Preparation, Purification and Regioselective Functionalization of Protoescigenin—The Main Aglycone of Escin Complex

 , and
, and
Abstract
:1. Introduction

2. Results and Discussion
2.1. General
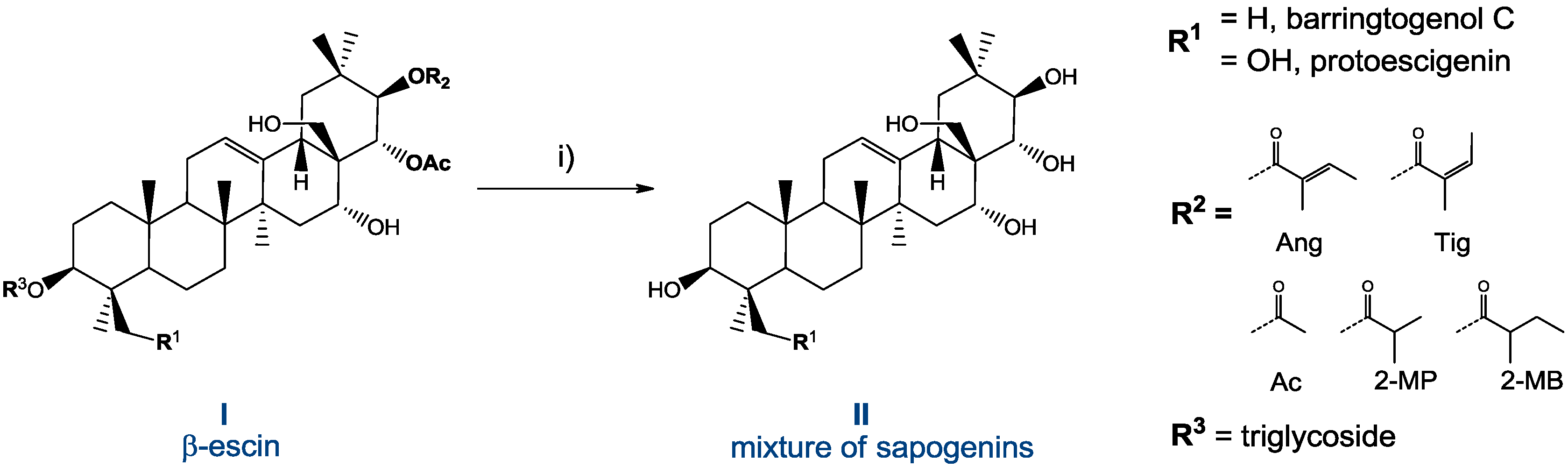
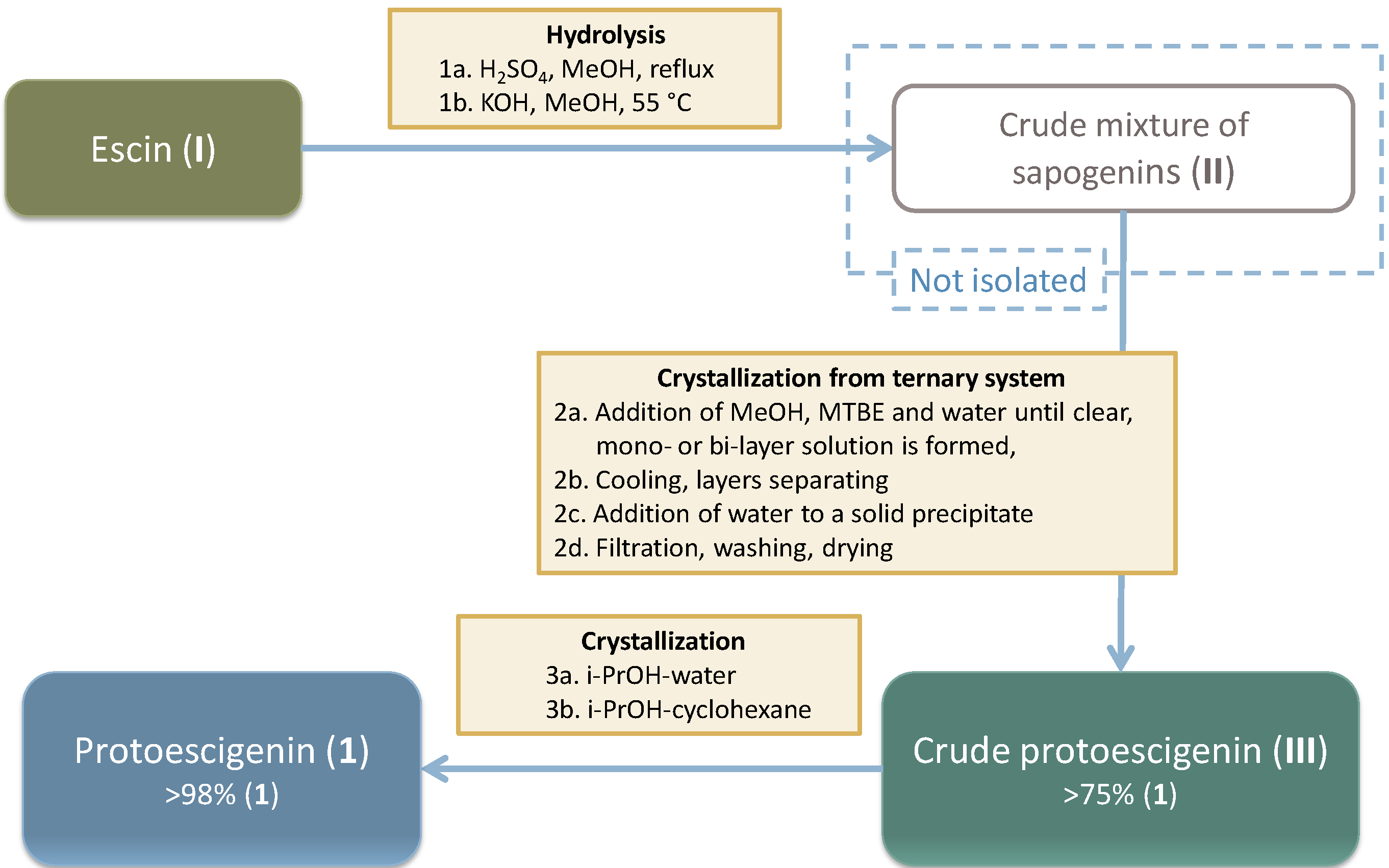
2.2. Preparation and Isolation of 1


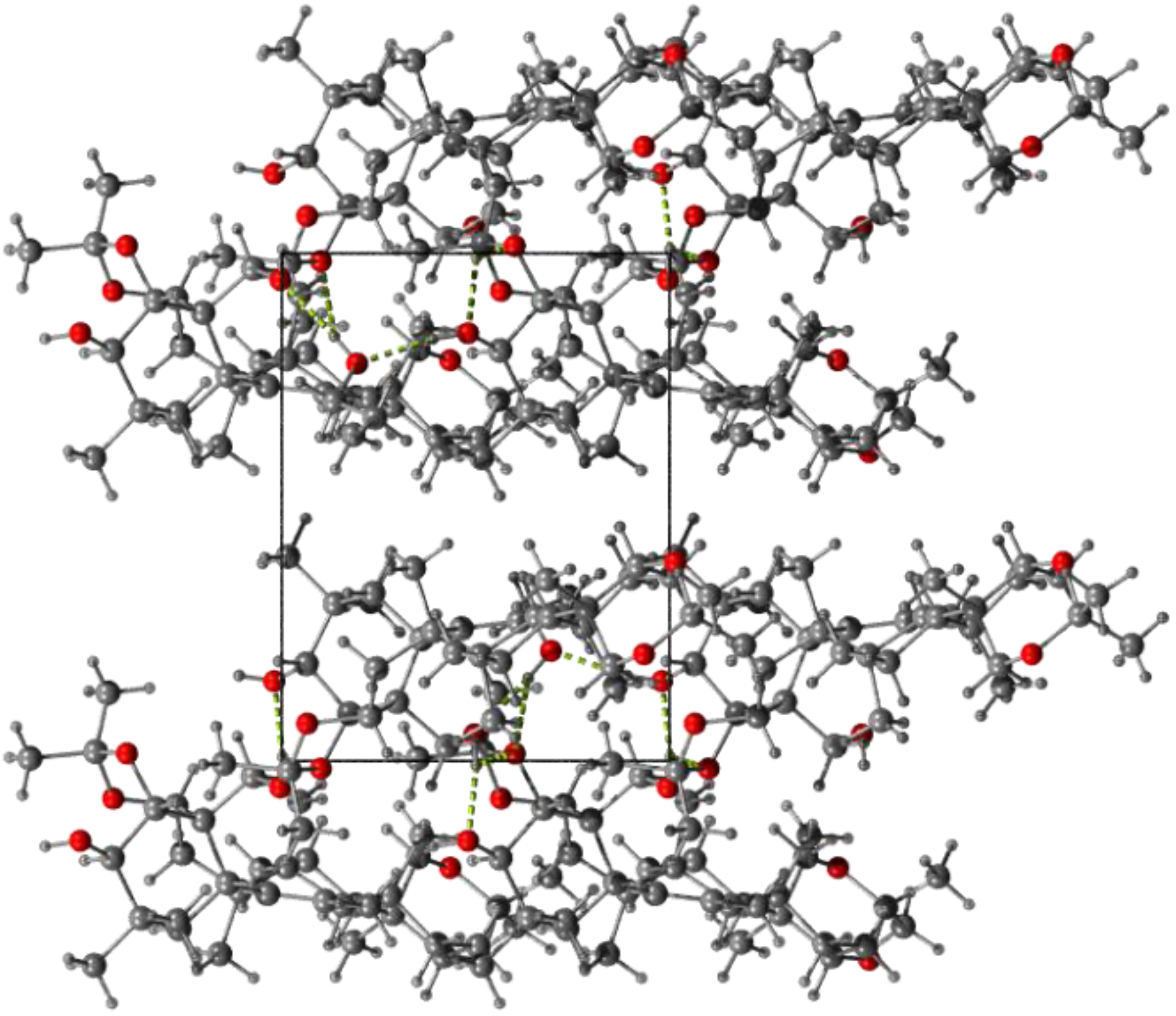
2.3. Characteristics of Solid Forms of 1


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 2θ [°] | |||
|---|---|---|---|
| form II | form III | form IV | form VI |
| 6.16 | 6.85 | 6.00 | 3.08 |
| 6.92 | 8.58 | 6.53 | 6.29 |
| 8.85 | 11.30 | 7.97 | 7.42 |
| 9.76 | 13.04 | 10.33 | 9.37 |
| 10.04 | 13.76 | 11.94 | 12.74 |
| 11.22 | 14.75 | 12.33 | 13.43 |
| 12.72 | 15.35 | 13.00 | 14.03 |
| 13.88 | 16.64 | 14.10 | 14.80 |
| 14.50 | 17.11 | 14.34 | 15.51 |
| 15.54 | 18.96 | 15.04 | 17.76 |
| 16.23 | 19.15 | 15.92 | 18.28 |
| 18.77 | 20.30 | 16.09 | 18.82 |
| 19.18 | 20.68 | 16.99 | 19.32 |
| 20.23 | 21.15 | 22.43 | 21.44 |
| 21.84 | 22.65 | 23.42 | |


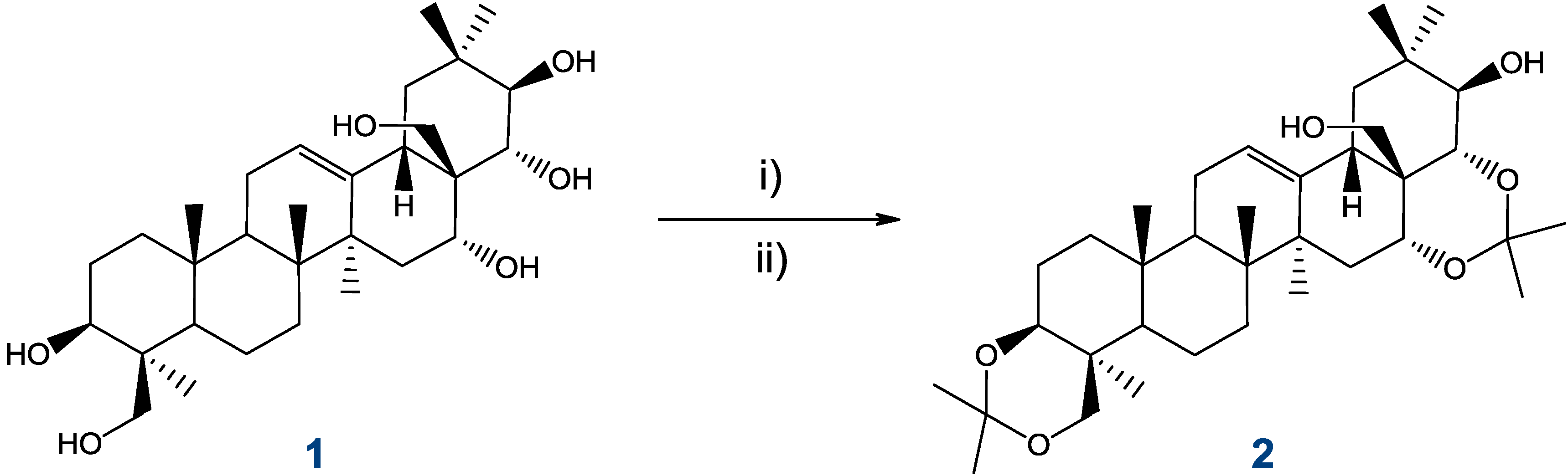
2.4. Preparation and Isolation of 2


2.5. Structural Data


3. Experimental
3.1. General
3.2. Preparation of Protoescigenin (1): Materials and Operations Shown in Scheme 2

3.3. Preparation of 3,24;16,22-di-O,O-isopropylideneprotoescigenin (2)
3.4. Single Crystal X-ray Diffraction
4. Conclusions
Acknowledgments
References
