Coordination Programming of Photofunctional Molecules

Abstract
:1. 2-Pyridylpyrimidine-Cu Complexes: Visible Light-Induced Pyrimidine Ring Rotation and Reversible CuII/CuI Electrochemical Potential Switching
1.1. Introduction


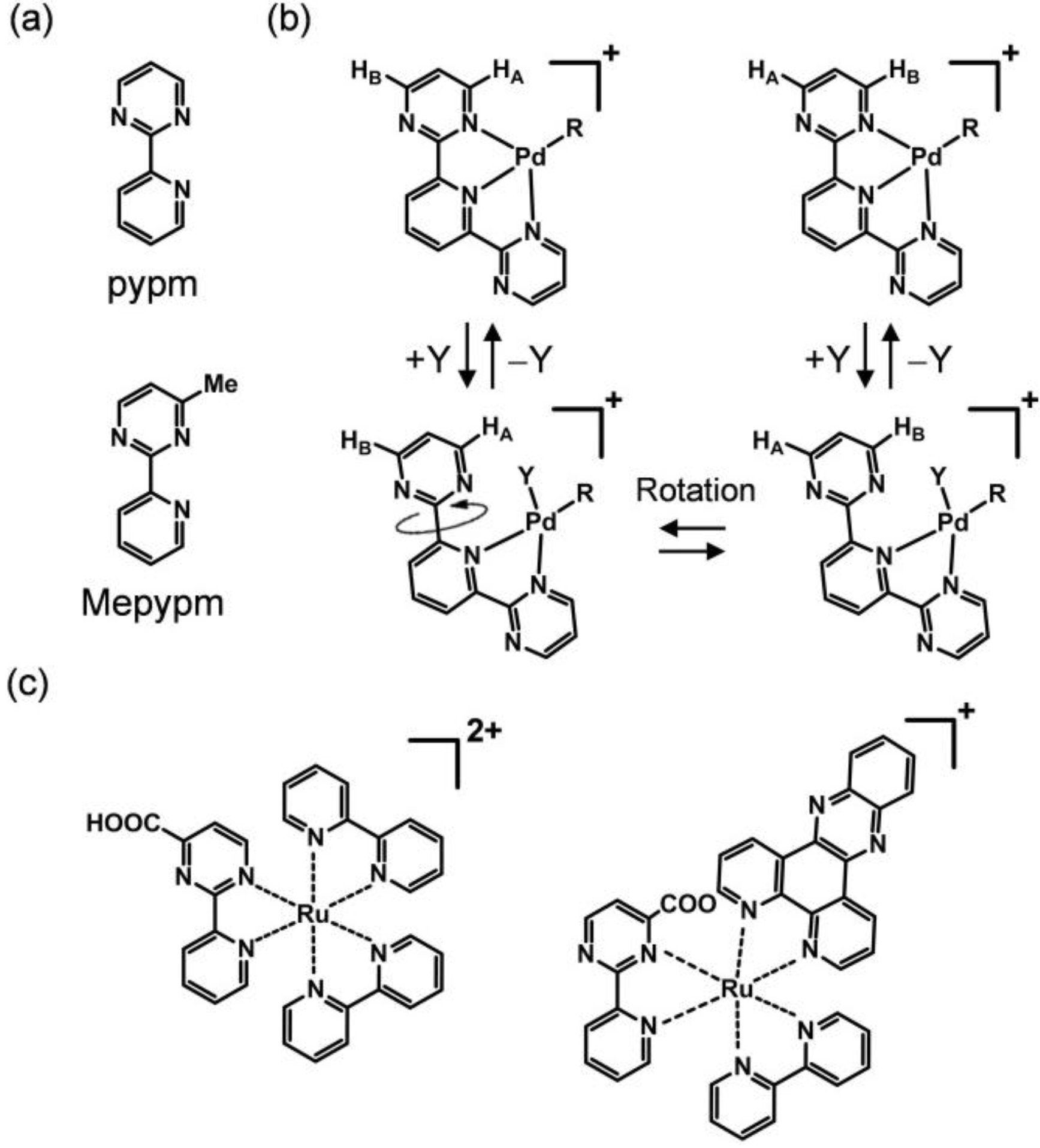
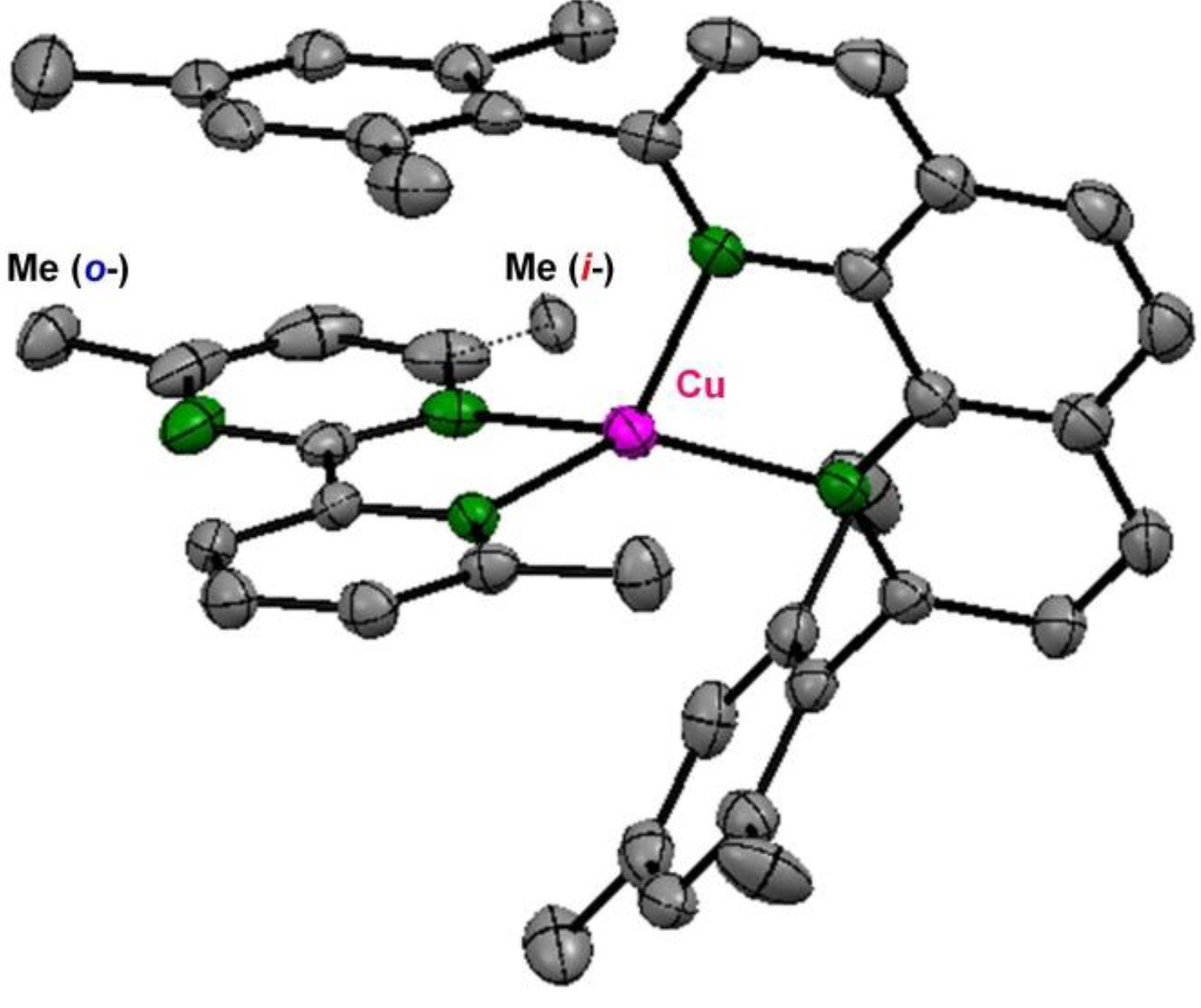
1.2. Ligand Design, Structure, and Electrochemical Ring Rotation


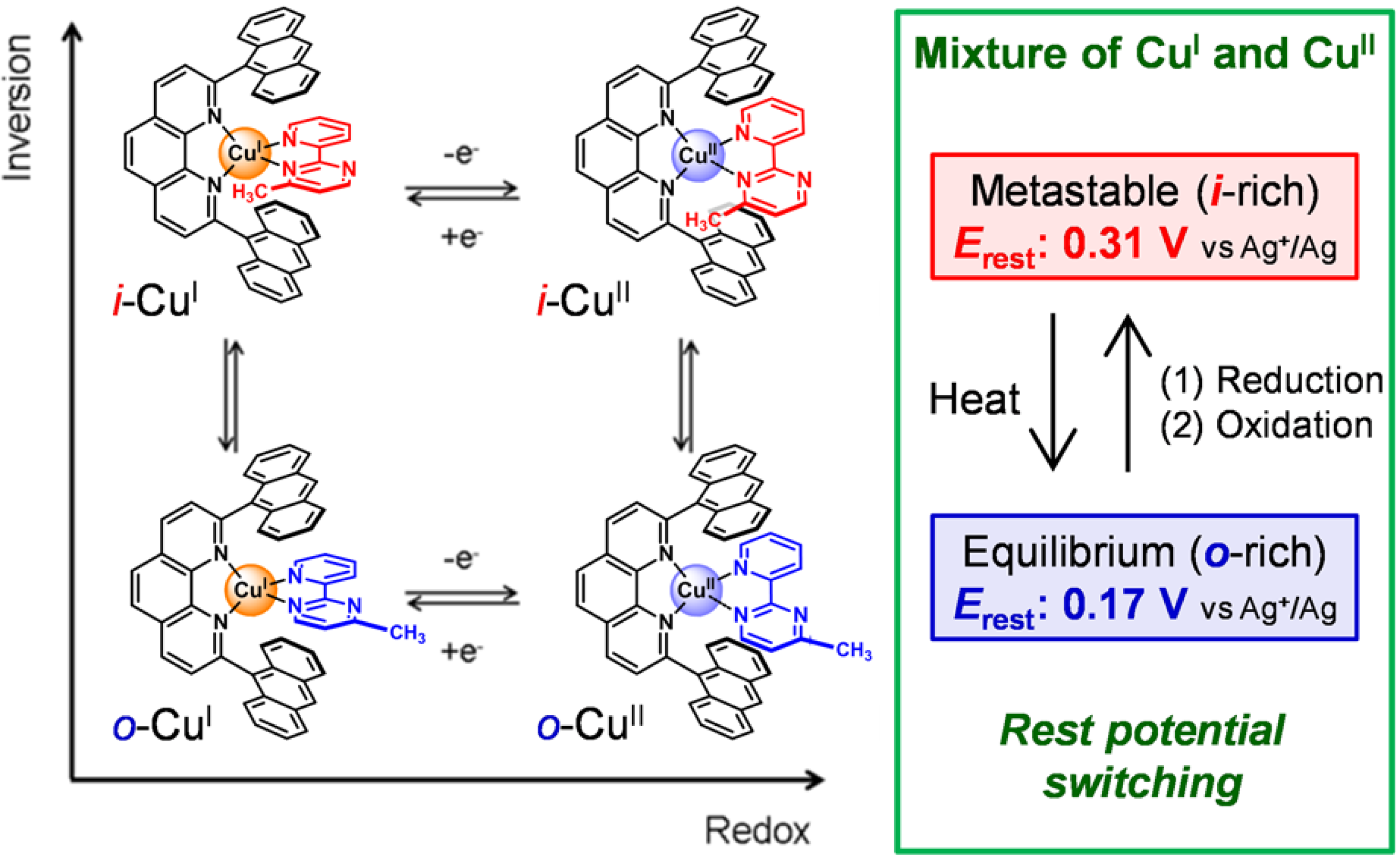
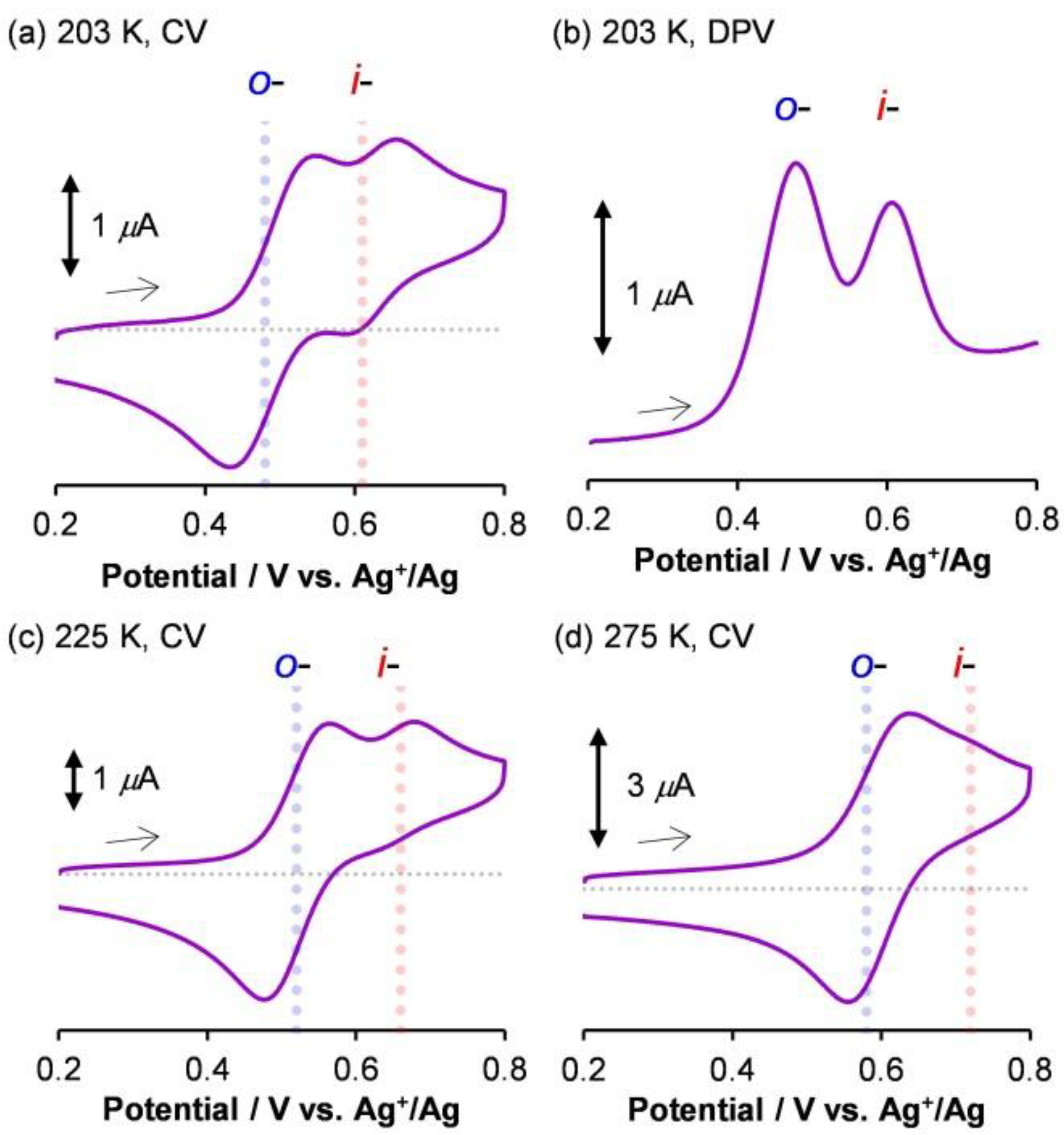
1.3. Electrochemistry


