34.2, 12.2; ESI-MS: m/z 448 [M+1]+.
3-(2-Chlorophenyl)-1-[3-(3-ethylmethylamino-2-hydroxypropyl)-2-hydroxy-4,6-dimethoxyphenyl] prop-2-en-1-one (9e). Yield: 76%, m.p. 115–118 °C; IR (KBr): 3448 (OH), 2924, 2856 (CH3, CH2), 1628 (C=O), 1563 (C=C), 1271 (Ar-O), 1120 (C-O), 749 cm−1; 1H-NMR (CDCl3) δ: 8.12 (d, 1H, J = 15.6 Hz, H-β), 7.82 (d, 1H, J = 15.6 Hz, H-α), 7.70 (m, 1H, H-6), 7.44 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.02 (s, 1H, H-5'), 3.95 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 2.88 (dd, 1H, J1 =13.2Hz, J2 = 5.2 Hz, H-1"α), 2.74 (dd, 1H, J1 = 13.6 Hz, J2 = 4.8 Hz, H-1"β), 2.41 (m, 4H, H-3" and NCH2), 2.24 (s, 3H, NCH3), 1.04 (t, 3H, NCH2CH3). ESI-MS: m/z 434 [M+1]+.
3-(2-Chlorophenyl)-1-[3-(3-dimethylamino-2-hydroxypropyl)-2-hydroxy-4,6-dimethoxyphenyl] prop-2-en-1-one (9f). Yield: 75%, m.p. 127–128 °C; IR (KBr): 3458 (OH), 2914, 2866 (CH3, CH2), 1629 (C=O), 1611, 1566 (C=C), 1256 (Ar-O), 1137 (C-O), 769 cm−1; 1H-NMR (CDCl3) δ: 14.03 (s, 1H, OH-2'), 8.12 (d, 1H, J = 15.6 Hz, H-α), 7.82 (d, 1H, J = 15.6 Hz, H-β), 7.69 (m, 1H, H-6), 7.43 (m, 1H, H-3), 7.30 (m, 2H, H-4 and 5), 6.02 (s, 1H, H-5'), 3.95 (s, 3H, OCH3), 3.92 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 2.87 (dd, 1H, J1 = 