- Pittler, M.H.; Ernst, E. Horse chesnut seed extract for chronic venous insufficiency. Cochrane Db. Syst. Rev. 2012, CD003230. [Google Scholar]
- Sirtori, C.R. Aescin: Pharmacology, Pharmacokinetics and therapeutic profile. Pharmacol. Res. 2001, 44, 183–193. [Google Scholar] [CrossRef]
- Zhang, Z.; Li, S.; Lian, X.-Y. An overview of genus Aesculus. L.: ethnobotany, phytochemistry, and pharmacological activities. Pharmac. Crops 2010, 1, 24–51. [Google Scholar]
- Persson, I.A.L.; Persson, K. Horse chesnut (Aesculus. hippocastanum L.). Recent Prog. Med. Plants 2010, 28, 159–171. [Google Scholar]
- Zhou, X.Y.; Fu, F.H.; Li, Z.; Dong, Q.J.; He, J.; Wang, C.H. Escin, a natural mixture of triterpene saponins, exhibits antitumor activity against hepatocellular carcinoma. Planta. Med. 2009, 75, 1580–1585. [Google Scholar] [CrossRef]
- Dinda, B.; Debnath, S.; Mohanta, B.C.; Harigaya, Y. Naturally occurring triterpenoid saponins. Chem. Biodivers. 2010, 7, 2327–2579. [Google Scholar] [CrossRef]
- Jäger, S.; Trojan, H.; Kopp, T.; Laszczyk, M.N.; Scheffler, A. Pentacyclic triterpene distribution in various plants–rich sources for a new group of multi-potent plant extracts. Molecules 2009, 14, 2016–2031. [Google Scholar] [CrossRef]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef]
- Beutler, J.A. Natural products as a foundation for drug discovery. Curr. Protoc. Pharmacol. 2009, 46, 9.11.1–9.11.21. [Google Scholar]