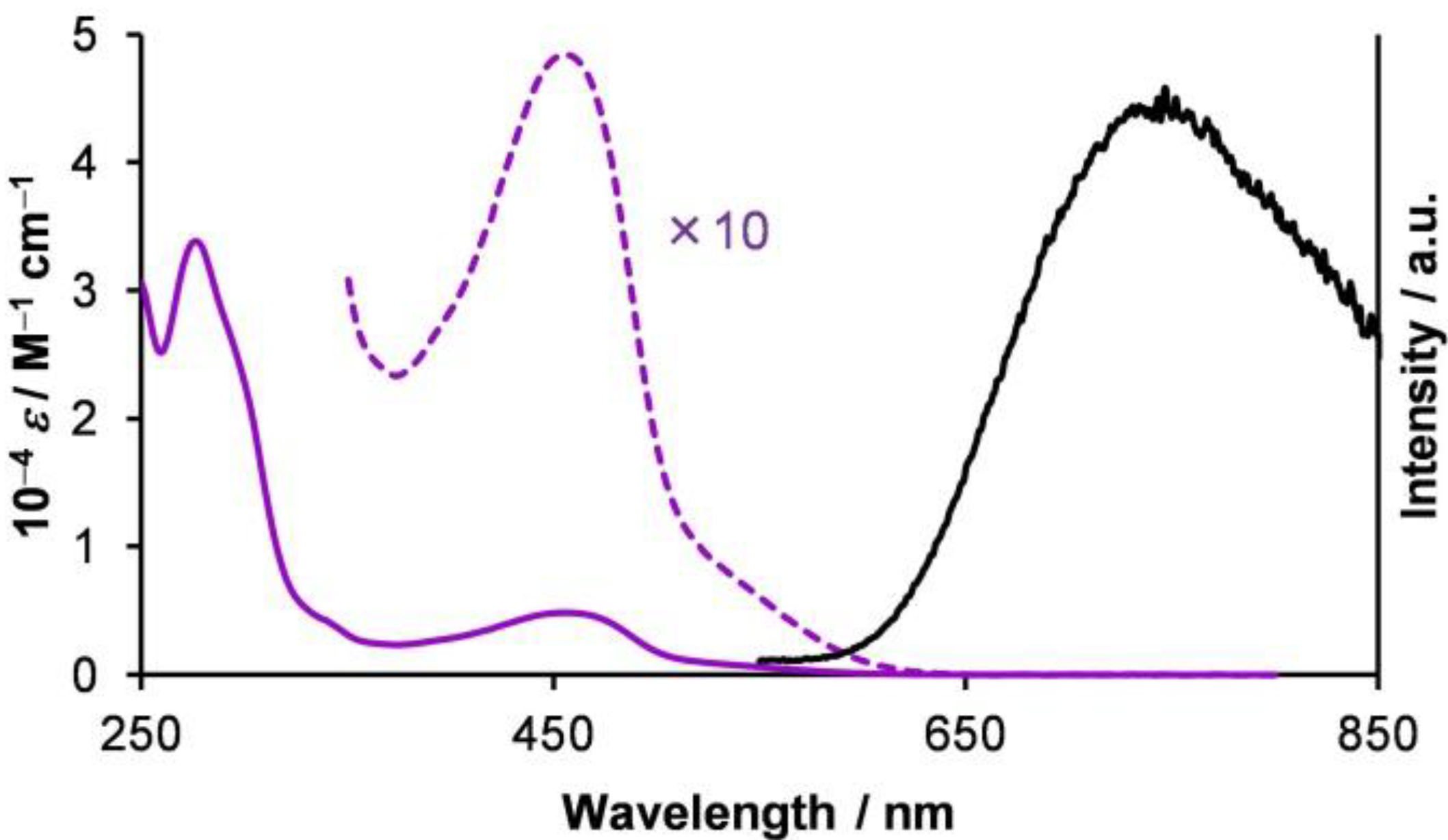
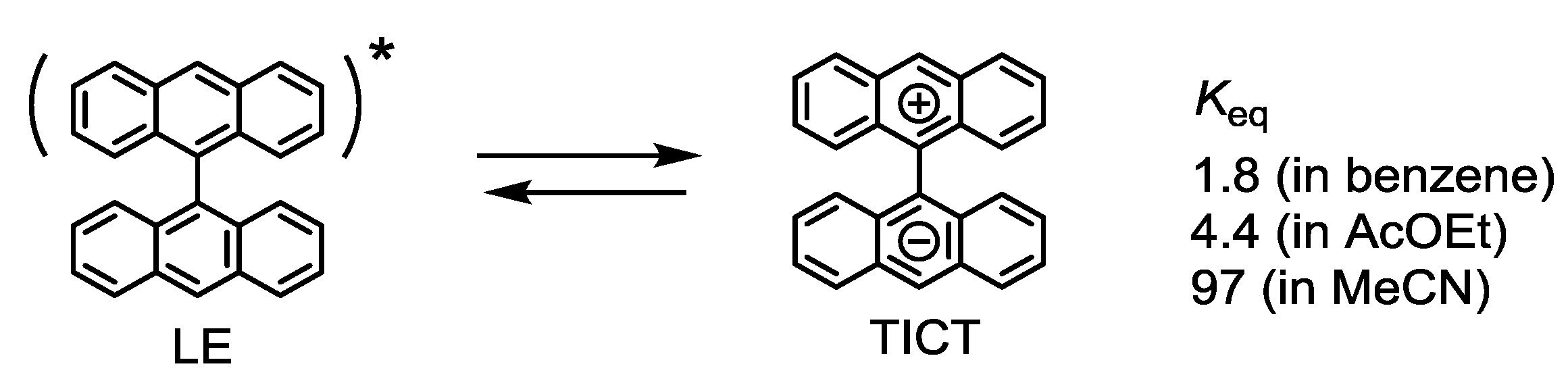
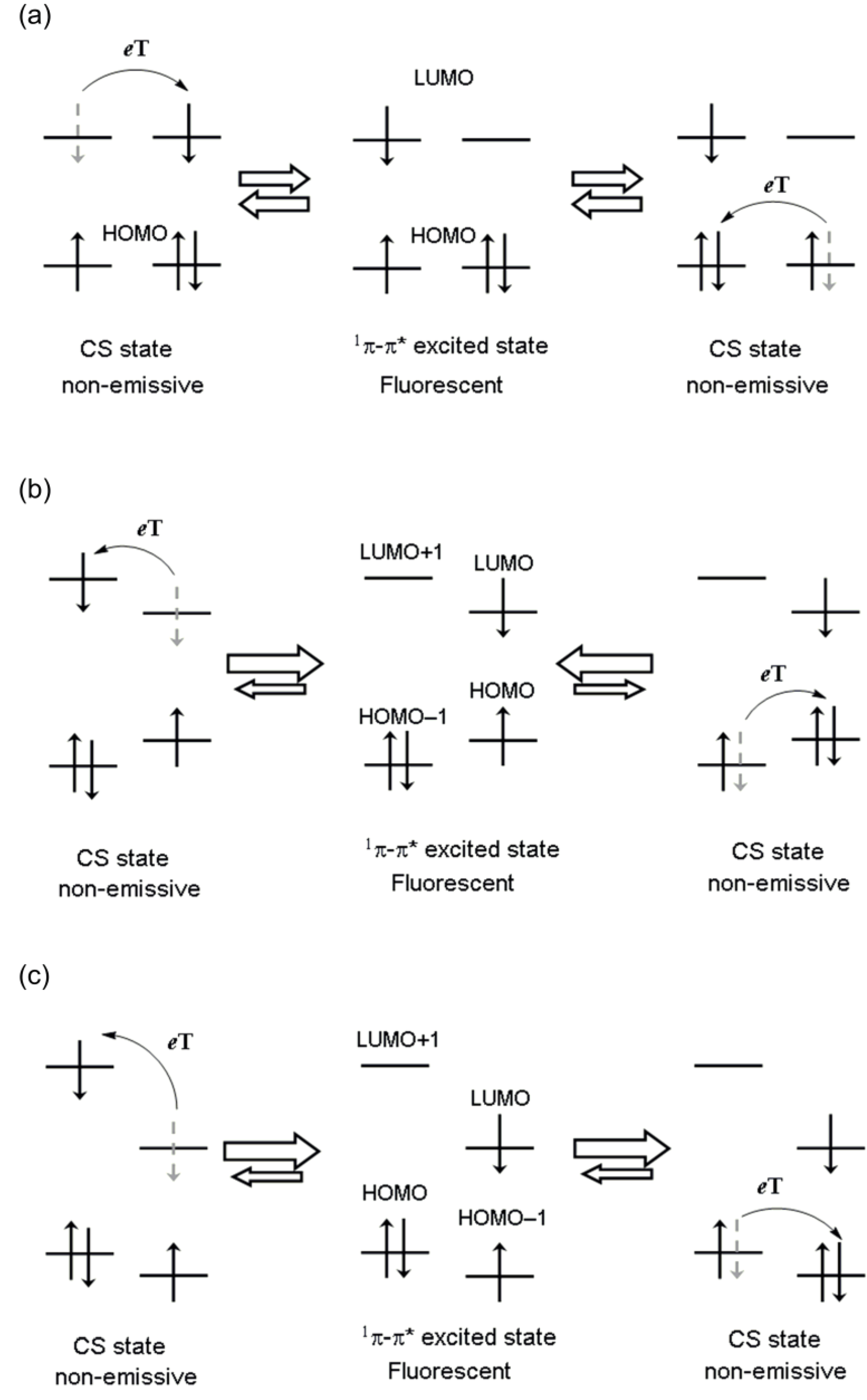
1.4. Photophysical Properties


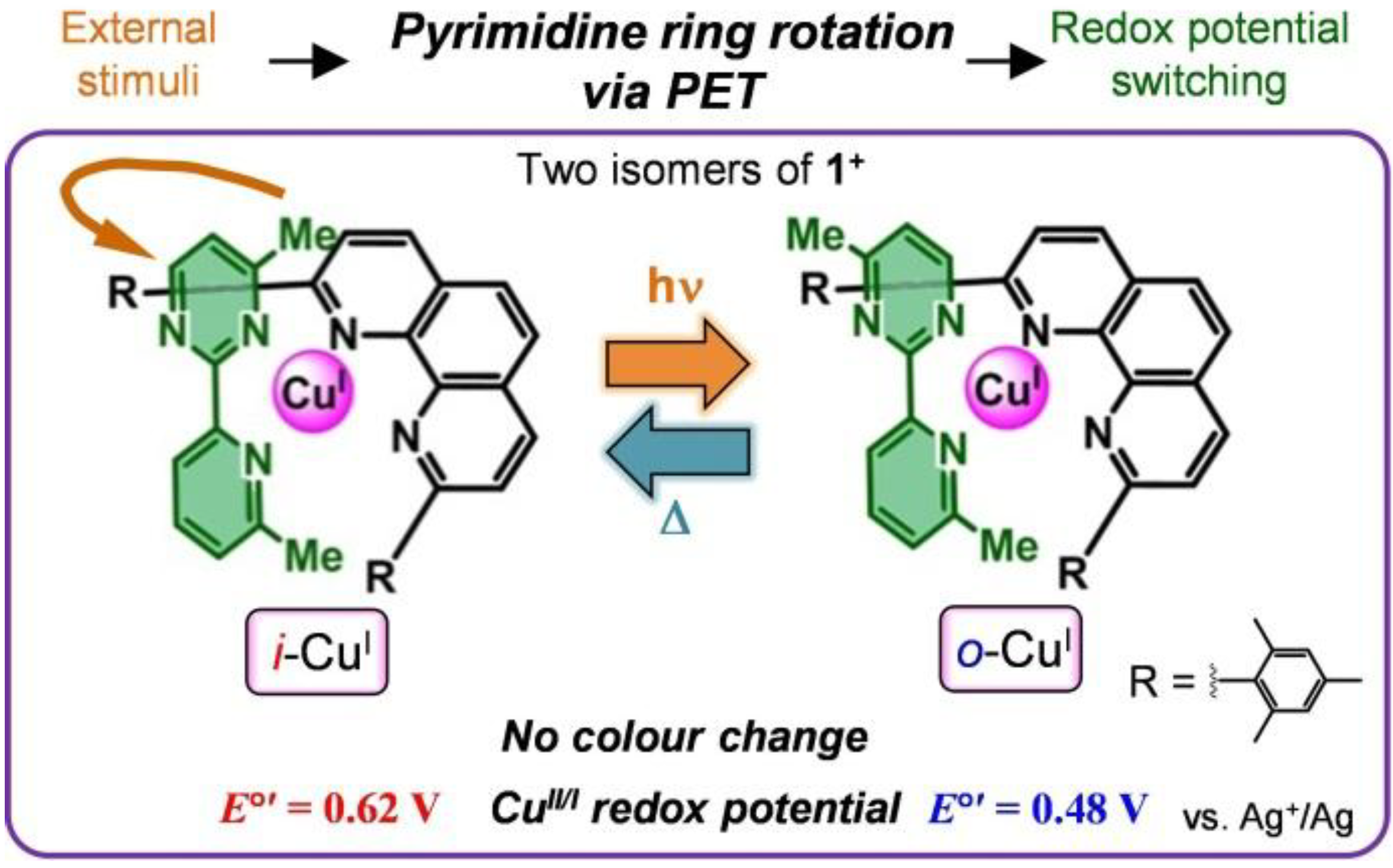
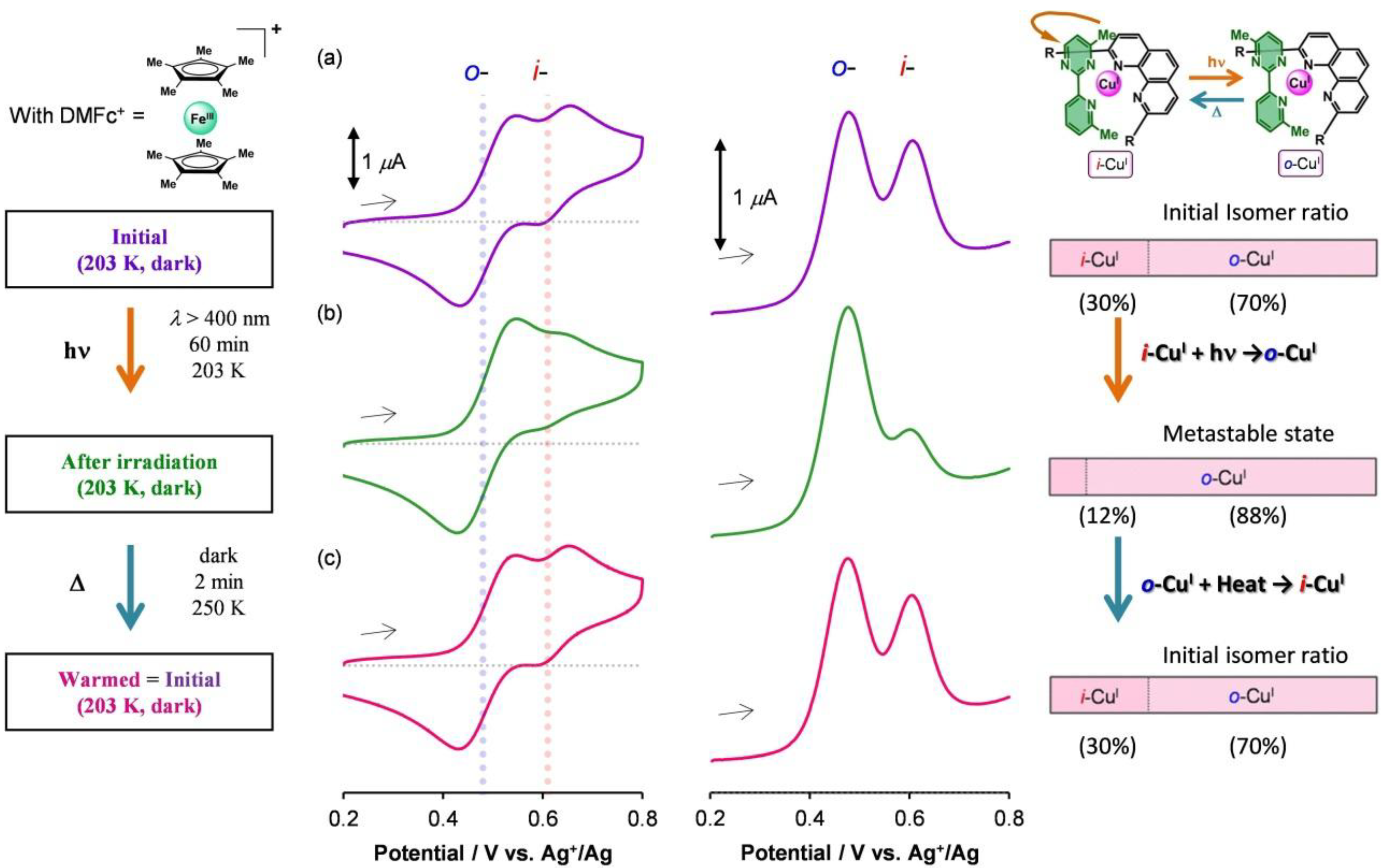
1.5. Photodriven Rotation of the Pyrimidine Ring