13.6Hz, J2 = 7.2 Hz, H-1"α), 2.75 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"β), 2.44 (t, 1H, J = 12.6 Hz, H-3"α), 2.26 (s, 6H, NCH3, CH3 × 2), 2.21 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-3"β). ESI-MS: m/z 420 [M+1]+.
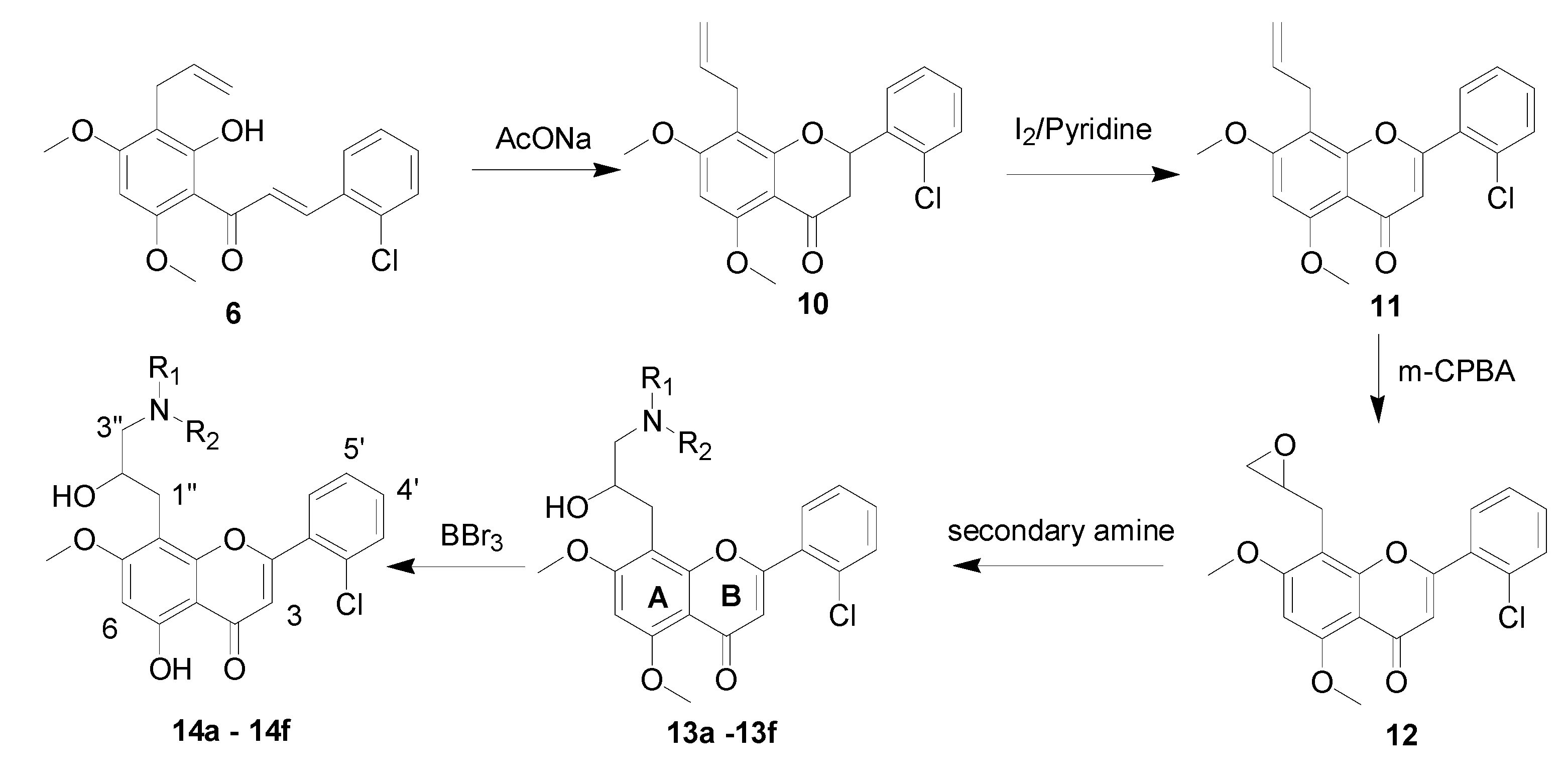
3.1.7. 8-Allyl-2-(2-chlorophenyl)-5,7-dimethoxy-2,3-dihydro-4H-1-benzopyran-4-one (10)
To a solution of 6 (4.52 g, 12.6 mmol) in 95% EtOH (136 mL) was added NaOAc (12.90, 157 mmol). The mixture was heated at reflux for 24 h, then EtOH was removed in vacuo, H2O was added and the mixture was extracted with CH2Cl2 (3 × 50 mL). The combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (5:1) as eluent to afford pure flavanone 10 (3.84 g, 85%) as a colorless solid, m.p. 131–133 °C. IR (KBr): 3006 (C=CH), 2942, 2879 (CH3, CH2), 1676 (C=O), 1602, 1567 (C=C), 1265 (Ar-O), 1126 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.70 (m, 1H, H-6'), 7.26 (m, 3H, H-3', 4' and 5'), 6.15 (s, 1H, H-6), 5.90 (m, 1H, H-2"), 5.73 (dd, 1H, J1 = 13.2 Hz, J2 = 3.2 Hz, H-2), 4.95 (m, 2H, H-2"), 3.96 (s, 3H, OCH3), 3.91 (s, 3H, OCH3), 3.35 (m, 2H, H-1"), 2.99 (dd, 1H, J1 = 16.8 Hz, J2 = 3.6 Hz, H-3'α), 2.75 (dd, 1H, J1 = 16.4 Hz, J2 = 4.0 Hz, H-3'β). ESI-MS: m/z 359 [M+1]+.