- Sporn, M.B.; Liby, K.T.; Yore, M.M.; Fu, L.; Lopchuk, J.M.; Gribble, G.W. New synthetic triterpenoids: potent agents for prevention and treatment of tissue injury caused by inflammatory and oxidative stress. J. Nat. Prod. 2011, 74, 537–545. [Google Scholar] [CrossRef]
- Sheng, H.; Sun, H. Synthesis, Biology and clinical significance of pentacyclic triterpenes: A multi-target approach to prevention and treatment of metabolic and vascular diseases. Nat. Prod. Rep. 2011, 28, 543–593. [Google Scholar] [CrossRef]
- Kuhn, R.; Löw, I. Über die protoäscigeninester aus Äscin. Tetrahedron 1966, 22, 1899–1906. [Google Scholar] [CrossRef]
- Wulff, G.; Tschesche, R. Über triterpene-XXVI: Über die struktur der rosskastaniensaponnie (Äscin) und die aglykone verwandter glykoside. Tetrahedron 1969, 25, 415–436. [Google Scholar] [CrossRef]
- Agrawal, P.K.; Thakur, R.S.; Shoolery, J.N. Application of 2D NMR spectroscopy to the structural establishment of the major hydrolysis product of escin. J. Nat. Prod. 1991, 54, 1394–1396. [Google Scholar] [CrossRef]
- Gruza, M.M.; Zegrocka-Stendel, O.; Giller, T.; Grynkiewicz, G.; Giller, T.; Łaszcz, M.; Jatczak, K. Preparation of protoescigenin from escin. PCT/PL2012/000102; filed 10 October 2012,
