Synthesis and Relaxivity Studies of a DOTA-Based Nanomolecular Chelator Assembly Supported by an Icosahedral Closo-B122− -Core for MRI: A Click Chemistry Approach

Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction

2. Results and Discussion
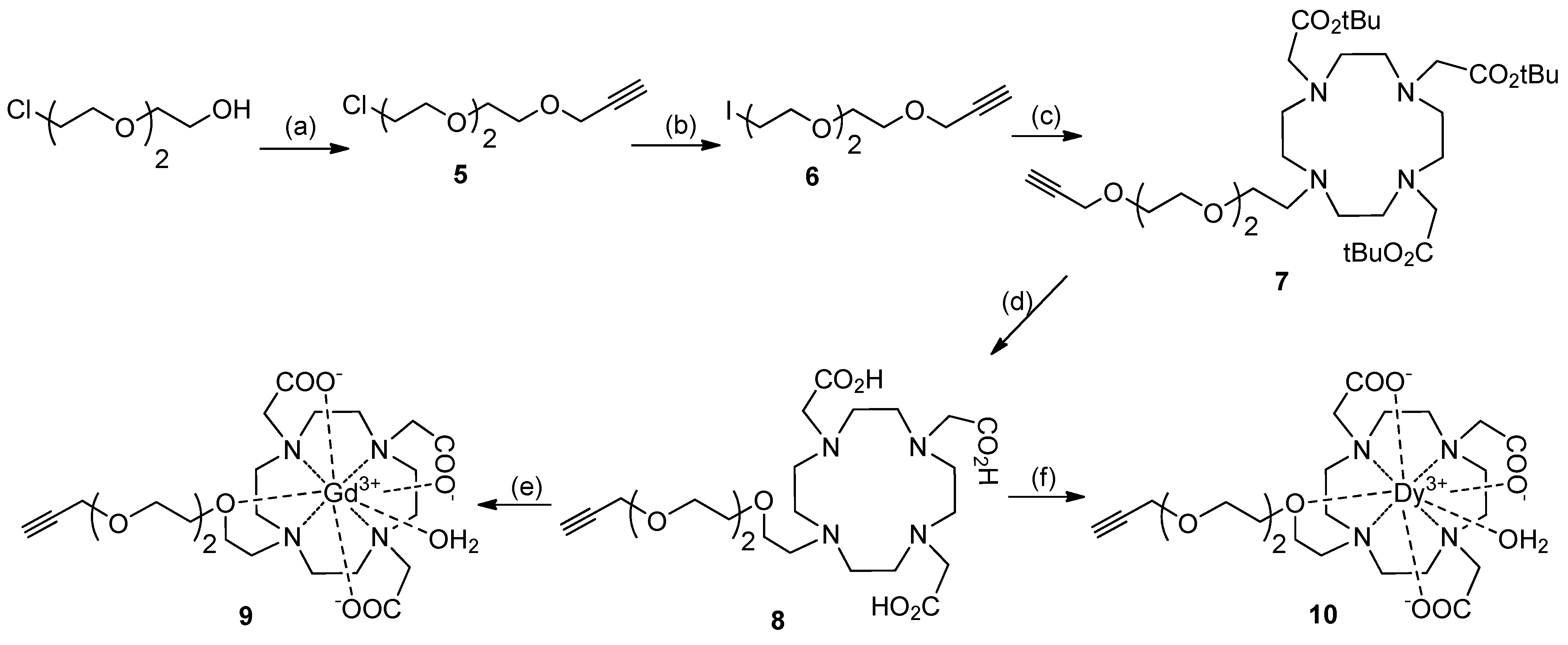
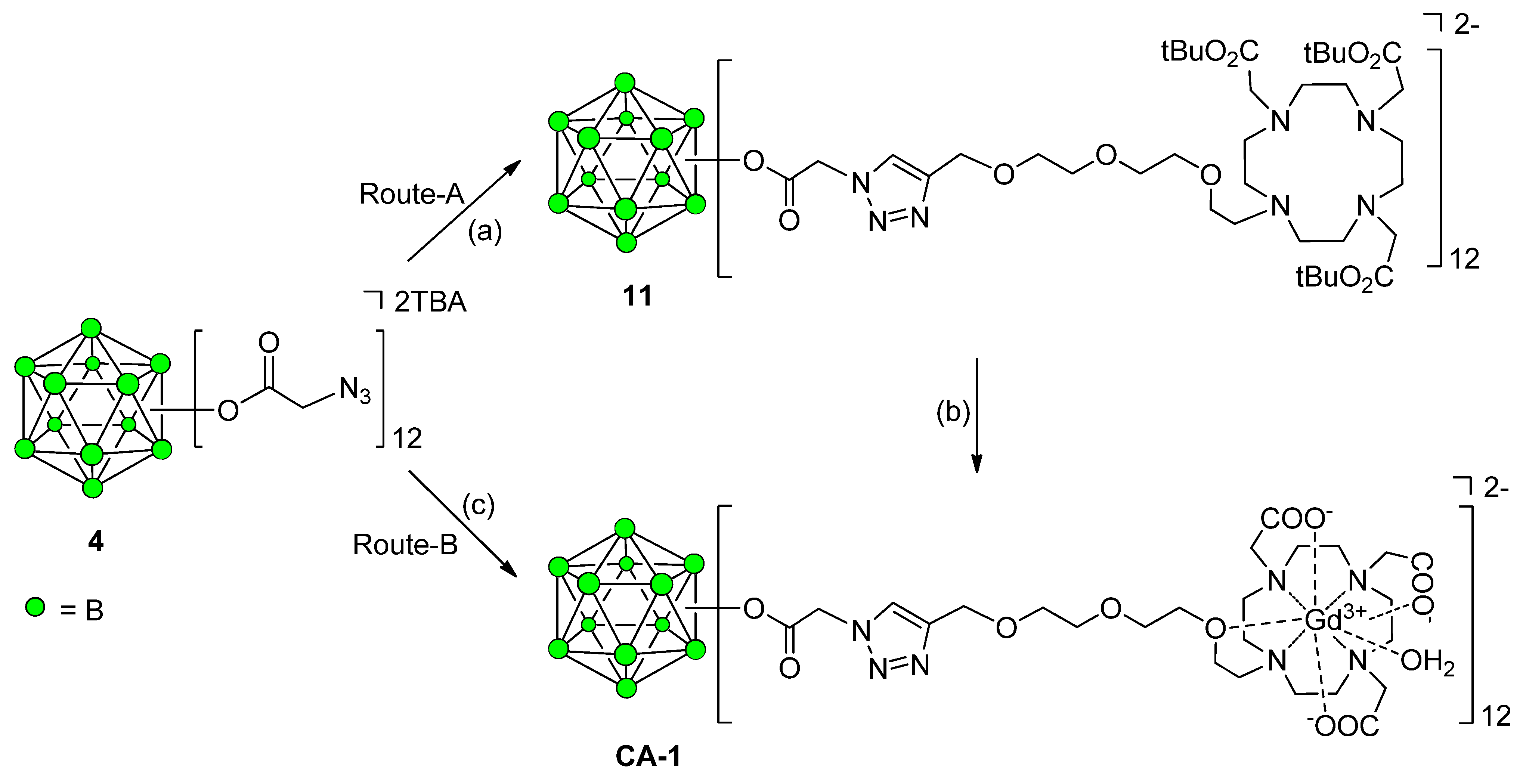
Synthesis of 12-Fold Azidoacetate Closomer 4

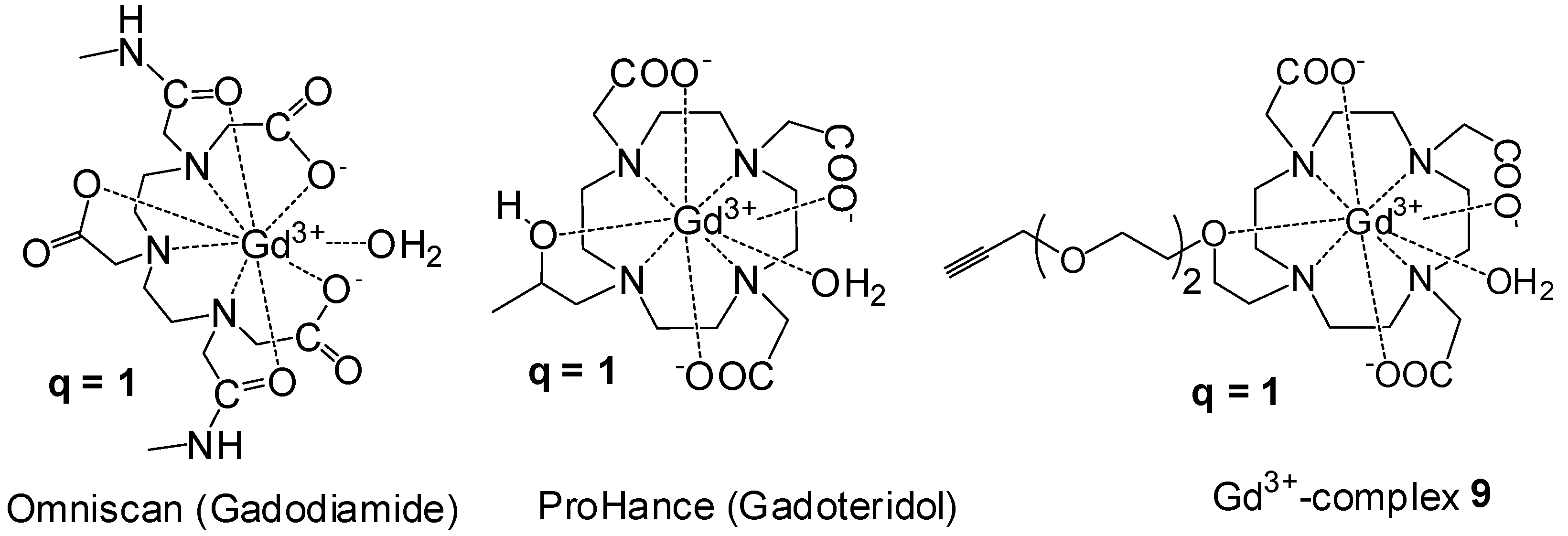
Synthesis of DO3A Ligands
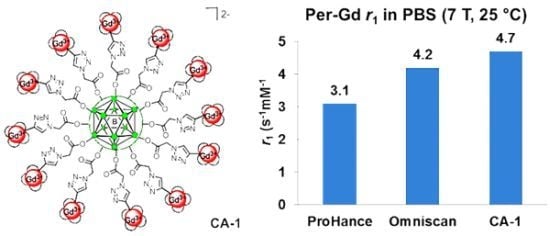
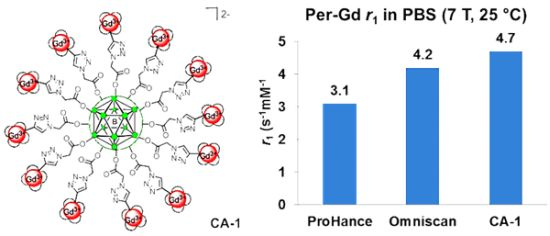
Synthesis of Closomer Contrast Agents CA-1


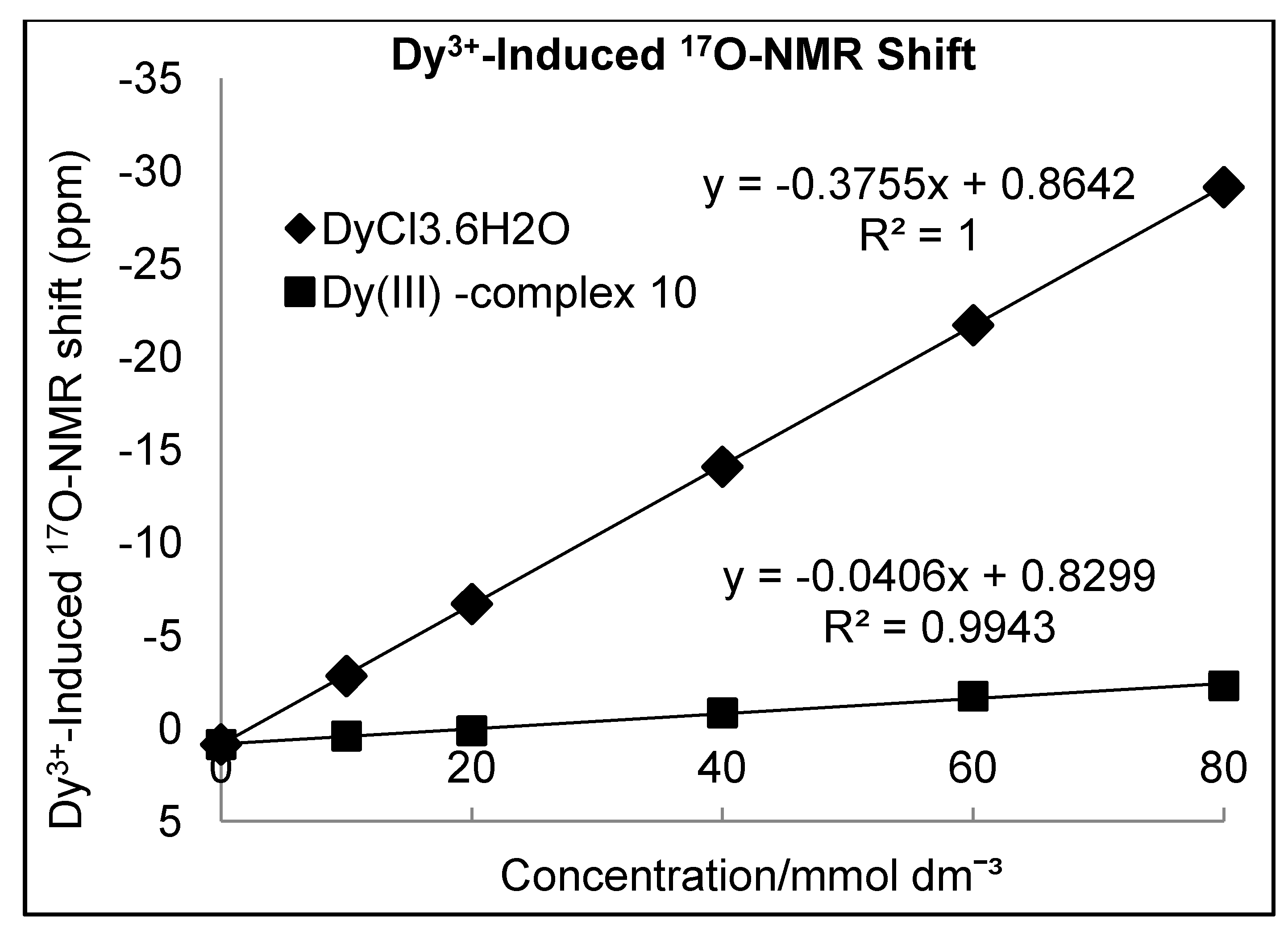
Determination of Hydration Number (q)

Relaxivity Studies



3. Experimental
3.1. General