3.1.8. 8-Allyl-2-(2-chlorophenyl)-5,7-dimethoxy-4H-1-benzopyran-4-one (11)
To a stirred solution of 10 (2.1 g, 5.85 mmol) in pyridine (42 mL) was added I2 (1.49 g, 5.86 mmol). The mixture was heated to 90 °C for 6 h, and then pyridine was removed in vacuo, followed by addition of solid Na2SO3 until fading of the color. The mixture was extracted with CH2Cl2 (3 × 50 mL); the combined organic extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (2:1) as eluent to give pure flavone 11 (1.00 g, 49%) as a colorless solid, m.p. 181–183 °C. IR (KBr): 3015 (C=CH), 2943, 2879 (CH3, CH2), 1663 (C=O), 1602, 1573 (C=C), 1217 (Ar-O), 1063 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.63 (m, 1H, H-6'), 7.52 (m, 1H, H-3'), 7.39 (m, 2H, H-4' and 5'), 6.54 (s, 1H, H-3), 6.46 (s, 1H, H-6), 5.92 (m, 1H, H-2"), 4.95 (m, 2H, H-3"), 4.02 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.54 (m, 2H, H-1"). ESI-MS: m/z 357 [M+1]+.
3.1.9. 2-(2-Chlorophenyl)-5,7-dimethoxy-8-oxiranylmethyl-4H-1-benzopyran-4-one (12)
To a stirred solution of 11 (3.28 g, 10 mmol) in CH2Cl2 (40 mL) was added a solution of m-chloroperoxybenzoic acid (0.42 g, 2.5 mmol) in CH2Cl2 (80 mL) dropwise during 0.5 h, and the mixture was stirred at room temperature for 24 h, then the CH2Cl2 layer was washed successively with aqueous sodium bicarbonate and brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using petroleum ether/EtOAc (2:1) as eluent to give pure 12 (1.78 g, 68%) as colorless solid. m.p. 106–109 °C. IR (KBr): 2999, 2886 (CH3, CH2), 1645 (C=O), 1603, 1579 (C=C), 1265 (Ar-O), 1116 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.70 (m, 1H, H-6'), 7.28 (m, 3H, H-3', 4' and 5'), 6.52 (s, 1H, H-3), 6.47 (s, 1H, H-6), 4.01 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 3.30 (dd, 1H, J1 = 13.6 Hz, J2 = 6.0 Hz, H-1"α), 3.16 (m, 1H, H-2"), 2.90 (dd, 1H, J1 = 13.6 Hz, J2 = 6.4 Hz, H-1"β), 2.69 (t, 1H, J = 5.0 Hz, H-3"α), 2.51 (m, 1H, J1 = 4.8 Hz, J2 = 2.0 Hz, H-3"β). ESI-MS: m/z 373 [M+1]+.
3.1.10. General Procedure to Obtain Dimethoxyflavones 13a–f
To a stirred solution of 12 (0.19 g, 0.50 mmol) and a suitable secondary amine (5.0 mmol) in ethanol (2 mL), a catalytic amount of triethylamine (0.1 mL) was added. The resulting mixture was refluxed for 24 h. Then ethanol was removed in vacuo, and the residue was then extracted with CH2Cl2. The CH2Cl2 extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using EtOAc/EtOH/Et3N (100:2:2.5) as eluent to afford pure target chalcones 13a–f. In this manner, the following target dimethoxyflavones were obtained:
2-(2-Chlorophenyl)-8-[2-hydroxy-3-(morpholino-4-yl)propy]-5,7-dimethoxy-4H-1-benzopyran-4-one (13a). Yield: 75%, m.p. 145–147 °C; IR (KBr): 3427 (OH), 2939, 2874 (CH3, CH2), 1645 (C=O), 1612, 1601 (C=C), 1285 (Ar-O), 1066 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.68 (m, 1H, H-6'), 7.54 (m, 1H, H-3'), 7.39 (m, 2H, H-4' and 5'), 6.52 (s, 1H, H-3), 6.49 (s, 1H, H-6), 5.00 (brs, 1H, OH-2''), 4.04 (s, 3H, OCH3), 3.98 (s, 3H, OCH3), 4.01 (m, 1H, H-2"), 3.69 (m, 4H, CH2O, CH2 × 2), 3.10 (dd, 1H, J1 =13.6 Hz, J2 =7.2 Hz, H-1"α), 2.91 (dd, 1H, J1 = 13.2 Hz, J2 = 7.2 Hz, H-1"β), 2.49 (m, 2H, H-3"), 2.24 (m, 4H, NCH2, CH2 × 2). ESI-MS: m/z 460 [M+1]+.