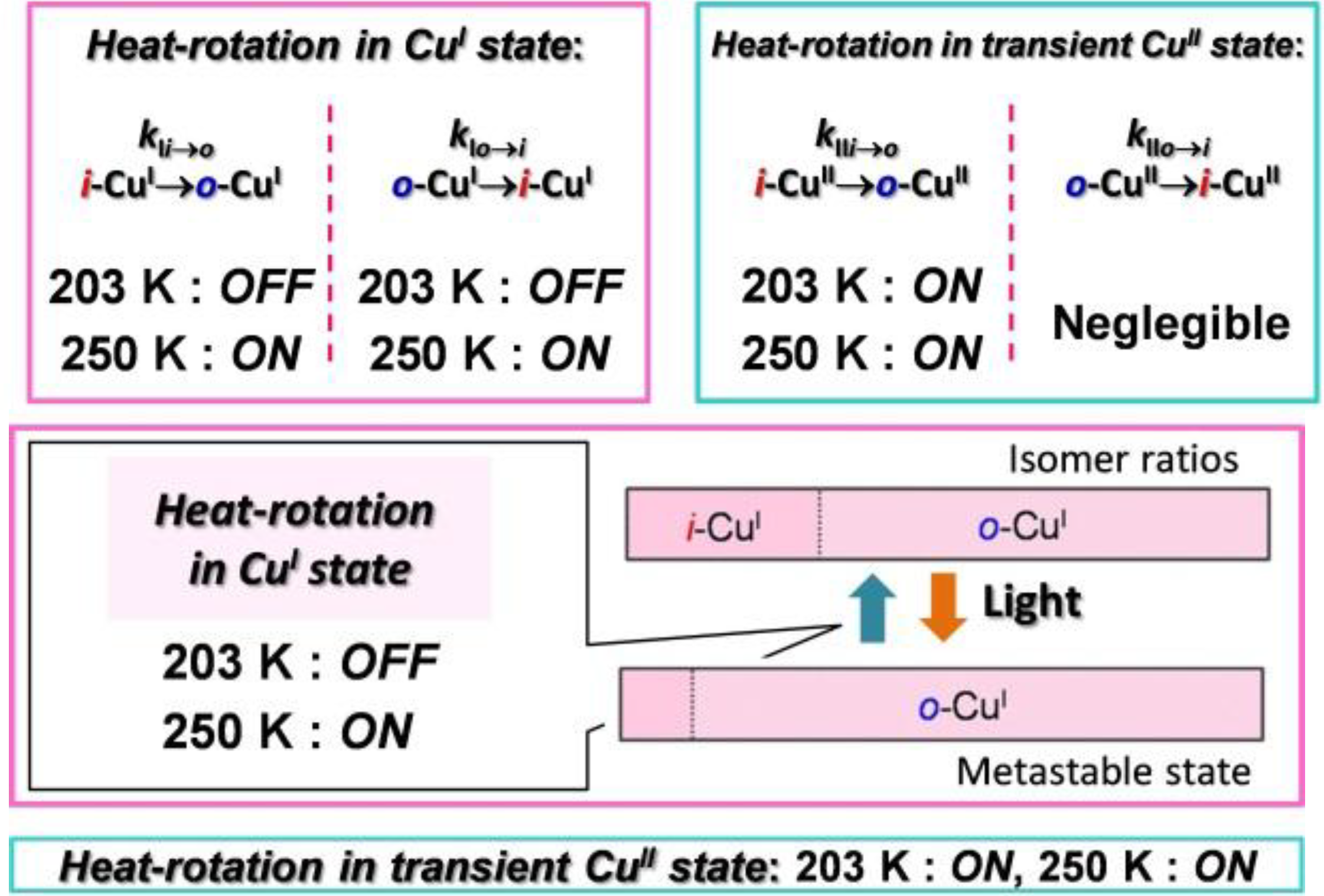
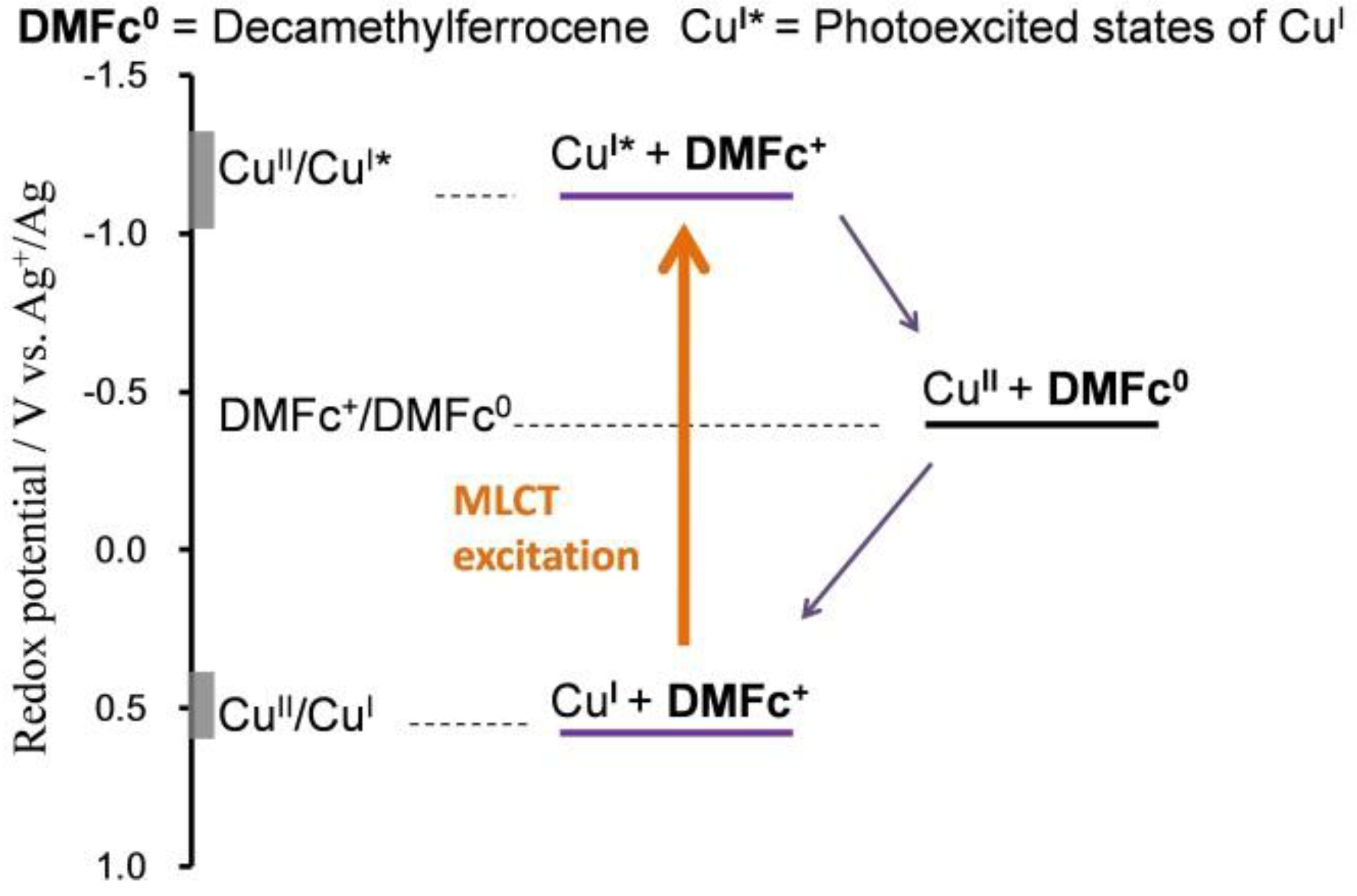
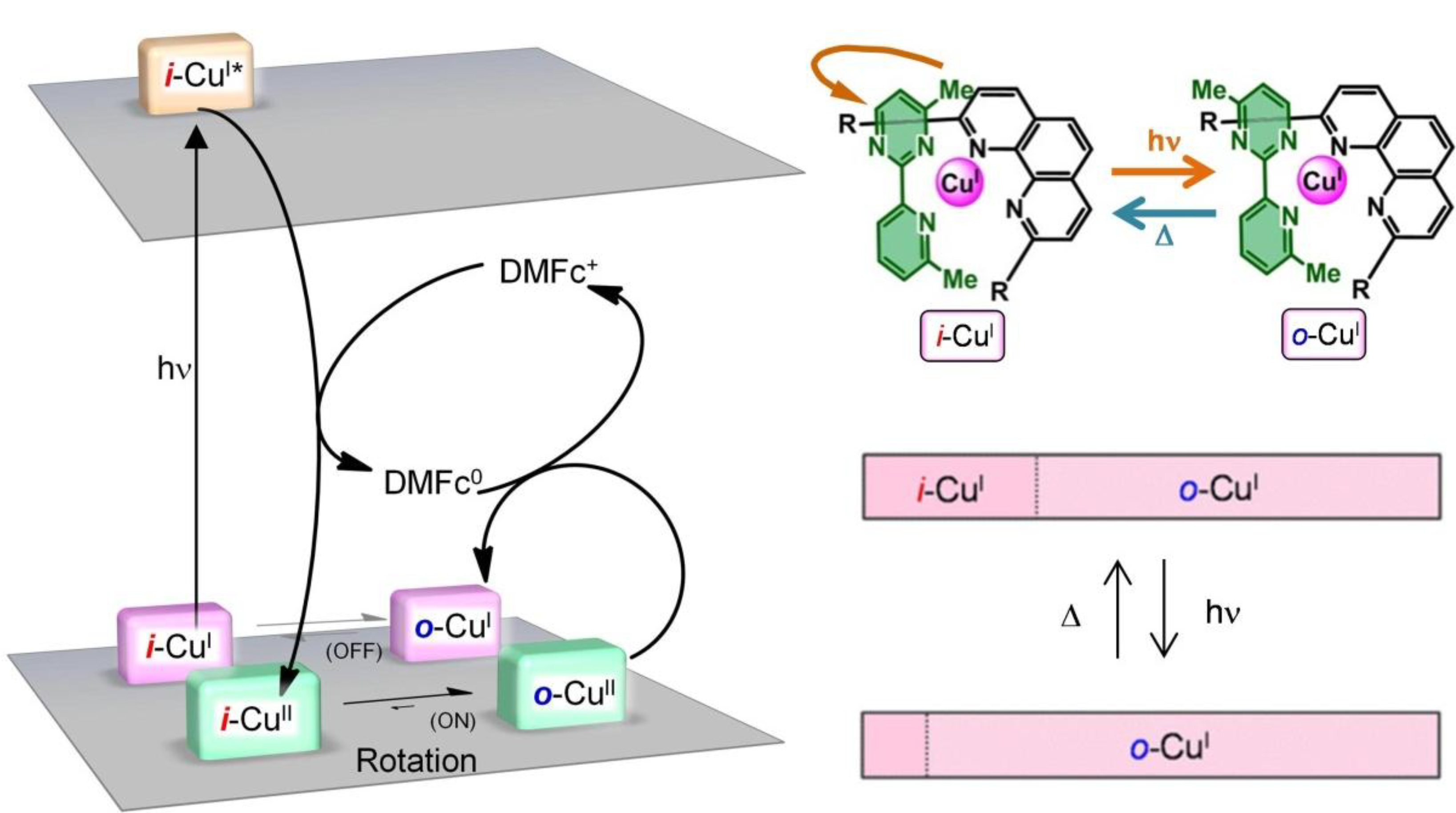
1.5. Mechanism for Photodriven Rotation of the Pyrimidine Ring