- Wagner, J.; Hoffmann, H.; Löw, I. Über Inhaltsstoffe des Roβkastaniensamens, VII. O-Isopropyliden-Derivate von Protoäscigenin, Barringtogenol C und von deren 21-bzw. 28-Angelika(Tiglin)-säureestern. Liebigs Ann. Chem. 1969, 729, 205–212. [Google Scholar] [CrossRef]
- Gruza, M.M.; Jatczak, K.; Łaszcz, M.; Grynkiewicz, G.; Giller, T.; Witkowska, A.B. Sposób wytwarzania 3,24;16,22-O,O;O,O-diizopropylideno-protoescygeniny oraz jej postacie krystaliczne. PL/P-402306; filed 30 December 2012,
- Tsuda, Y.; Kiuchi, F.; Liu, H.-M. Establishment of the structure of gymnemagenin by x-ray analysis and the structure of deacylgymnemic acid. Tetrahedron Lett. 1989, 30, 361–362. [Google Scholar] [CrossRef]
- Stöcklin, W. Gymnemagenin, vermutliche struktur Glykoside und Aglykone, 289. Mitteilung. Helv. Chim. Acta 1967, 50, 491–503. [Google Scholar] [CrossRef]
- Kuhn, R.; Löw, I. Über Protoäscigenin, die Stammsubstanz der Äscine. Liebigs. Ann. Chem. 1963, 669, 183–188. [Google Scholar] [CrossRef]
- Spek, A. Structure validation in chemical crystallography. Acta Crystallogr. D 2009, 65, 148–155. [Google Scholar] [CrossRef]
- Van der Sluis, P.; Spek, A.L. BYPASS: an effective method for the refinement of crystal structures containing disordered solvent regions. Acta Crystallogr. A 1990, 46, 194–201. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1 and 2 subject to individual enquiry.