2-(2-Chlorophenyl)-8-[2-hydroxy-3-(piperidin-1-yl)propyl]-5,7-dimethoxy-4H-1-benzopyran-4-one (13b). Yield: 78%, m.p. 149–153 °C; IR (KBr): 3434 (OH), 2932, 2847 (CH3, CH2), 1655 (C=O), 1612, 1600 (C=C), 1284 (Ar-O), 1120 (C-O), 770 cm−1; 1H-NMR (CDCl3) δ: 7.66 (m, 1H, H-6'), 7.52 (m, 1H, H-3'), 7.39 (m, 2H, H-4' and 5'), 6.52 (s, 1H, H-3), 6.46 (s, 1H, H-6), 4.00 (s, 3H, OCH3), 3.96 (s, 3H, OCH3), 3.95 (m, 1H,H-2"), 3.07 (dd, 1H, J1 =13.6 Hz, J2 = 6.4 Hz, H-1"α), 2.86 (dd, 1H, J1 = 13.2 Hz, J2 =6.8 Hz, H-1"β), 2.50 (brs, 1H, OH-2''), 2.32 (m, 2H, H-3"), 2.25 (m, 4H, NCH2, CH2 × 2), 1.52 (m, 4H, NCH2CH2, CH2 × 2), 1.39 (m, 2H, CH2CH2CH2). ESI-MS: m/z 458 [M+1]+.
2-(2-Chlorophenyl)-8-[2-hydroxy-3-(pyrrolidin-1-yl)propyl]-5,7-dimethoxy-4H-1-benzopyran-4-one (13c). Yield: 74%, m.p. 149–153 °C; IR (KBr): 3428 (OH), 2963, 2933 (CH3, CH2), 1643 (C=O), 1602 (C=C), 1376 (Ar-O), 1127 (C-O) cm−1; 1H-NMR (CDCl3) δ: 7.67 (m, 1H, H-6'), 7.52(m, 1H, H-3'), 7.40 (m, 2H, H-4' and'), 6.52 (s, 1H, H-3), 6.46 (s, 1H, H-6), 4.04 (s, 3H, OCH3), 4.00 (s, 3H, OCH3), 4.02 (m, 1H, H-2"), 3.06 (dd, 1H, J1 =13.6 Hz, J2 = 6.8 Hz, H-1"α), 2.92 (dd, 1H, J1 = 13.6 Hz, J2 =6.8 Hz, H-1"β), 2.31 (m, 6H, NCH2 × 2 and H-3"), 1.73 (s, 4H, NCH2CH2, CH2 × 2). ESI-MS: m/z 444 [M+1]+.
2-(2-Chlorophenyl)-8-(3-diethylamino-2-hydroxypropyl)-5,7-dimethoxy-4H-1-benzopyran-4-one (13d). Yield: 80%, m.p. 149–153 °C; IR (KBr): 3435 (OH), 2964, 2931 (CH3, CH2), 1654 (C=O), 1621, 1602 (C=C), 1384 (Ar-O), 1118 (C-O), 772 cm−1; 1H-NMR (CDCl3) δ: 7.69 (m, 1H, H-6'), 7.52 (m, 1H, H-3'), 7.36 (m, 2H, H-4' and 5'), 6.53 (s, 1H, H-3), 6.42 (s, 1H, H-6), 4.01 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.90 (m, 1H, H-2"), 3.07 (dd, 1H, J1 = 13.2 Hz, J2 = 6.4 Hz, H-1"α), 2.84 (dd, 1H, J1 = 13.2 Hz, J2 = 6.4 Hz, H-1"β), 2.39 (m, 6H, H-3" and NCH2), 0.94 (t, 6H, NCH2CH3, CH3 × 2); 13C-NMR (DMSO-d6) δ: 179.3, 165.3, 164.2, 161.9, 157.0, 134.2, 132.9, 130.1, 129.8, 128.0, 126.5, 125.2, 112.5, 107.1, 95.8, 70.8, 61.0, 56.2, 55.9, 47.5, 35.8, 12.6; ESI-MS: m/z 446 [M+1]+.