1.6. Conclusions
2. A Brightly Luminescent Heteroleptic bis(dipyrrinato)zinc(II) Complex
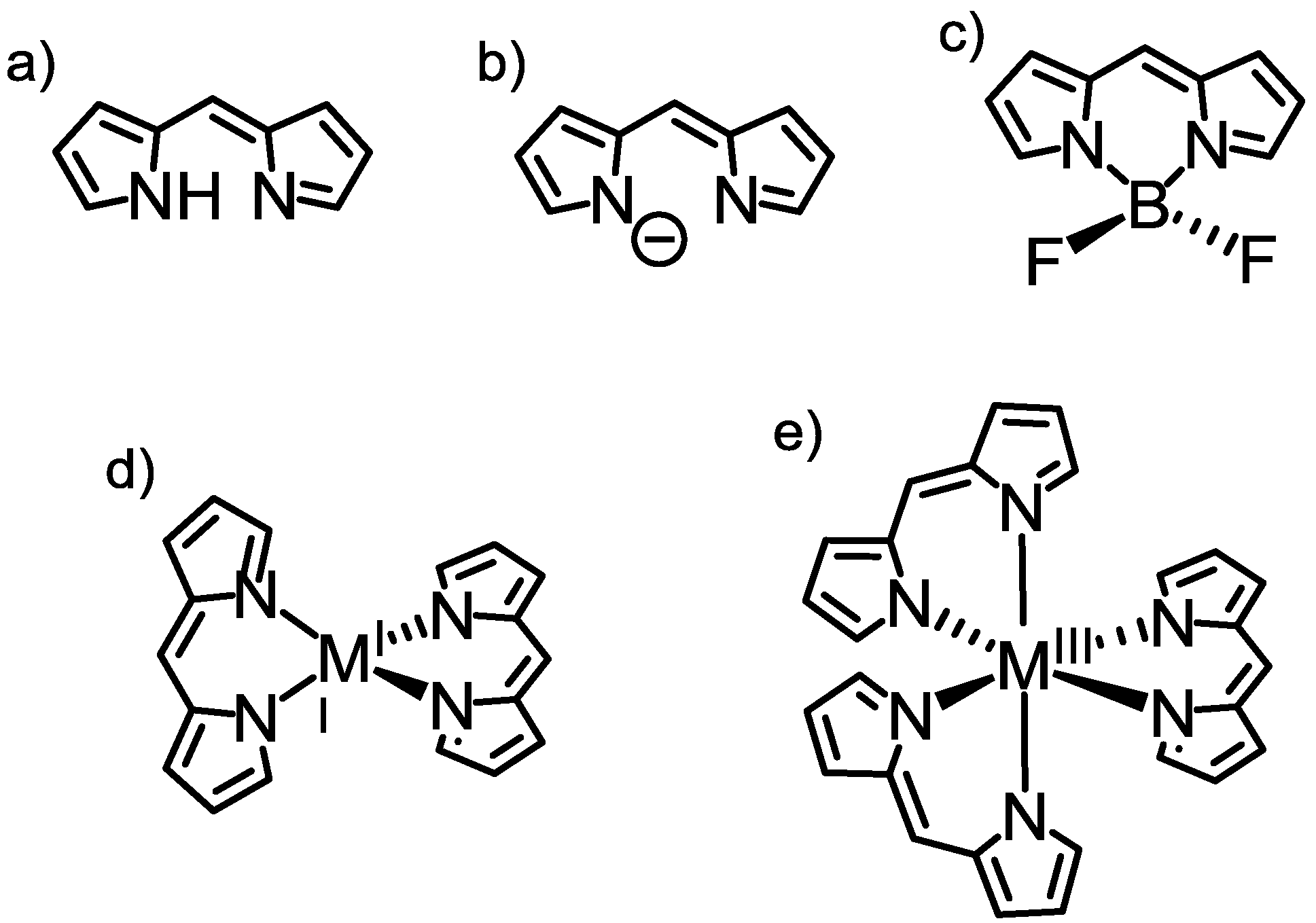
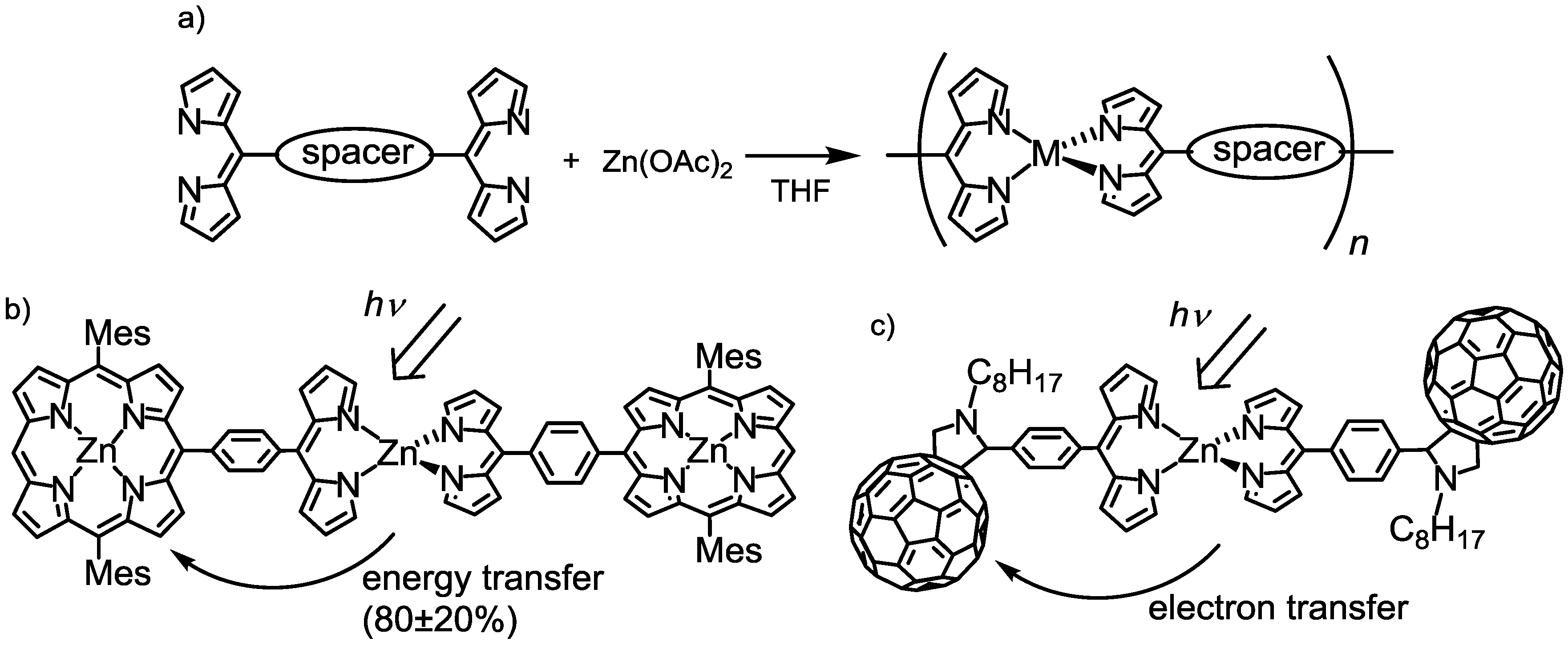
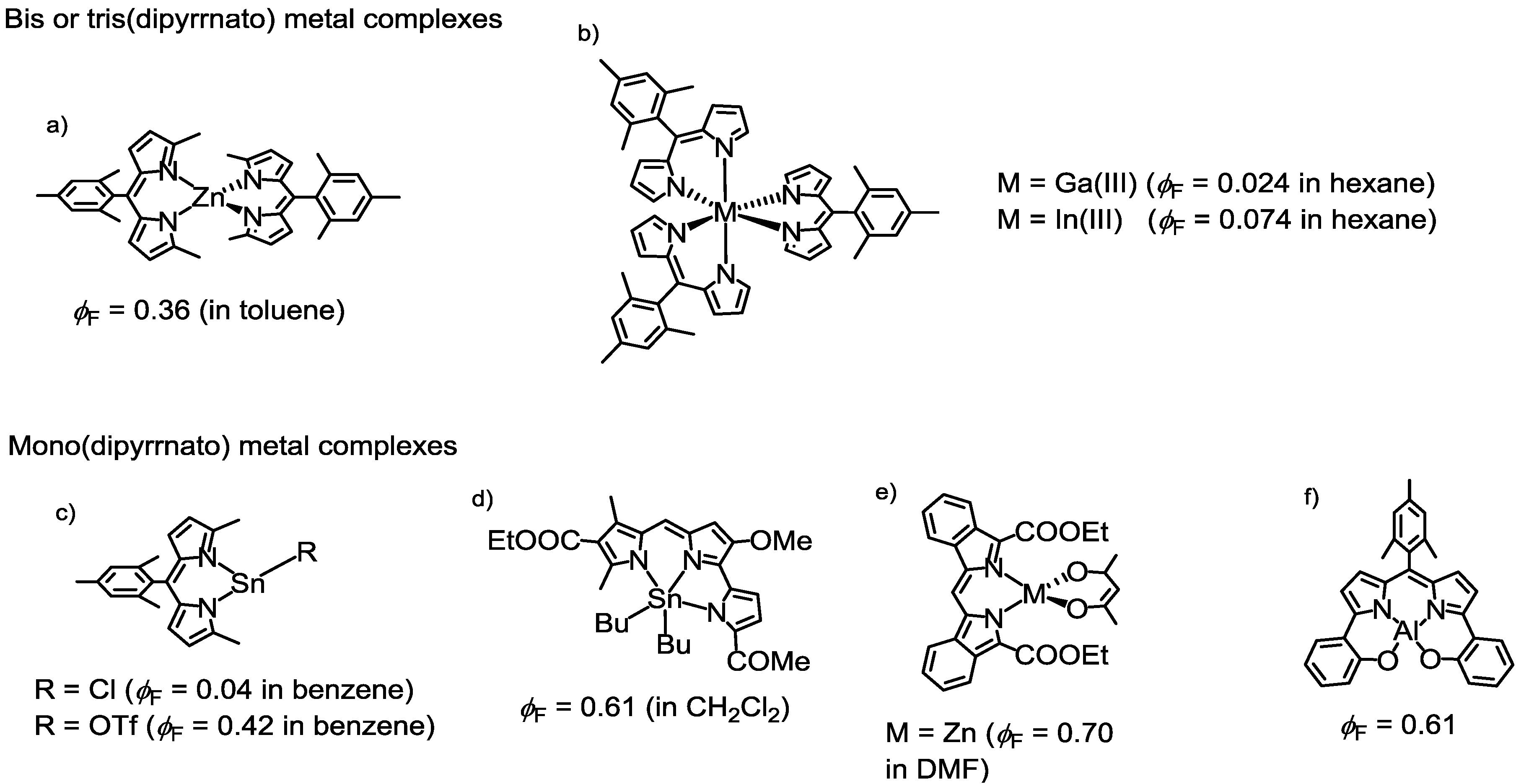
2.1. Introduction


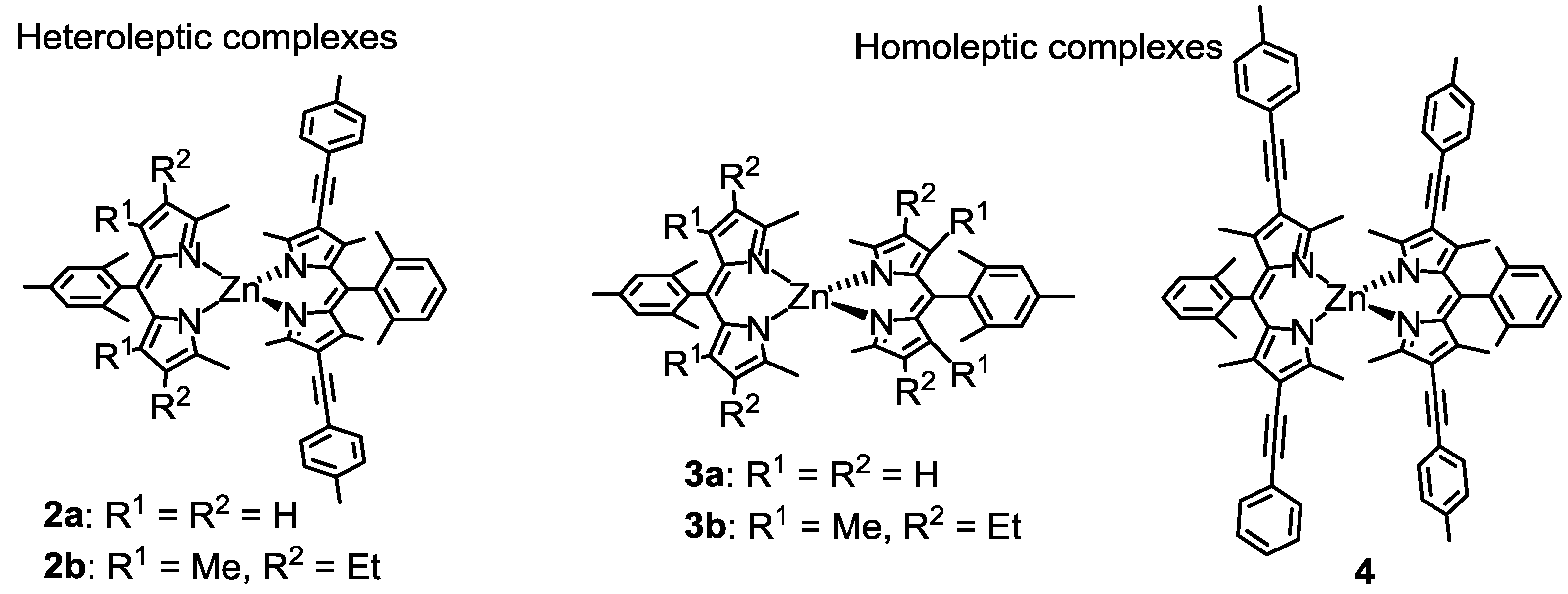
2.2. Strategy to Improve the Fluorescence Quantum Yield of bis(dipyrrinato)zinc(II) Complexes




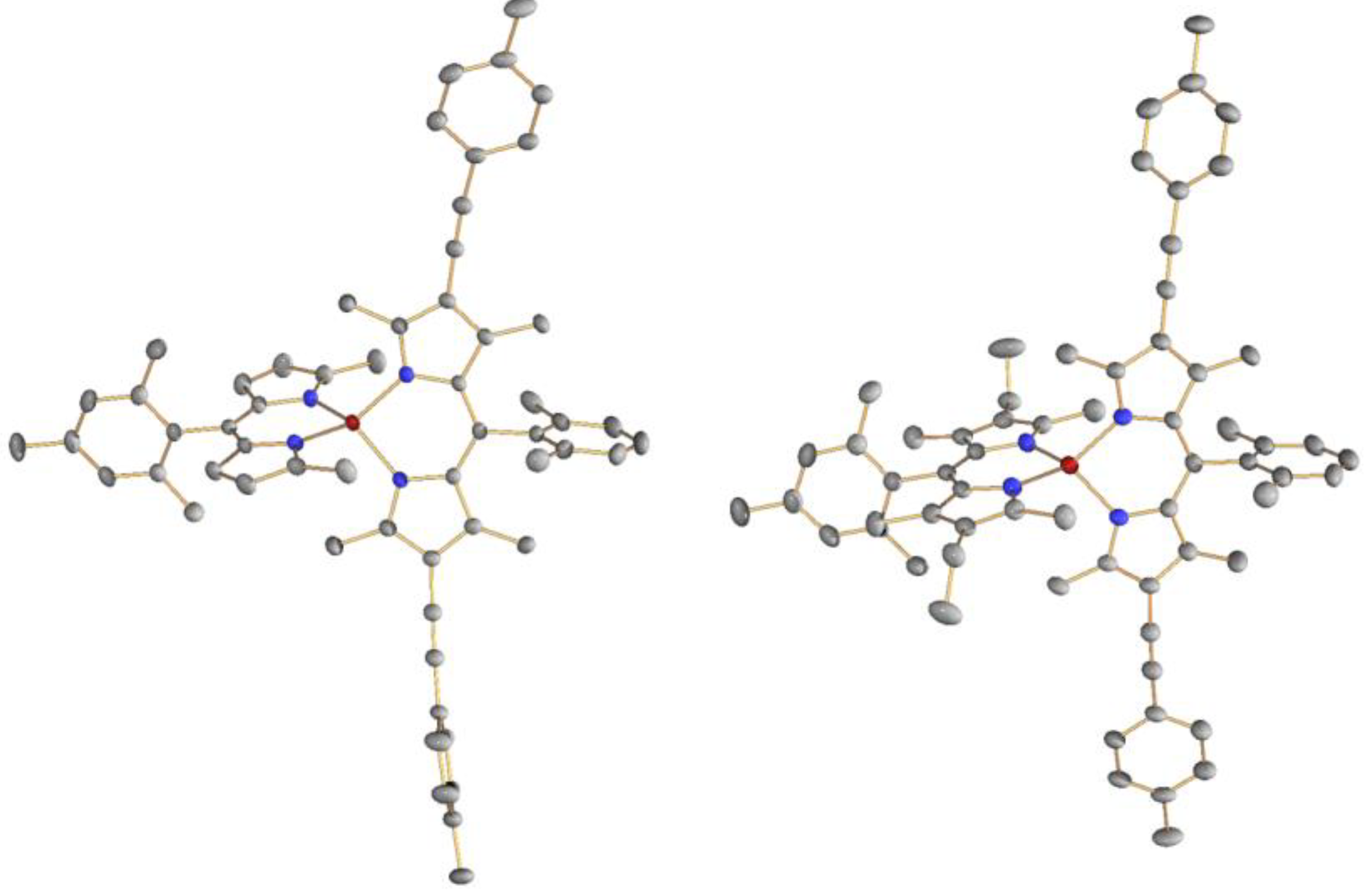
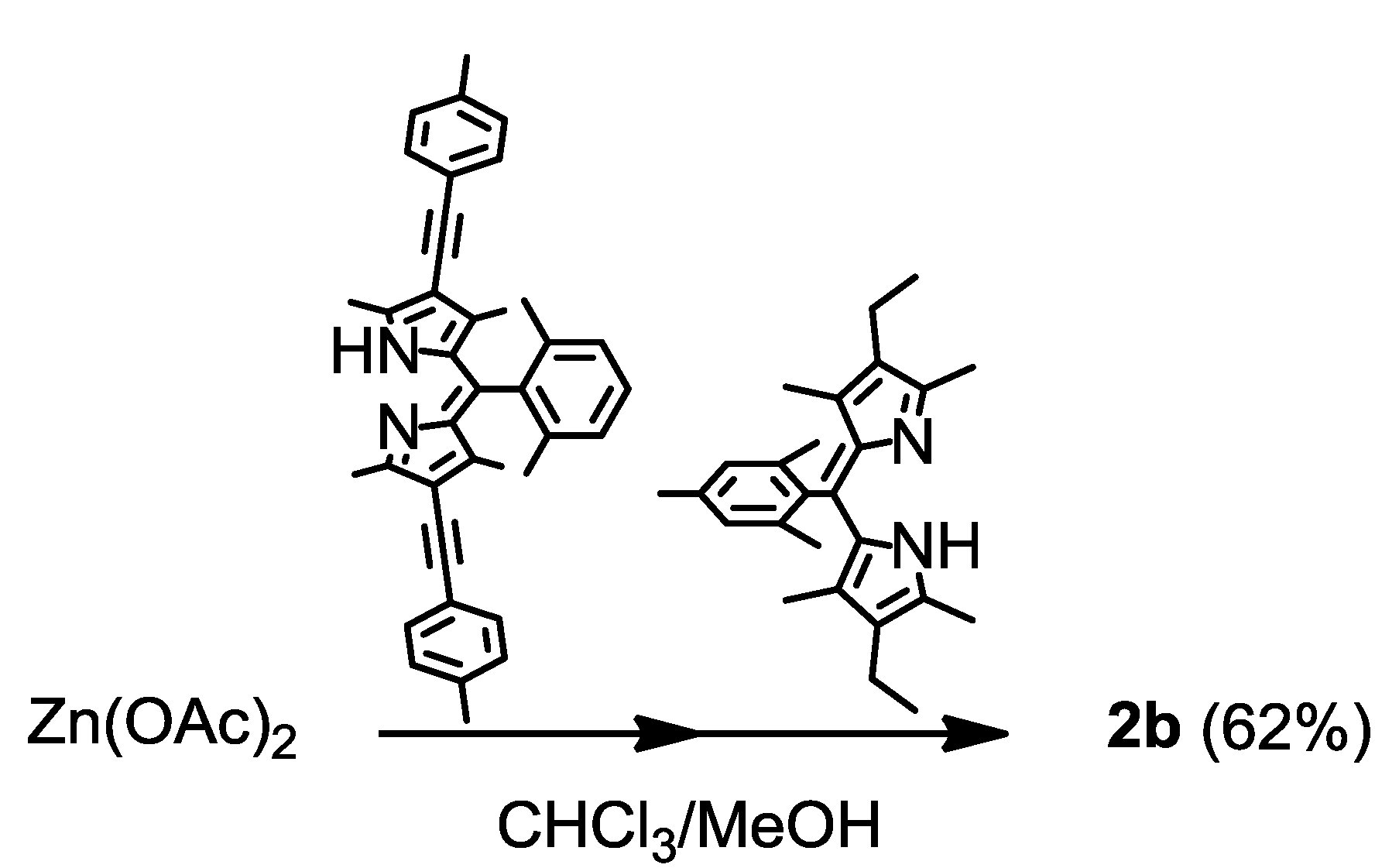
2.3. Synthesis, Structure, and Thermal Stability