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gruza, M.M.; Jatczak, K.; Zagrodzki, B.; Łaszcz, M.; Koziak, K.; Malińska, M.; Cmoch, P.; Giller, T.; Zegrocka-Stendel, O.; Woźniak, K.; et al. Preparation, Purification and Regioselective Functionalization of Protoescigenin—The Main Aglycone of Escin Complex. Molecules 2013, 18, 4389-4402. https://doi.org/10.3390/molecules18044389
Gruza MM, Jatczak K, Zagrodzki B, Łaszcz M, Koziak K, Malińska M, Cmoch P, Giller T, Zegrocka-Stendel O, Woźniak K, et al. Preparation, Purification and Regioselective Functionalization of Protoescigenin—The Main Aglycone of Escin Complex. Molecules. 2013; 18(4):4389-4402. https://doi.org/10.3390/molecules18044389
Chicago/Turabian StyleGruza, Mariusz M., Kamil Jatczak, Bogdan Zagrodzki, Marta Łaszcz, Katarzyna Koziak, Maura Malińska, Piotr Cmoch, Tomasz Giller, Oliwia Zegrocka-Stendel, Krzysztof Woźniak, and et al. 2013. "Preparation, Purification and Regioselective Functionalization of Protoescigenin—The Main Aglycone of Escin Complex" Molecules 18, no. 4: 4389-4402. https://doi.org/10.3390/molecules18044389
APA StyleGruza, M. M., Jatczak, K., Zagrodzki, B., Łaszcz, M., Koziak, K., Malińska, M., Cmoch, P., Giller, T., Zegrocka-Stendel, O., Woźniak, K., & Grynkiewicz, G. (2013). Preparation, Purification and Regioselective Functionalization of Protoescigenin—The Main Aglycone of Escin Complex. Molecules, 18(4), 4389-4402. https://doi.org/10.3390/molecules18044389