2-(2-Chlorophenyl)-8-(3-ethylmethylamino-2-hydroxypropyl)-5,7-dimethoxy-4H-1-benzopyran-4-one (13e). Yield: 82%, m.p. 149–153 °C; IR (KBr): 3424 (OH), 2968, 2938 (CH3, CH2), 1653 (C=O), 1602, 1578 (C=C), 1328 (Ar-O), 1120 (C-O), 763 cm−1; 1H-NMR (CDCl3) δ: 7.67 (m, 1H, H-6'), 7.51 (m, 1H, H-3'), 7.36 (m, 2H, H-4' and 5'), 6.52 (s, 1H, H-3), 6.45 (s, 1H, H-6), 4.01 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 3.07 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"α), 2.85 (dd, 1H, J1 = 13.2 Hz, J2 = 6.8 Hz, H-1"β), 2.41 (m, 4H, H-3" and NCH2), 2.17 (s, 3H, NCH3), 0.96 (t, 3H, NCH2CH3). ESI-MS: m/z 432 [M+1]+.
2-(2-Chlorophenyl)-8-(3-dimethylamino-2-hydroxypropyl)-5,7-dimethoxy-4H-1-benzopyran-4-one (13f). Yield: 87%, m.p. 149–153 °C; IR (KBr): 3441 (OH), 2939 (CH3, CH2), 1653 (C=O), 1623, 1602 (C=C), 1327 (Ar-O), 1120 (C-O), 762 cm−1; 1H-NMR (CDCl3) δ: 7.66 (m, 1H, H-6'), 7.51 (m, 1H, H-3'), 7.38 (m, 2H, H-4' and 5'), 6.51 (s, 1H, H-3), 6.46 (s, 1H, H-6), 4.00 (s, 3H, OCH3), 3.97 (s, 3H, OCH3), 3.92 (m, 1H, H-2"), 3.05 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"α), 2.86 (dd, 1H, J1 = 13.2 Hz, J2 = 3.8 Hz, H-1"β), 2.37 (t, 1H, J =11.8 Hz, H-3"α), 2.18 (s, 6H, NCH3, CH3 × 2), 2.11 (dd, 1H, J1 = 12.4 Hz, J2 = 3.2 Hz, H-3"β). ESI-MS: m/z 418 [M+1]+.
3.1.11. General Procedure to Obtain Monomethoxyflavones 14a–f
To a stirred solution of dimethoxyflavone (0.2 mmol) in 1,2-dichloroethane (2 mL) at room temperature was added a solution of BBr3 (1 mol/L, 0.4 mL, 0.4 mmol). The resulting mixture was stirred at room temperature for 3 h. It was cooled to 0 °C, and quenched by water (10 mL). It was made alkaline with Na2CO3 solution and extracted with CH2Cl2. The CH2Cl2 extracts were washed with brine, dried over anhydrous Na2SO4, and concentrated under reduced pressure. The residue was purified by silica gel column chromatography using EtOAc/petroleum ether/Et3N (100:50:2.5) as eluent to give pure monomethoxyflavones 14a–f. In this manner, the following targets were obtained:
2-(2-Chlorophenyl)-5-hydroxy-8-[2-hydroxy-3-(morpholin-4-yl)propyl]-7-methoxy-4H-1-benzopyran-4-one (14a). Yield: 88%, m.p. 139–151 °C; IR (KBr): 3424 (OH), 2934, 2870 (CH3, CH2), 1658 (C=O), 1620, 1601 (C=C), 1278 (Ar-O), 1166 (C-O) cm−1; 1H-NMR (CDCl3) δ: 12.80 (s, 1H, OH-5), 7.66 (m, 1H, H-6'), 7.54 (m, 1H, H-3'), 7.42 (m, 2H, H-4' and 5'), 6.54 (s, 1H, H-6), 6.45 (s, 1H, H-3), 3.98 (m, 1H, H-2"), 3.92 (s, 3H, OCH3), 3.64 (m, 4H, -CH2-O, CH2 × 2), 3.03 (dd, 1H, J1 = 13.6 Hz, J2 = 6.8 Hz, H-1"α), 2.85 (dd, 1H, J1 = 13.6 Hz, J2 = 5.6 Hz, H-1"β), 2.56 (m, 2H, H-3"), 2.30 (m, 4H, -N-CH2, CH2 × 2). ESI-MS: m/z 446 [M+1]+.