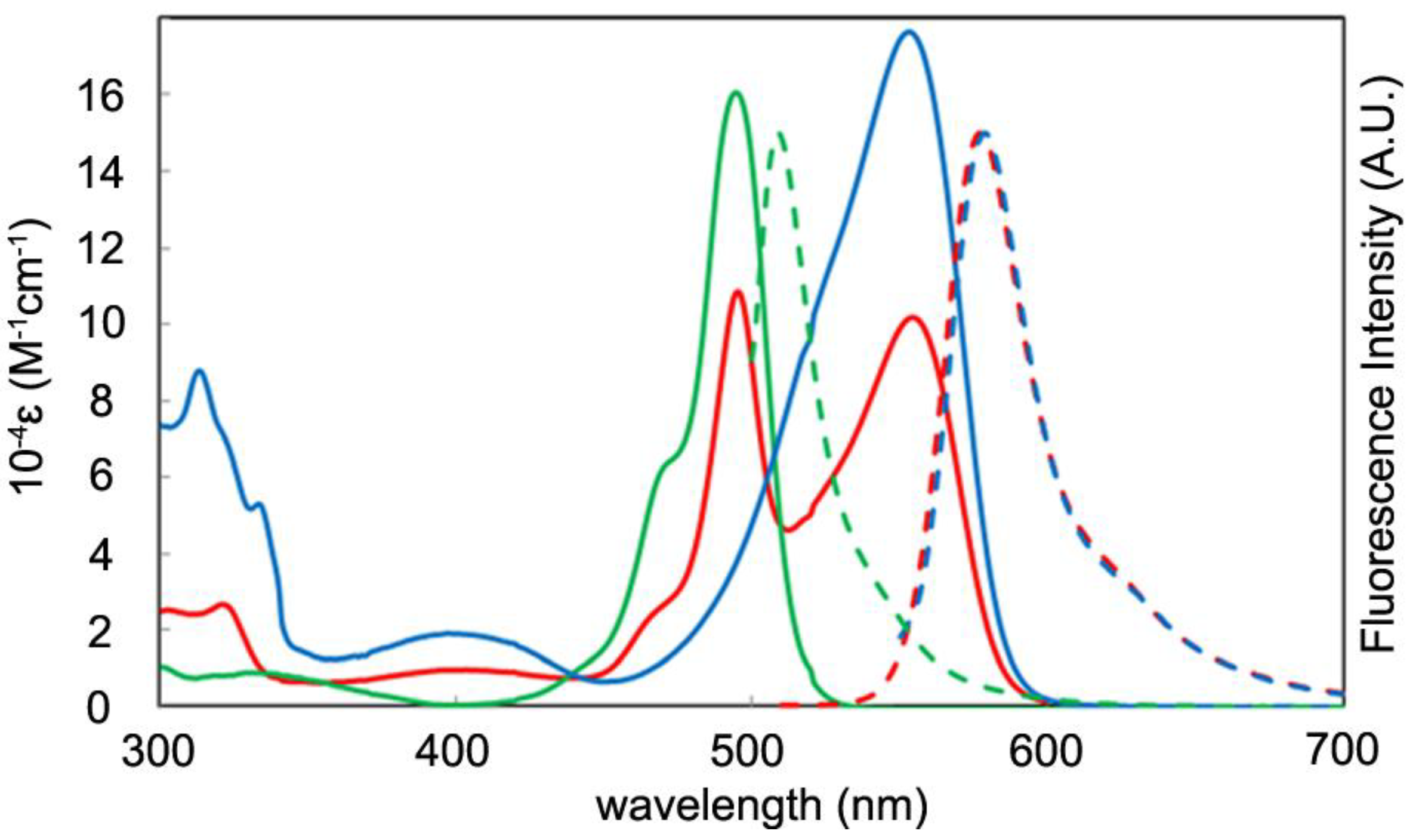
2.4. Optical Properties


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 10−4ε (M−1 cm−1) | λabs (nm) | λem (nm) | φF (in Toluene) | φF (in CH2Cl2) | |
|---|---|---|---|---|---|
| 2a | 11, 10 | 495, 553 | 578 | 0.76, 0.75 | 0.53, 0.51 |
| 2b | 11, 10 | 508, 556 | 579 | 0.07, 0.08 | 0.01, 0.03 |
| 3a | 16 | 495 | 509 | 0.28 | 0.00 |
| 3b | 14 | 508 | 532 | 0.20 | 0.05 |
| 4 | 18 | 553 | 579 | 0.72 | 0.31 |

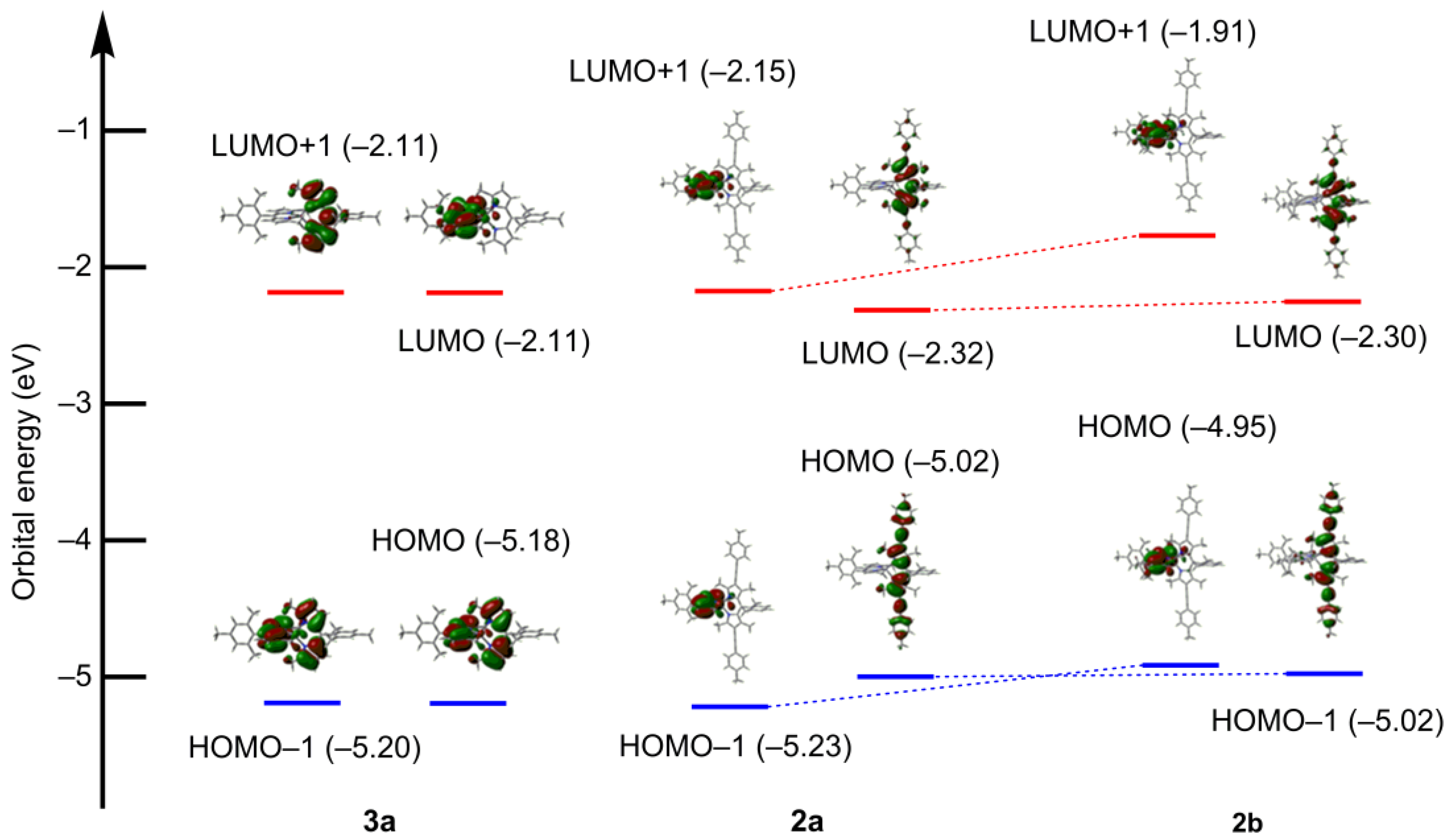
2.5. DFT Calculations

2.6. Conclusions
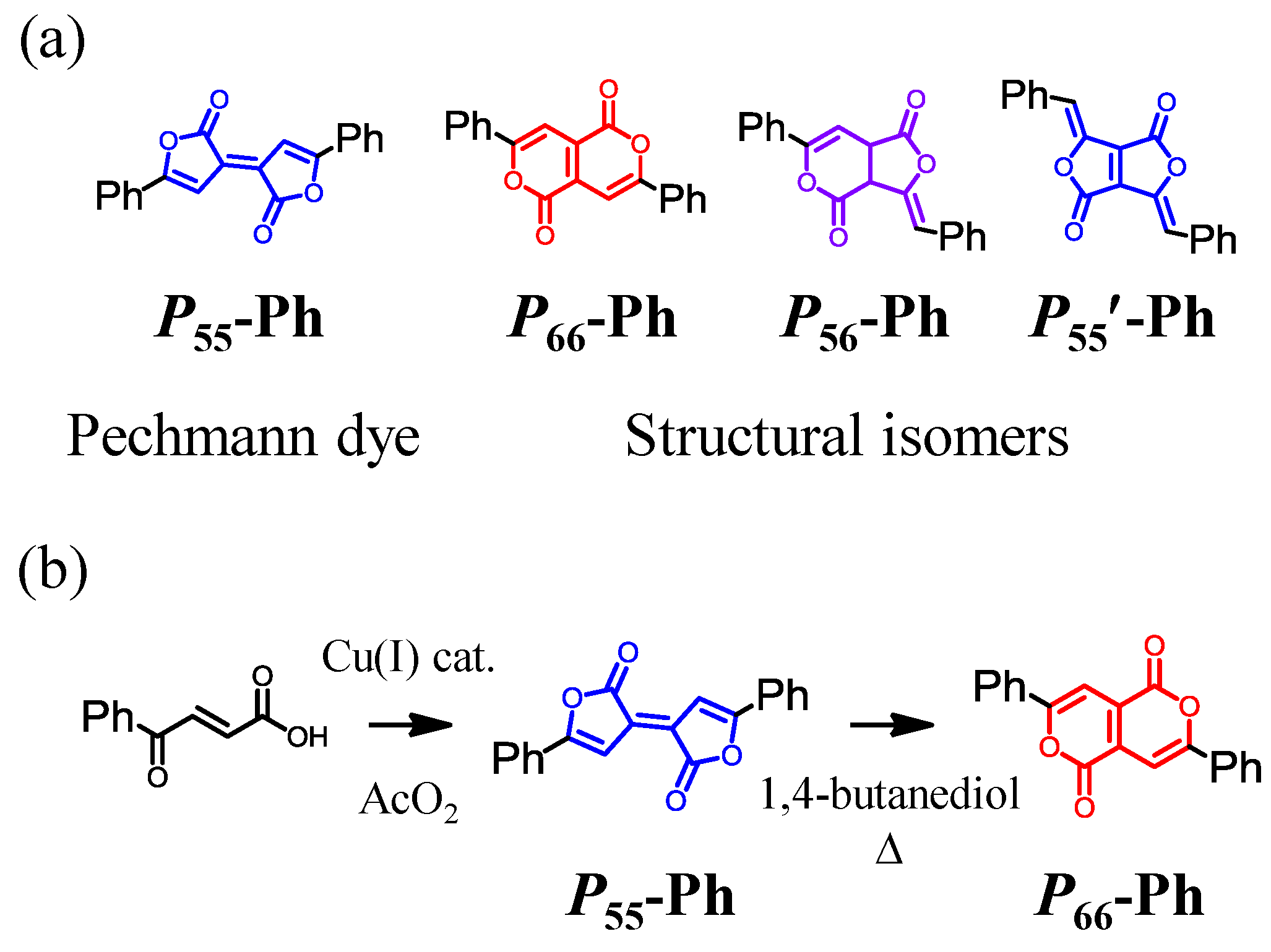
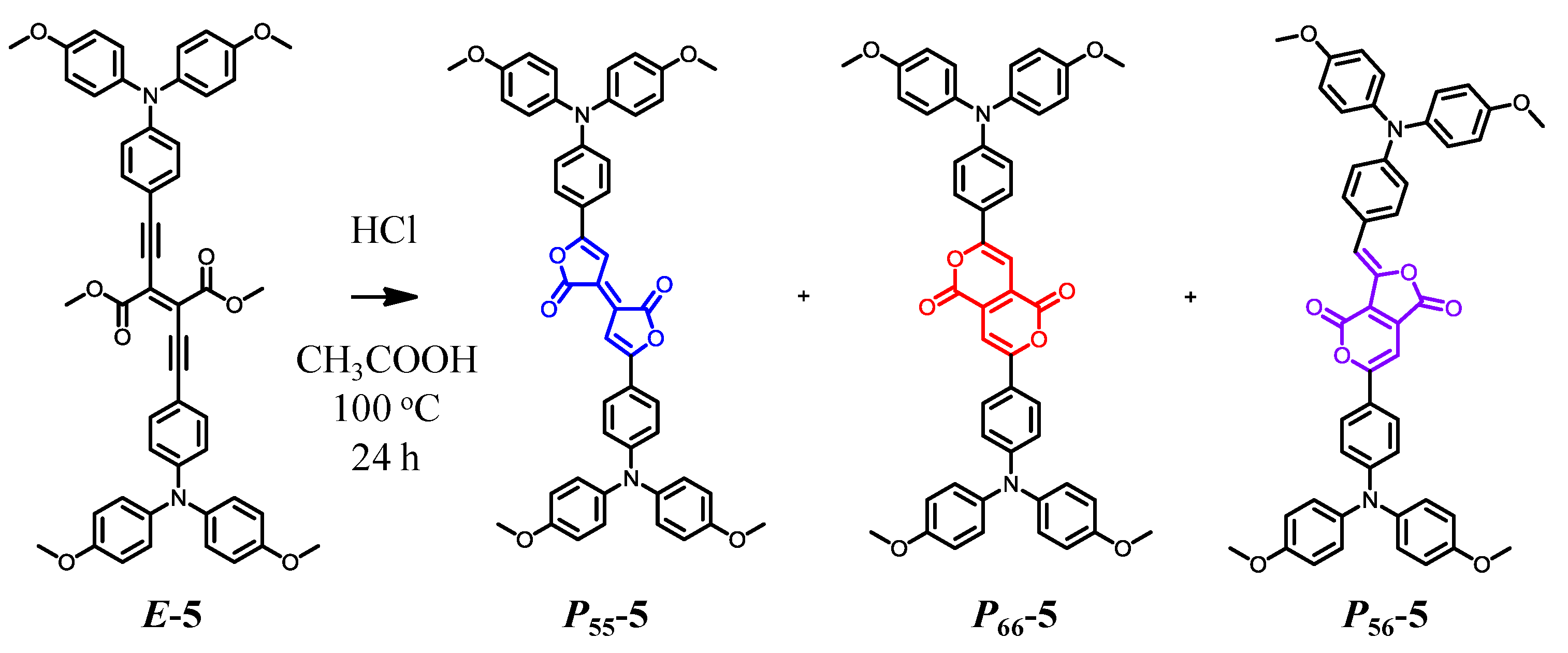
3. New Discoveries for Pechmann Dyes: An Alternative Synthesis, a Missing Structural Isomer, and Applications for Organic Electronics
3.1. Introduction