2-(2-Chlorophenyl)-5-hydroxy-8-[2-hydroxy-3-(piperidin-1-yl)propyl]-7-methoxy-4H-1-benzopyran-4-one (14b). Yield: 85%, m.p. 127–128 °C; IR (KBr): 3430 (OH), 2930, 2845 (CH3, CH2), 1659 (C=O), 1612, 1600 (C=C), 1280 (Ar-O), 1121(C-O), 771 cm−1; 1H-NMR (CDCl3) δ: 12.80 (s, 1H, OH-5), 7.68 (m, 1H, H-6'), 7.51(m, 1H, H-3'), 7.42 (m, 2H, H-4' and 5'), 6.53 (s, 1H, H-3), 6.42 (s, 1H, H-6), 3.89 (m, 4H, OCH3 and H-2"), 3.56 (brs, 1H, OH-2"), 3.00 (dd, 1H, J1 =13.6 Hz, J2 = 6.0 Hz, H-1"α), 2.79 (dd, 1H, J1 = 13.2 Hz, J2 =6.0 Hz, H-1"β), 2.47 (m, 2H, H-3"), 2.23 (m, 4H, NCH2, CH2 × 2), 1.48 (m, 4H, NCH2CH2, CH2 × 2), 1.37 (m, 2H, CH2CH2CH2). ESI-MS: m/z 444 [M+1]+.
2-(2-Chlorophenyl)-5-hydroxy-8-[2-hydroxy-3-(pyrrolidin-1-yl)propyl]-7-methoxy-4H-1-benzopyran-4-one (14c). Yield: 72%, m.p. 134–137 °C; IR (KBr): 3424 (OH), 2963, 2933 (CH3, CH2), 1659 (C=O), 1602 (C=C), 1384 (Ar-O), 1117 (C-O), 765 cm−1; 1H-NMR (CDCl3) δ: 12.79 (s, 1H, OH-5), 7.67 (m, 1H, H-6'), 7.51 (m, 1H, H-3'), 7.37 (m, 2H, H-4' and 5'), 6.53 (s, 1H, H-3), 6.42 (s, 1H, H-6), 3.88 (s, 3H, OCH3), 3.78 (m, 1H, H-2"), 3.54 (brs, 1H, OH-2"), 3.00 (dd, 1H, J1 = 13.6 Hz, J2 = 4.8 Hz, H-1"α), 2.78 (dd, 1H, J1 = 13.6 Hz, J2 = 4.8 Hz, H-1"β), 2.48 (m, 2H, H-3"), 2.22 (s, 4H, NCH2, CH2 × 2), 1.73 (s, 4H, NCH2CH2, CH2 × 2). ESI-MS: m/z 430 [M+1]+.
2-(2-Chlorophenyl)-8-(3-diethylamino-2-hydroxypropyl)-5-hydroxy-7-methoxy-4H-1-benzopyran-4-one (14d). Yield: 77%, m.p. 114–115 °C; IR (KBr): 3437 (OH), 2962, 2929 (CH3, CH2), 1662 (C=O), 1619, 1600 (C=C), 1294 (Ar-O), 1114 (C-O), 775 cm−1; 1H-NMR (CDCl3) δ: 12.79 (s, 1H, OH-5), 7.69 (m, 1H, H-6'), 7.53 (m, 1H, H-3'), 7.40 (m, 2H, H-4' and 5'), 6.53 (s, 1H, H-3), 6.42 (s, 1H, H-6), 4.01 (m, 1H, H-2"), 3.90 (s, 3H, OCH3), 3.06 (dd, 1H, J1 =13.4 Hz, J2 = 6.4 Hz, H-1"α), 2.84 (dd, 1H, J1 = 13.2 Hz, J2 =6.4 Hz, H-1"β), 2.39 (m, 6H, H-3" and NCH2), 0.94 (t, 6H, NCH2CH3, CH3 × 2); 13C-NMR (DMSO-d6) δ: 183.3, 166.3, 165.2, 162.9, 157.0, 134.2, 132.9, 131.1, 130.8, 129.0, 126.9, 125.2, 108.5, 106.1, 95.8, 70.4, 60.8, 56.2, 55.9, 47.6, 36.2, 12.4; ESI-MS: m/z 432 [M+1]+.
2-(2-Chlorophenyl)-8-(3-ethylmethylamino-2-hydroxypropyl)-5-hydroxy-7-methoxy-4H-1-benzopyran-4-one (14e). Yield: 70%, m.p. 119–123 °C; IR (KBr): 3425 (OH), 2965, 2945 (CH3, CH2), 1662 (C=O), 1617, 1583 (C=C), 1332 (Ar-O), 1114 (C-O) cm−1; 1H-NMR (CDCl3) δ: 12.80 (s, 1H, OH-5), 7.70 (m, 1H, H-6'), 7.54 (m, 1H, H-3'), 7.44 (m, 2H, H-4' and 5'), 6.56 (s, 1H, H-3), 6.45 (s, 1H, H-6), 3.92 (s, 3H, OCH3), 3.90 (m, 1H, H-2"), 3.02 (dd, 1H, J1 = 12.6 Hz, J2 = 6.4 Hz, H-1"α), 2.82 (dd, 1H, J1 = 13.0 Hz, J2 = 6.4 Hz, H-1"β), 2.42 (m, 4H, H-3" and NCH2), 2.17 (s, 3H, NCH3), 0.98 (t, 3H, NCH2CH3). ESI-MS: m/z 418 [M+1]+.
2-(2-Chlorophenyl)-8-(3-dimethylamino-2-hydroxypropyl)-5-hydroxy-7-methoxy-4H-1-benzopyran-4-one (14f). Yield: 80%, m.p. 149–153 °C; IR (KBr): 3435 (OH), 2937 (CH3, CH2), 1662 (C=O), 1617, 1583 (C=C), 1287 (Ar-O), 1120 (C-O) cm−1; 1H-NMR (CDCl3) δ: 12.82 (s, 1H, OH-5), 7.69 (m, 1H, H-6'), 7.54 (m, 1H, H-3'), 7.45 (m, 2H, H-4' and 5'), 6.55 (s, 1H, H-3), 6.45 (s, 1H, H-6), 3.92 (s, 3H, OCH3), 3.91 (m, 1H, H-2"), 3.02 (dd, 1H, J1 = 13.6 Hz, J2 = 3.8 Hz, H-1"α), 2.83 (dd, 1H, J1 = 13.2 Hz, J2 = 3.2 Hz, H-1"β), 2.36 (t, 1H, J = 12.0 Hz, H-3"α), 2.20 (s, 6H, NCH3, CH3 × 2), 2.12 (dd, 1H, J1 = 12.0 Hz, J2 = 3.2 Hz, H-3"β); 13C-NMR (DMSO-d6) δ: 183.5, 166.0, 165.0, 162.8, 157.1, 134.6, 133.2, 131.4, 130.8, 128.4, 126.9, 124.8, 107.5, 105.7, 95.6, 70.2, 63.8, 56.0, 44.6, 36.0; ESI-MS: m/z 404 [M+1]+.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}