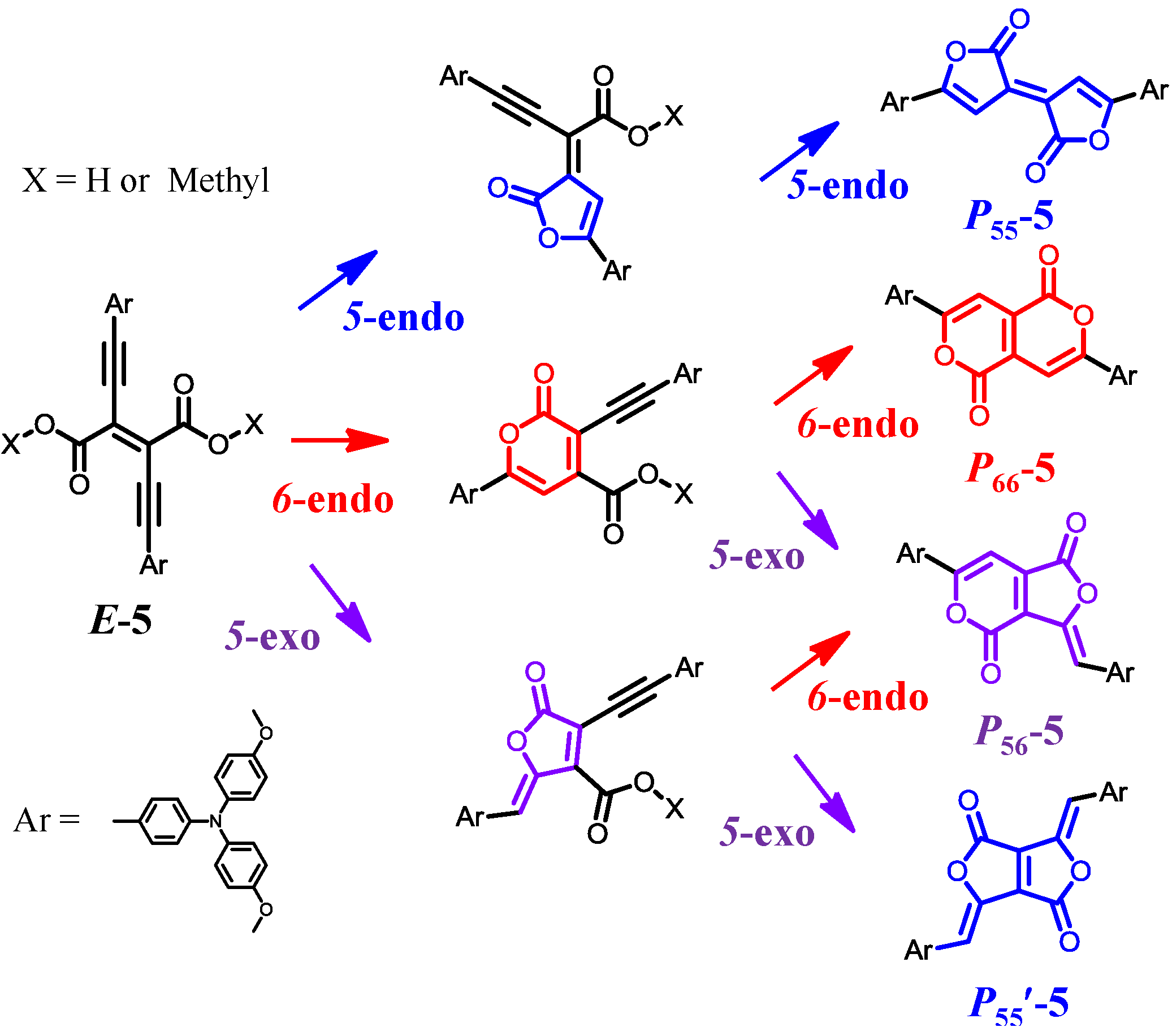
3.2. Synthesis, Identification, and Reaction Mechanism



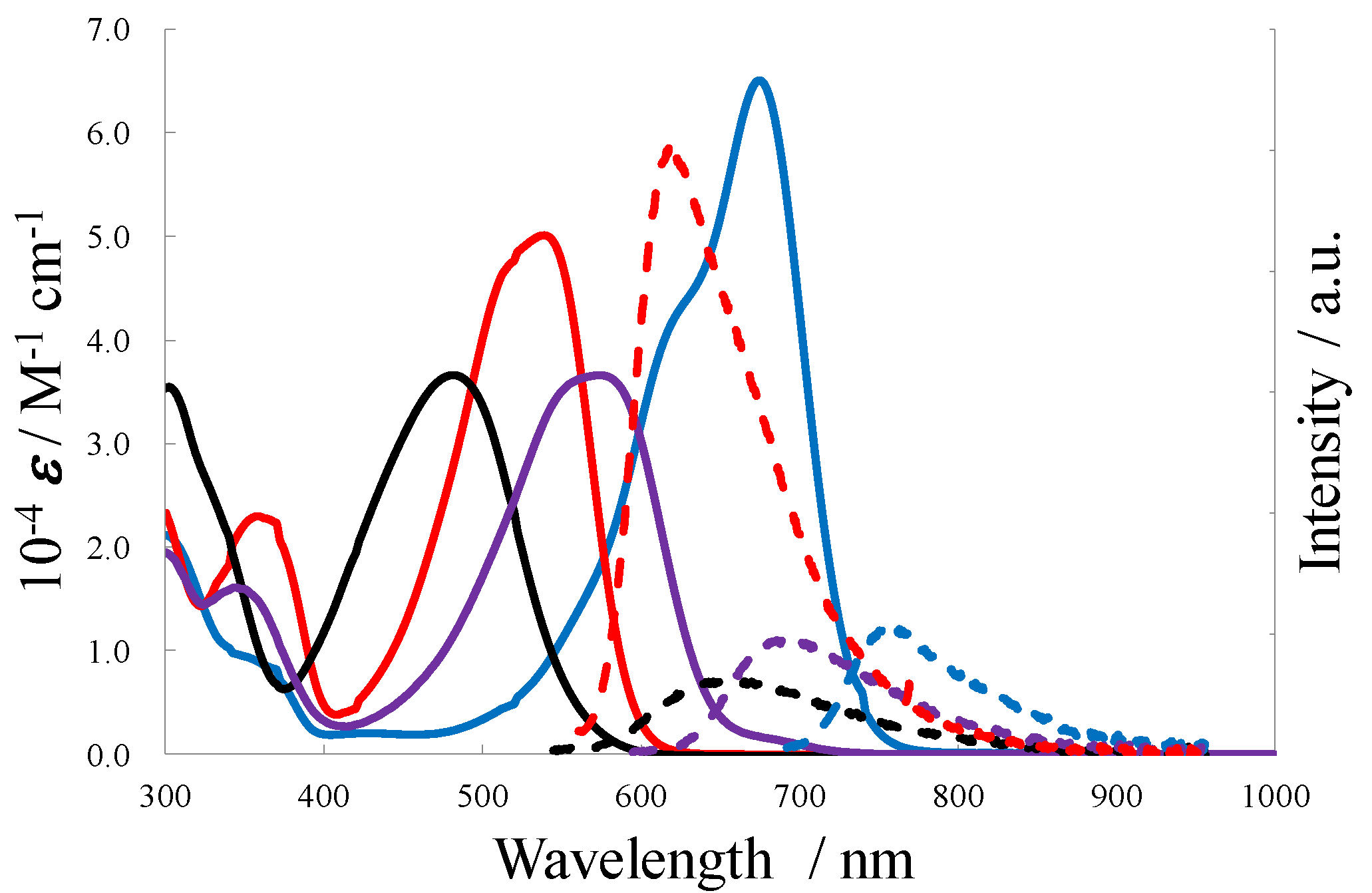
3.3. Photochemical Properties

| 10−4ε (M−1 cm−1) | λabs (nm) | λem (nm) | φF | |
|---|---|---|---|---|
| P55-5 | 65 | 674 | 753 | 0.27 |
| P56-5 | 37 | 574 | 692 | 0.20 |
| P66-5 | 50 | 538 | 617 | 0.82 |
| E-5 | 36 | 482 | 660 | 0.15 |
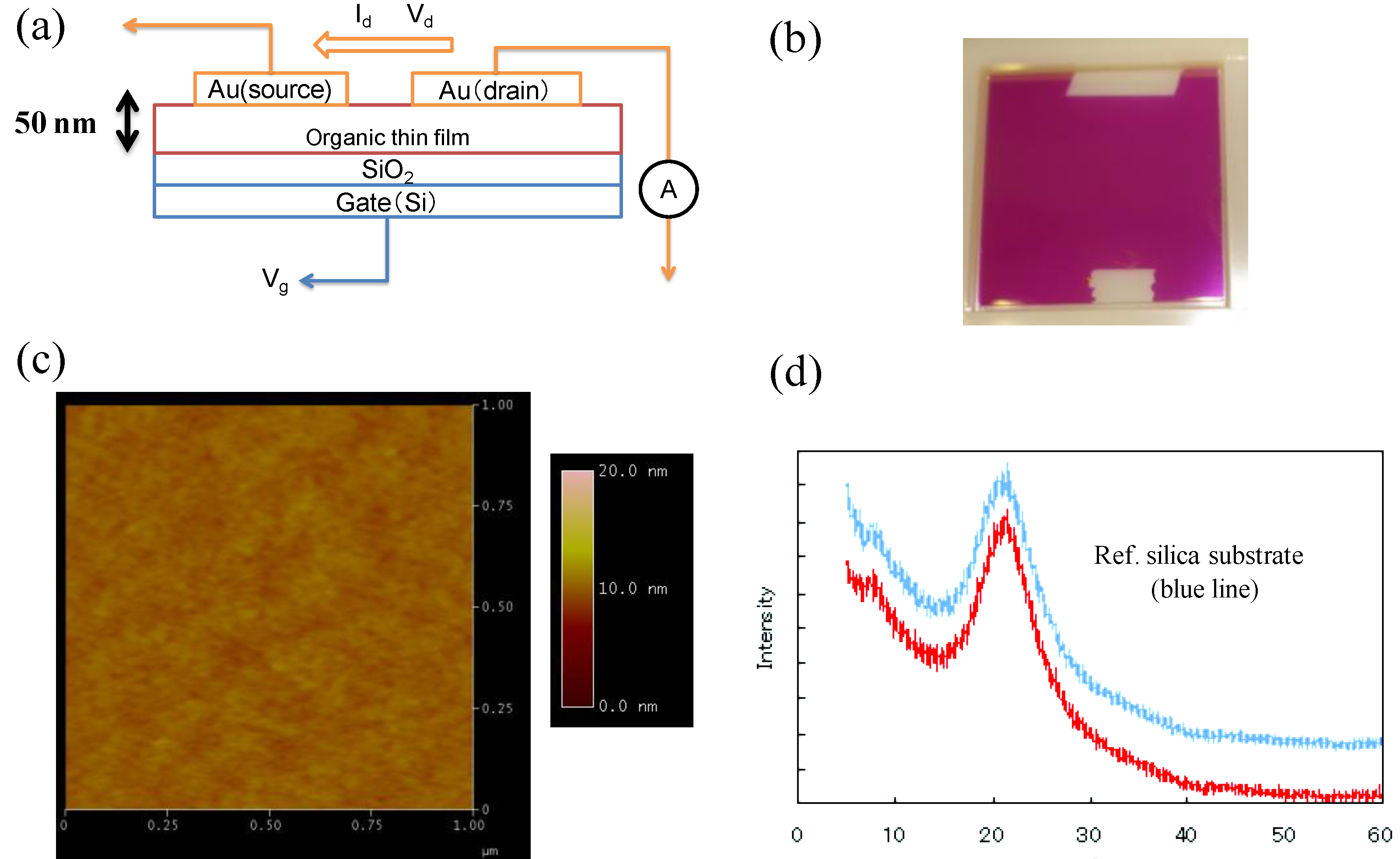
3.4. Application to Organic Electronics

4. Conclusions
Acknowledgments
References
- Addison, A.W. Is ligand topology an influence on the redox potentials of copper complexes? Inorg. Chim. Acta 1989, 162, 217–220. [Google Scholar] [CrossRef]
- Ambundo, E.A.; Deydier, M.-V.; Grall, A.J.; Aguera-Vega, N.; Dressel, L.T.; Cooper, T.H.; Heeg, M.J.; Ochrymowycz, L.A.; Rorabacher, D.B. Influence of Coordination Geometry upon Copper(II/I) Redox Potentials. Physical Parameters for Twelve Copper Tripodal Ligand Complexes. Inorg. Chem. 1999, 38, 4233–4242. [Google Scholar] [CrossRef]
- Ruthkosky, M.; Kelly, C.A.; Castellano, F.N.; Meyer, G.J. Electron and energy transfer from CuI MLCT excited states. Coord. Chem. Rev. 1998, 171, 309–322. [Google Scholar] [CrossRef]
- Scaltrito, D.V.; Thompson, D.W.; O’Callaghan, J.A.; Meyer, G.J. MLCT excited states of cuprous bis-phenanthroline coordination compounds. Coord. Chem. Rev. 2000, 208, 243–266. [Google Scholar] [CrossRef]
- Ruthkosky, M.; Castellano, F.N.; Meyer, G.J. Photodriven Electron and Energy Transfer from Copper Phenanthroline Excited States. Inorg. Chem. 1996, 35, 6406–6412. [Google Scholar] [CrossRef]
- Miller, M.T.; Gantzel, P.K.; Karpishin, T.B. Structures of the Copper(I) and Copper(II) Complexes of 2,9-Diphenyl-1,10-phenanthroline: Implications for Excited-State Structural Distortion. Inorg. Chem. 1998, 37, 2285–2290. [Google Scholar] [CrossRef]
- Rorabacher, D.B. Electron Transfer by Copper Centers. Chem. Rev. 2004, 104, 651–697. [Google Scholar] [CrossRef]
- Le Poul, N.; Campion, M.; Douziech, B.; Rondelez, Y.; Le Clainche, L.; Reinaud, O.; le Mest, Y. Monocopper Center Embedded in a Biomimetic Cavity: From Supramolecular Control of Copper Coordination to Redox Regulation. J. Am. Chem. Soc. 2007, 129, 8801–8810. [Google Scholar] [CrossRef]
- Munakata, M.; Kitagawa, S.; Asahara, A.; Masuda, H. Crystal Structure of Bis(2,2′-bipyridine)copper(I) Perchlorate. Bull. Chem. Soc. 1987, 60, 1927–1929. [Google Scholar] [CrossRef]
- Meyer, M.; Albrecht-Gary, A.M.; Dietrich-Buchecker, C.O.; Sauvage, J.-P. π−π Stacking-Induced Cooperativity in Copper(I) Complexes with Phenanthroline Ligands. Inorg. Chem. 1999, 38, 2279–2287. [Google Scholar] [CrossRef]
- Munakata, M.; Endicott, J.F. Oxidation-reduction reactions of complexes with macrocyclic ligands. Role of kinetic factors in distinguishing mechanistic pathways in reactions of copper(I) complexes with coordinated dioxygen. Inorg. Chem. 1984, 23, 3693–3698. [Google Scholar] [CrossRef]
- Federlin, P.; Kern, J.-M.; Rastegar, A.; Dietrich-Buchecker, C.; Marnot, P.A.; Sauvage, J.-P. Electrochemical properties of copper(I) catenates and some of their open chain analogues; Correlation with their photophysical properties. New. J. Chem. 1990, 14, 9–12. [Google Scholar]
- Solomon, E.I.; Szilagyi, R.K.; George, S.D.; Basumallick, L. Electronic Structures of Metal Sites in Proteins and Models: Contributions to Function in Blue Copper Proteins. Chem. Rev. 2004, 104, 419–458. [Google Scholar] [CrossRef]
- Lewis, E.A.; Tolman, W.B. Reactivity of Dioxygen−Copper Systems. Chem. Rev. 2004, 104, 1047–1076. [Google Scholar] [CrossRef]
- Suzuki, M. Ligand Effects on Dioxygen Activation by Copper and Nickel Complexes: Reactivity and Intermediates. Acc. Chem. Res. 2007, 40, 609–617. [Google Scholar] [CrossRef]
- Livoreil, A.; Dietrich-Buchecker, C.O.; Sauvage, J.-P. Electrochemically Triggered Swinging of a [2]-Catenate. J. Am. Chem. Soc. 1994, 116, 9399–9400. [Google Scholar] [CrossRef]
- Dietrich-Buchecker, C.O.; Sauvage, J.-P. Interlocking of molecular threads: from the statistical approach to the templated synthesis of catenands. Chem. Rev. 1987, 87, 795–810. [Google Scholar] [CrossRef]
- Raehm, L.; Kern, J.-M.; Sauvage, J.-P. A Transition Metal Containing Rotaxane in Motion: Electrochemically Induced Pirouetting of the Ring on the Threaded Dumbbell. Chem. Eur. J. 1999, 5, 3310–3317. [Google Scholar] [CrossRef]
- Poleschak, I.; Kern, J.M.; Sauvage, J.-P. A copper-complexed rotaxane in motion: pirouetting of the ring on the millisecond timescale. Chem. Commun. 2004, 474–476. [Google Scholar] [CrossRef]
- Durola, F.; Lux, J.; Sauvage, J.-P. A Fast-Moving Copper-Based Molecular Shuttle: Synthesis and Dynamic Properties. Chem. Eur. J. 2009, 15, 4124–4134. [Google Scholar] [CrossRef]
- Sauvage, J.-P. From Chemical Topology To Molecular Machines: The Transition Metal Approach. Bull. Jpn. Soc. Coord. Chem. 2010, 55, 3–18. [Google Scholar] [CrossRef]
- Armaroli, N.; Balzani, V.; Collin, J.-P.; Gavin, P.; Sauvage, J.-P. Rotaxanes Incorporating Two Different Coordinating Units in Their Thread: Synthesis and Electrochemically and Photochemically Induced Molecular Motions. J. Am. Chem. Soc. 1999, 121, 4397–4408. [Google Scholar] [CrossRef]
- Livoreil, A.; Sauvage, J.-P.; Armaroli, N.; Balzani, V.; Flamigni, L.; Ventura, B. Electrochemically and Photochemically Driven Ring Motions in a Disymmetrical Copper [2]-Catenate. J. Am. Chem. Soc. 1997, 119, 12114–12124. [Google Scholar] [CrossRef]
- Collin, J.-P.; Dietrich-Buchecker, C.; Gaviña, P.; Jiménez-Molero, M.C.; Sauvage, J.-P. Shuttles and Muscles: Linear Molecular Machines Based on Transition Metals. Acc. Chem. Res. 2001, 34, 477–487. [Google Scholar] [CrossRef]
- Sauvage, J.-P. Transition Metal-Containing Rotaxanes and Catenanes in Motion: Toward Molecular Machines and Motors. Acc. Chem. Res. 1998, 31, 611–619. [Google Scholar] [CrossRef]
- Zahn, S.; Canary, J.W. Cu(I/II) Redox Control of Molecular Conformation and Shape in Chiral Tripodal Ligands: Binary Exciton-Coupled Circular Dichroic States. J. Am. Chem. Soc. 2002, 124, 9204–9211. [Google Scholar] [CrossRef]
- Mortezaei, S.; Catarineu, N.R.; Canary, J.W. A Redox-Reconfigurable, Ambidextrous Asymmetric Catalyst. J. Am. Chem. Soc. 2012, 134, 8054–8057. [Google Scholar] [CrossRef]
- Kawanishi, Y.; Kitamura, N.; Tazuke, S. Dependence of spectroscopic, electrochemical, and excited-state properties of tris chelate ruthenium(II) complexes on ligand structure. Inorg. Chem. 1989, 28, 2968–2975. [Google Scholar] [CrossRef]
- Groen, J.H.; van Leeuwen, P.W.N.M.; Vrieze, K. Synthesis, characterisation and dynamic behaviour of palladium complexes containing the novel terdentate nitrogen ligand 2,6-bis(pyrimidin-2-yl)pyridine. J. Chem. Soc. Dalton Trans. 1998, 113–117. [Google Scholar]
- Nickita, N.; Gasser, G.; Pearson, P.; Belousoff, M.J.; Goh, L.Y.; Bond, A.M.; Deacon, G.B.; Spiccia, L. Ruthenium(II) Complexes Incorporating 2-(2′-Pyridyl)pyrimidine-4-carboxylic Acid. Inorg. Chem. 2008, 48, 68–81. [Google Scholar]
- Pearson, R.G.; Henry, P.M.; Bergmann, J.G.; Basolo, F. Mechanism of Substitution Reactions of Complex Ions. VI. Formation of Nitrito- and Nitrocobalt(III) Complexes. O-Nitrosation1,2,3. J. Am. Chem. Soc. 1954, 76, 5920–5923. [Google Scholar] [CrossRef]
- Basolo, F.; Hammaker, G.S. Synthesis and Isomerization of Nitritopentammine Complexes of Rhodium(III), Iridium(III), and Platinum(IV). Inorg. Chem. 1962, 1, 1–5. [Google Scholar] [CrossRef]
- Murmann, R.K.; Taube, H. The Mechanism of the Formation and Rearrangement of Nitritocobalt(III) Ammines1. J. Am. Chem. Soc. 1956, 78, 4886–4890. [Google Scholar] [CrossRef]
- Murmann, R.K. The requirement of triphosphopyridine nucleotide in fatty acid synthesis. J. Am. Chem. Soc. 1955, 77, 5190–5192. [Google Scholar] [CrossRef]
- Mares, M.; Palmer, D.A.; Kelm, H. Activation volumes for the linkage isomerization reactions of nitritopentaammine complexes of cobalt(III), rhodium(III) and iridium(III) in aqueous solution. Inorg. Chim. Acta. 1978, 27, 153–156. [Google Scholar] [CrossRef]
- Nomoto, K.; Kume, S.; Nishihara, H. A Single Molecular System Gating Electron Transfer by Ring Inversion of a Methylpyridylpyrimidine Ligand on Copper. J. Am. Chem. Soc. 2009, 131, 3830–3831. [Google Scholar] [CrossRef]
- Kume, S.; Nishihara, H. Tuning-up and driving a redox-active rotor. Chem. Commun. 2011, 47, 415–417. [Google Scholar] [CrossRef]
- Kume, S.; Nomoto, K.; Kusamoto, T.; Nishihara, H. Intramolecular Electron Arrangement with a Rotative Trigger. J. Am. Chem. Soc. 2009, 131, 14198–14199. [Google Scholar]
- Kume, S.; Nishihara, H. Synchronized motion and electron transfer of a redox-active rotor. Dalton Trans. 2011, 40, 2299–2305. [Google Scholar] [CrossRef]
- Nishikawa, M.; Nomoto, K.; Kume, S.; Inoue, K.; Sakai, M.; Fujii, M.; Nishihara, H. Dual Emission Caused by Ring Inversion Isomerization of a 4-Methyl-2-pyridyl-pyrimidine Copper(I) Complex. J. Am. Chem. Soc. 2010, 132, 9579–9581. [Google Scholar]
- Nishikawa, M.; Nomoto, K.; Kume, S.; Nishihara, H. Solvated-Ion-Pairing-Sensitive Molecular Bistability Based on Copper(I)-Coordinated Pyrimidine Ring Rotation. Inorg. Chem. 2013, 52, 369–380. [Google Scholar]
- Nishikawa, M.; Nomoto, K.; Kume, S.; Nishihara, H. Reversible Copper(II)/(I) Electrochemical Potential Switching Driven by Visible Light-Induced Coordinated Ring Rotation. J. Am. Chem. Soc. 2012, 134, 10543–10553. [Google Scholar]
- Tran, D.; Ryu, C.K.; Ford, P.C. Diffusion Limited Quenching of the Cluster Centered Excited State of the Copper(I) Cluster Cu4I4(py)4 by Ferrocenium Ion in CH2Cl2 Solution. Inorg. Chem. 1994, 33, 5957–5959. [Google Scholar] [CrossRef]
- Armaroli, N.; Accorsi, G.; Cardinali, F.; Listorti, A. Photochemistry and Photophysics of Coordination Compounds: Copper. Top. Curr. Chem. 2007, 280, 69–115. [Google Scholar] [CrossRef]
- Lavie-Cambot, A.; Cantuel, M.; Leydet, Y.; Jonusauskas, G.; Bassani, D.M.; McClenaghan, N.D. Improving the photophysical properties of copper(I) bis(phenanthroline) complexes. Coord. Chem. Rev. 2008, 252, 2572–2584. [Google Scholar] [CrossRef]
- McMillin, D.R.; McNett, K.M. Photoprocesses of Copper Complexes That Bind to DNA. Chem. Rev. 1998, 98, 1201–1219. [Google Scholar] [CrossRef]
- Everly, R.M.; Ziessel, R.; Suffert, J.; McMillin, D.R. Steric influences on the photoluminescence from copper(I) phenanthrolines in rigid media. Inorg. Chem. 1991, 30, 559–561. [Google Scholar] [CrossRef]
- Cunningham, C.T.; Cunningham, K.L.H.; Michalec, J.F.; McMillin, D.R. Cooperative Substituent Effects on the Excited States of Copper Phenanthrolines. Inorg. Chem. 1999, 38, 4388–4392. [Google Scholar] [CrossRef]
- Cuttell, D.G.; Kuang, S.M.; Fanwick, P.E.; McMillin, D.R.; Walton, R.A. Simple Cu(I) Complexes with Unprecedented Excited-State Lifetimes. J. Am. Chem. Soc. 2002, 124, 6–7. [Google Scholar]
- Kuang, S.M.; Cuttell, D.G.; McMillin, D.R.; Fanwick, P.E.; Walton, R.A. Synthesis and Structural Characterization of Cu(I) and Ni(II) Complexes that Contain the Bis[2-(diphenylphosphino)phenyl]ether Ligand. Novel Emission Properties for the Cu(I) Species. Inorg. Chem. 2002, 41, 3313–3322. [Google Scholar] [CrossRef]
- Vorontsov, I.I.; Graber, T.; Kovalevsky, A.Y.; Novozhilova, I.V.; Gembicky, M.; Chen, Y.-S.; Coppens, P. Capturing and Analyzing the Excited-State Structure of a Cu(I) Phenanthroline Complex by Time-Resolved Diffraction and Theoretical Calculations. J. Am. Chem. Soc. 2009, 131, 6566–6573. [Google Scholar] [CrossRef]
- Siddique, Z.A.; Yamamoto, Y.; Ohno, T.; Nozaki, K. Structure-Dependent Photophysical Properties of Singlet and Triplet Metal-to-Ligand Charge Transfer States in Copper(I) Bis(diimine) Compounds. Inorg. Chem. 2003, 42, 6366–6378. [Google Scholar] [CrossRef]
- Schmittel, M.; Ganz, A. Stable mixed phenanthroline copper(i) complexes. Key buildingblocks for supramolecular coordination chemistry. Chem. Commun. 1997, 999–1000. [Google Scholar] [CrossRef]
- Schmittel, M.; Michel, C.; Liu, S.-X.; Schildbach, D.; Fenske, D. New Sterically Encumbered 2,9-Diarylphenanthrolines for the Selective Formation of Heteroleptic Bis(phenanthroline)copper(I) Complexes. Eur. J. Inorg. Chem. 2001, 1155–1166. [Google Scholar]
- Schmittel, M.; Lüning, U.; Meder, M.; Ganz, A.; Michel, C.; Herderich, M. Synthesis of Sterically Encumbered 2,9-Diaryl Substituted Phenantrolines. Key Building Blocks for the Preparation of Mixed (Bis-Heteroleptic) Phenanthroline Copper(I) Complexes. Heterocycl. Commun. 1997, 3, 493–498. [Google Scholar]
- Bard, A.J.; Faulkner, L.R. Electrochemical Methods, Fundamentals and Applications, 2nd ed.; Wiley: New York, NY, USA, 2001. [Google Scholar]
- Jacq, J. Schema carre: Etablissement et Discussion de L'equation Generale de la Courbe Intensite-Potentiel en Regime Stationnaire et Diffusion Convective. J. Electroanal. Chem. 1971, 29, 149–180. [Google Scholar] [CrossRef]
- Carano, M.; Echegoyen, L. Mechanisms of Electrochemically-Induced Retro-Cyclopropanation Reactions of Fullerene Derivatives Using Digital Simulations. Chem. Eur. J. 2003, 9, 1974–1981. [Google Scholar] [CrossRef]
- Lerke, S.A.; Evans, D.H.; Feldberg, S.W. Digital simulation of the square scheme in cyclic voltammetry: A comparison of methods. J. Electroanal. Chem. 1990, 296, 299–315. [Google Scholar] [CrossRef]
- Juris, A.; Balzani, V.; Barigelletti, F.; Campagna, S.; Belser, P.; von Zelewsky, A. Ru(II) polypyridine complexes: photophysics, photochemistry, eletrochemistry, and chemiluminescence. Coord. Chem. Rev. 1988, 84, 85–277. [Google Scholar] [CrossRef]
- Durr, H. Photochromism: Molecules and Systems; Elsevier: Amsterdam, The Netherlands, 1990. [Google Scholar]
- Fischer, H.; Orth, H. Die Chemie des Pyrrols; Akademische Verlagsgesellschaft: Leipzig, Germany, 1937. [Google Scholar]
- Wood, T.E.; Thompson, A. Advances in the Chemistry of Dipyrrins and Their Complexes. Chem. Rev. 2007, 107, 1831–1861. [Google Scholar] [CrossRef]
- Treibs, A.; Kreuzer, F.-H. Difluorboryl-Komplexe von Di- und Tripyrrylmethenen. Justus Liebigs Ann. Chem. 1968, 718, 208–223. [Google Scholar] [CrossRef]
- Shah, M.; Thangaraj, K.; Soong, M.L.; Wolford, L.; Boyer, J.H.; Politzer, I.R.; Pavlopoulos, T.G. Pyrromethene–BF2 complexes as laser dyes:1. Heteroat. Chem. 1990, 1, 389–399. [Google Scholar] [CrossRef]
- Boyer, J.H.; Haag, A.M.; Sathyamoorthi, G.; Soong, M.L.; Thangaraj, K.; Pavlopoulos, T.G. Pyrromethene–BF2 complexes as laser dyes: 2. Heteroat. Chem. 1993, 4, 39–49. [Google Scholar] [CrossRef]
- Hermes, R.E.; Allik, T.H.; Chandra, S.; Hutchinson, J.A. High – efficiency pyrromethene doped solid – state dye lasers. Appl. Phys. Lett. 1993, 63, 877–879. [Google Scholar] [CrossRef]
- Coskun, A.; Akkaya, E.U. Ion Sensing Coupled to Resonance Energy Transfer: A Highly Selective and Sensitive Ratiometric Fluorescent Chemosensor for Ag(I) by a Modular Approach. J. Am. Chem. Soc. 2005, 127, 10464–10465. [Google Scholar] [CrossRef]
- Coskun, A.; Akkaya, E.U. Signal Ratio Amplification via Modulation of Resonance Energy Transfer: Proof of Principle in an Emission Ratiometric Hg(II) Sensor. J. Am. Chem. Soc. 2006, 128, 14474–14475. [Google Scholar] [CrossRef]
- Yuan, M.; Li, Y.; Li, J.; Li, C.; Liu, X.; Lv, J.; Xu, J.; Liu, H.; Wang, S.; Zhu, D. A Colorimetric and Fluorometric Dual-Modal Assay for Mercury Ion by a Molecule. Org. Lett. 2007, 9, 2313–2316. [Google Scholar] [CrossRef]
- Ekmekci, Z.; Ylimaz, M.D.; Akkaya, E.U. A Monostyryl-boradiazaindacene (BODIPY) Derivative as Colorimetric and Fluorescent Probe for Cyanide Ions. Org. Lett. 2008, 10, 461–464. [Google Scholar] [CrossRef]
- Gabe, Y.; Urano, Y.; Kikuchi, K.; Kojima, H.; Nagano, T. Highly Sensitive Fluorescence Probes for Nitric Oxide Based on Boron Dipyrromethene ChromophoreRational Design of Potentially Useful Bioimaging Fluorescence Probe. J. Am. Chem. Soc. 2004, 126, 3357–3367. [Google Scholar] [CrossRef]
- Wu, Y.; Peng, X.; Guo, B.; Fan, J.; Zhang, Z.; Wang, J.; Cui, A.; Gao, Y. Boron dipyrromethene fluorophore based fluorescence sensor for the selective imaging of Zn(II) in living cells. Org. Biomol. Chem. 2005, 3, 1387–1392. [Google Scholar] [CrossRef]
- Verdoes, M.; Florea, B.I.; Menendez-Benito, V.; Maynard, C.J.; Witte, M.D.; van der Linden, W.A.; van den Nieuwendijk, A.M.C.H.; Hofmann, T.; Berkers, C.R.; van Leeuwen, F.W.B.; et al. A Fluorescent Broad-Spectrum Proteasome Inhibitor for Labeling Proteasomes in vitro and in vivo. Chem. Biol. 2006, 13, 1217–1226. [Google Scholar] [CrossRef]
- Sun, Z.N.; Wang, H.L.; Liu, F.Q.; Chen, Y.; Kwong, P.; Tam, H.; Yang, D. BODIPY-Based Fluorescent Probe for Peroxynitrite Detection and Imaging in Living Cells. Org. Lett. 2009, 11, 1887–1890. [Google Scholar] [CrossRef]
- Middleton, R.J.; Briddon, S.J.; Cordeaux, Y.; Yates, A.S.; Cale, C.L.; George, M.W.; Baker, J.G.; Hill, S.J.; Kellam, B. New Fluorescent Adenosine A1-Receptor Agonists That Allow Quantification of Ligand−Receptor Interactions in Microdomains of Single Living Cells. J. Med. Chem. 2007, 50, 782–793. [Google Scholar] [CrossRef]
- Erten-Ela, S.; Ylimaz, M.D.; Icil, B.; Dede, Y.; Icli, S.; Akkaya, E.U. A Panchromatic Boradiazaindacene (BODIPY) Sensitizer for Dye-Sensitized Solar Cells. Org. Lett. 2008, 10, 3299–3302. [Google Scholar] [CrossRef]
- Kumaresan, D.; Thummel, R.P.; Bura, T.; Ulrich, G.; Ziessel, R. Color Tuning in New Metal-Free Organic Sensitizers (Bodipys) for Dye-Sensitized Solar Cells. Chem. Eur. J. 2009, 15, 6335–6339. [Google Scholar] [CrossRef]
- Kolemen, S.; Cakmak, Y.; Erten-Ela, S.; Altay, Y.; Brendel, J.; Thelakkat, M.; Akkaya, E.U. Solid-State Dye-Sensitized Solar Cells Using Red and Near-IR Absorbing Bodipy Sensitizers. Org. Lett. 2010, 12, 3812–3815. [Google Scholar]
- Maeda, H.; Hasegawa, M.; Hashimoto, T.; Kakimoto, T.; Nishio, S.; Nakanishi, T. Nanoscale Spherical Architectures Fabricated by Metal Coordination of Multiple Dipyrrin Moieties. J. Am. Chem. Soc. 2006, 128, 10024–10025. [Google Scholar]
- Yu, L.; Muthukumaran, K.; Sazanovich, I.V.; Kirmaier, C.; Hindin, E.; Diers, J.R.; Boyle, P.D.; Bocian, D.F.; Holten, D.; Lindsey, J.S. Excited-State Energy-Transfer Dynamics in Self-Assembled Triads Composed of Two Porphyrins and an Intervening Bis(dipyrrinato)metal Complex. Inorg. Chem. 2003, 42, 6629–6647. [Google Scholar] [CrossRef]
- Rio, Y.; Sánchez-García, D.; Seitz, W.; Torres, T.; Sessler, J.L.; Guldi, D.M. A Bisfullerene–Bis(dipyrrinato)zinc Complex: Electronic Coupling and Charge Separation in an Easy-to-Assemble Synthetic System. Chem. Eur. J. 2009, 15, 3956–3959. [Google Scholar] [CrossRef]
- Sazanovich, I.V.; Kirmaier, C.; Hindin, E.; Yu, L.; Bocian, D.F.; Lindsey, J.S.; Holten, D. Structural Control of the Excited-State Dynamics of Bis(dipyrrinato)zinc Complexes: Self-Assembling Chromophores for Light-Harvesting Architectures. J. Am. Chem. Soc. 2004, 126, 2664–2665. [Google Scholar]
- Thoi, V.S.; Stork, J.R.; Magde, D.; Cohen, S.M. Luminescent Dipyrrinato Complexes of Trivalent Group 13 Metal Ions. Inorg. Chem. 2006, 45, 10688–10697. [Google Scholar] [CrossRef]
- Kobayashi, J.; Kushida, T.; Kawashima, T. Synthesis and Reversible Control of the Fluorescent Properties of a Divalent Tin Dipyrromethene. J. Am. Chem. Soc. 2009, 131, 10836–10837. [Google Scholar] [CrossRef]
- Crawford, S.M.; Ali, A.A.; Cameron, T.S.; Thompson, A. Synthesis and Characterization of Fluorescent Pyrrolyldipyrrinato Sn(IV) Complexes. Inorg. Chem. 2011, 50, 8207–8213. [Google Scholar] [CrossRef]
- Filatov, M.A.; Lebedev, A.Y.; Mukhin, S.N.; Vinogradov, S.A.; Cheprakov, A.V. π-Extended Dipyrrins Capable of Highly Fluorogenic Complexation with Metal Ions. J. Am. Chem. Soc. 2010, 132, 9552–9554. [Google Scholar] [CrossRef]
- Ikeda, C.; Ueda, S.; Nabeshima, T. Aluminium complexes of N2O2-type dipyrrins: the first hetero-multinuclear complexes of metallo-dipyrrins with high fluorescence quantum yields. Chem. Commun. 2009, 2544–2546. [Google Scholar] [CrossRef]
- Zander, M.; Rettig, W. Fluorescence studies on solvent-induced intramolecular charge separation in symmetric biaryls. Chem. Phys. Lett. 1984, 110, 602–608. [Google Scholar] [CrossRef]
- Kang, T.J.; Kahlow, M.A.; Giser, D.; Swallen, S.; Nargarajan, V.; Jarzeba, W.; Barbara, P.F. Dynamic solvent effects in the electron-transfer kinetics of S1 bianthryls. J. Phys. Chem. 1988, 92, 6800–6807. [Google Scholar] [CrossRef]
- Rettig, W. Charge Separation in Excited States of Decoupled Systems—TICT Compounds and Implications Regarding the Development of New Laser Dyes and the Primary Process of Vision and Photosynthesis. Angew. Chem. Int. Ed. Engl. 1986, 25, 971–988. [Google Scholar] [CrossRef]
- Vauthey, E. Photoinduced Symmetry-Breaking Charge Separation. ChemPhysChem 2012, 13, 2001–2011. [Google Scholar] [CrossRef]
- Giaimo, J.V.; Gusev, A.V.; Wasielewski, M.R. Excited-State Symmetry Breaking in Cofacial and Linear Dimers of a Green Perylenediimide Chlorophyll Analogue Leading to Ultrafast Charge Separation. J. Am. Chem. Soc. 2002, 124, 8530–8531. [Google Scholar] [CrossRef]
- Banerji, N.; Fürstenberg, A.; Bhosale, S.; Sisson, A.L.; Sakai, N.; Matile, S.; Vauthey, E. Ultrafast Photoinduced Charge Separation in Naphthalene Diimide Based Multichromophoric Systems in Liquid Solutions and in a Lipid Membrane. J. Phys. Chem. B 2008, 112, 8912–8922. [Google Scholar] [CrossRef]
- Kusaka, S.; Sakamoto, R.; Kitagawa, Y.; Okumura, M.; Nishihara, H. An Extremely Bright Heteroleptic Bis(dipyrrinato)zinc(II) Complex. Chem. Asian J. 2012, 7, 907–910. [Google Scholar] [CrossRef]
- Sheldrick, W.; Engel, J. X-Ray crystal structure of the zinc complex of 1,2,3,7,8,12,13,17,18,19-decamethylbiladiene-a,c. J. Chem. Soc. Chem. Commun. 1980, 5–6. [Google Scholar] [CrossRef]
- Pechmann, H.V. Ueber Condensationsprodukte zweibasischer Fettsäuren. Ber. Dtsch. Chem. Ges. 1882, 15, 881. [Google Scholar] [CrossRef]
- Silver, J.; Ahmet, M.T.; Bowden, K.; Miller, J.R.; Rahmat, S.; Reynolds, C.A.; Bashall, A.; McPathlkin, M.; Trotter, J. Electrochromic behaviour and X-ray structure analysis of a Pechmann dye, (E)-5,5′-diphenyl-3,3′-bifuranylidene-2,2′-dione. J. Mater. Chem. 1994, 4, 1201–1204. [Google Scholar] [CrossRef]
- Bergley, M.J.; Crombie, L.; Griffiths, G.L.; Jones, R.C.F.; Rahmani, M. Charge-transfer and non-charge-transfer crystal forms of (E)-5,5′-dimesitylbifuranylidenediones : an X-ray structural investigation. Chem. Commun. 1981, 823–825. [Google Scholar]
- Fang, C.S.; Bergmann, W. Pechmann's Dye and Related Compounds. J. Org. Chem. 1951, 16, 1231–1237. [Google Scholar] [CrossRef]
- Irikawa, H.; Adachi, N.; Muraoka, H. Preparation and Absorption Spectral Properties of the Nitrogen Analogs of a Pechmann Dye and Its Isomeric Pyrano[4,3-c]pyran-1,5-dione. Heterocycles 1998, 48, 1415–1422. [Google Scholar] [CrossRef]
- Bowden, K.; Etemadi, R.; Ranson, R.J. Reactions of carbonyl compounds in basic solutions. Part 17. The alkaline hydrolysis of substituted (E)-5,5-diphenylbifuranylidenediones and 3,7-diphenylpyrano[4,3-c]pyran-1,5-diones. J. Chem. Soc. Perkin Trans. 2 1991, 743–746. [Google Scholar] [CrossRef]
- Didier, P.; Ulrich, G.; Mely, Y.; Ziessel, R. Improved push-pull-push E-Bodipy fluorophores for two-photon cell-imaging. Org. Biomol. Chem. 2009, 7, 3639–3642. [Google Scholar] [CrossRef]
- Andrade, C.D.; Yanez, C.O.; Rodriguez, L.; Belfield, K.D. A Series of Fluorene-Based Two-Photon Absorbing Molecules: Synthesis, Linear and Nonlinear Characterization, and Bioimaging. J. Org. Chem. 2010, 75, 3975–3982. [Google Scholar] [CrossRef]
- Velusamy, M.; Shen, J.; Lin, J.; Lin, Y.; Hsieh, C.; Lai, C.; Lai, C.; Ho, M.; Chen, Y.; Chou, P.; Hisao, J. A New Series of Quadrupolar Type Two-Photon Absorption Chromophores Bearing 11, 12-Dibutoxydibenzo[a,c]-phenazine Bridged Amines; Their Applications in Two-Photon Fluorescence Imaging and Two-Photon Photodynamic Therapy. Adv. Funct. Mater. 2009, 19, 2388–2397. [Google Scholar] [CrossRef]
- Zhou, G.; Wong, W.-Y.; Poon, S.-Y.; Ye, C.; Lin, Z. Symmetric Versus Unsymmetric Platinum(II) Bis(aryleneethynylene)s with Distinct Electronic Structures for Optical Power Limiting/Optical Transparency Trade-off Optimization. Adv. Funct. Mater. 2009, 19, 531–544. [Google Scholar] [CrossRef]
- Charlot, M.; Izard, N.; Mongin, O.; Riehl, D.; Blanchard-Desce, M. Optical limiting with soluble two-photon absorbing quadrupoles: Structure–property relationships. Chem. Phys. Lett. 2006, 417, 297–302. [Google Scholar] [CrossRef]
- Yang, Y.; Farley, R.T.; Steckler, T.T.; Eom, S.-H.; Reynolds, J.R.; Schanze, K.S.; Xue, J. Near infrared organic light-emitting devices based on donor-acceptor-donor oligomers. Appl. Phy. Lett. 2008, 93, 163305–163307. [Google Scholar]
- Goudreault, T.; He, Z.; Guo, Y.; Ho, C.-L.; Zhan, H.; Wang, Q.; Ho, K.Y.-F.; Wong, K.-L.; Fortin, D.; Yao, B.; et al. Synthesis, Light-Emitting, and Two-Photon Absorption Properties of Platinum-Containing Poly(arylene-ethynylene)s Linked by 1,3,4-Oxadiazole Units. Macromolecules 2010, 43, 7936–7949. [Google Scholar] [CrossRef]
- Lincker, F.; Kreher, D.; Attias, A.; Do, J.; Kim, E.; Hapiot, P.; Lemaitre, N.; Gettroy, B.; Ulrich, G.; Ziessel, R. Rodlike Fluorescent π-Conjugated 3,3′-Bipyridazine Ligand: Optical, Electronic, and Complexation Properties. Inorg. Chem. 2010, 49, 3991–4001. [Google Scholar] [CrossRef]
- Huang, F.; Chem, K.; Yip, H.; Hau, S.K.; Acton, O.; Zhang, Y.; Luo, J.; Jen, A.K. -Y. Development of New Conjugated Polymers with Donor−π-Bridge−Acceptor Side Chains for High Performance Solar Cells. J. Am. Chem. Soc. 2009, 131, 13886–13887. [Google Scholar]
- Zou, Y.; Najari, A.; Berrouard, P.; Beaupre, S.; Aich, B.R.; Tao, Y.; Leclerc, M. A Thieno[3,4-c]pyrrole-4,6-dione-Based Copolymer for Efficient Solar Cells. J. Am. Chem. Soc. 2010, 132, 5330–5331. [Google Scholar] [CrossRef]
- Kono, T.; Kumaki, D.; Nishida, J.; Tokito, S.; Yamashita, Y. Dithienylbenzobis(thiadiazole) based organic semiconductors with low LUMO levels and narrow energy gaps. Chem. Commun. 2010, 46, 3265–3267. [Google Scholar] [CrossRef]
- Le, Y.; Nitani, M.; Kawahara, M.; Tada, H.; Aso, Y. Air-Stable n-Type Organic Field-Effect Transistors Based on Carbonyl-Bridged Bithiazole Derivatives. Adv. Funct. Mater. 2010, 20, 907–913. [Google Scholar] [CrossRef]
- Hayashi, M.; Toshimitsu, F.; Sakamoto, R.; Nishihara, H. Double Lactonization in Triarylamine-Conjugated Dimethyl Diethynylfumarate: Formation of Intensely Colored and Luminescent Quadrupolar Molecules Including a Missing Structural Isomer of Pechmann Dyes. J. Am. Chem. Soc. 2011, 133, 14518–14521. [Google Scholar] [CrossRef]
- Sakamoto, R.; Murata, M.; Nishihara, H. Visible-Light Photochromism of Bis(ferrocenylethynyl)ethenes Switches Electronic Communication between Ferrocene Sites. Angew. Chem. Int. Ed. Engl. 2006, 45, 4793–4795. [Google Scholar] [CrossRef]
- Sakamoto, R.; Kume, S.; Nishihara, H. Visible-Light Photochromism of Triarylamine- or Ferrocene-Bound Diethynylethenes that Switches Electronic Communication between Redox Sites and Luminescence. Chem. Eur. J. 2008, 14, 6978–6986. [Google Scholar] [CrossRef]
- Hayashi, M.; Sakamoto, R.; Nishihara, H. Extremely Efficient and Reversible Visible-Light Photochromism and Accompanying Switch of Electronic Communication in N-Phenylcarbazole-Appended Diethynylethene. Chem. Eur. J. 2012, 18, 8610–8613. [Google Scholar] [CrossRef]
- Uchiyama, M.; Ozawa, H.; Takuma, K.; Matsumoto, Y.; Yonehara, M.; Hiroya, K.; Sakamoto, T. Regiocontrolled Intramolecular Cyclizations of Carboxylic Acids to Carbon−Carbon Triple Bonds Promoted by Acid or Base Catalyst. Org. Lett. 2006, 8, 5517–5520. [Google Scholar] [CrossRef]
- Hellal, M.; Bourguignon, J.-J.; Bihel, F.J.-J. 6-endo-dig Cyclization of heteroarylesters to alkynes promoted by Lewis acid catalyst in the presence of Brønsted acid. Tetrahedron Lett. 2008, 49, 62–65. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sakamoto, R.; Kusaka, S.; Hayashi, M.; Nishikawa, M.; Nishihara, H. Coordination Programming of Photofunctional Molecules. Molecules 2013, 18, 4091-4119. https://doi.org/10.3390/molecules18044091
Sakamoto R, Kusaka S, Hayashi M, Nishikawa M, Nishihara H. Coordination Programming of Photofunctional Molecules. Molecules. 2013; 18(4):4091-4119. https://doi.org/10.3390/molecules18044091
Chicago/Turabian StyleSakamoto, Ryota, Shinpei Kusaka, Mikihiro Hayashi, Michihiro Nishikawa, and Hiroshi Nishihara. 2013. "Coordination Programming of Photofunctional Molecules" Molecules 18, no. 4: 4091-4119. https://doi.org/10.3390/molecules18044091
APA StyleSakamoto, R., Kusaka, S., Hayashi, M., Nishikawa, M., & Nishihara, H. (2013). Coordination Programming of Photofunctional Molecules. Molecules, 18(4), 4091-4119. https://doi.org/10.3390/molecules18044091