3.2. Synthesis
Synthesis of CA-1
3.3. Relaxivity Determinations
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflicts of Interest
References
- Hermann, P.; Kotek, J.; Kubíček, V.; Lukeš, I. Gadolinium(III) complexes as MRI contrast agents: ligand design and properties of the complexes. Dalton Trans. 2008, 3027–3047. [Google Scholar]
- Caravan, P.; Ellison, J.J.; McMurry, T.J.; Lauffer, R.B. Gadolinium(III) Chelates as MRI Contrast Agents: Structure, Dynamics, and Applications. Chem. Rev. 1999, 99, 2293–2352. [Google Scholar] [CrossRef]
- Aime, S.; Botta, M.; Fasano, M.; Terreno, E. Lanthanide(III) chelates for NMR biomedical applications. Chem. Soc. Rev. 1998, 27, 19–29. [Google Scholar]
- Idée, J.M.; Port, M.; Medina, C.; Lancelot, E.; Fayoux, E.; Ballet, S.; Corot, C. Possible involvement of gadolinium chelates in the pathophysiology of nephrogenic systemic fibrosis: A critical review. Toxicology 2008, 248, 77–88. [Google Scholar] [CrossRef]
- Caravan, P. protein-targeted gadolinium-based magnetic resonance imaging (mri) contrast agents: Design and mechanism of action. Acc. Chem. Res. 2009, 42, 851–862. [Google Scholar] [CrossRef]
- Solomon, I. Relaxation processes in a system of two spins. Phys. Rev. 1955, 99, 559–565. [Google Scholar] [CrossRef]
- Bloembergen, N.J. Proton relaxation times in paramagnetic solutions. Chem. Phys. 1957, 27, 572–573. [Google Scholar]
- Botta, M.; Tei, L. Relaxivity enhancement in macromolecular and Nanosized GdIII-Based MRI contrast agents. Eur. J. Inorg. Chem. 2012, 2012, 1945–1960. [Google Scholar] [CrossRef]
- Langereis, S.; de Lussanet, Q.G.; van Genderen, M.H.P.; Backes, W.H.; Meijer, E.W. Multivalent contrast agents based on Gadolinium−Diethylenetriaminepentaacetic Acid-Terminated Poly(propylene imine) dendrimers for Magnetic Resonance Imaging. Macromolecules 2004, 37, 3084–3091. [Google Scholar] [CrossRef]
- Torchilin, V.; Babich, J.; Weissig, V. Liposomes and Micelles to Target the Blood Pool for Imaging Purposes. J. Liposome Res. 2000, 10, 483–499. [Google Scholar] [CrossRef]
- Floyd, W.C.; Klemm, P.J.; Smiles, D.E.; Kohlgruber, A.C.; Pierre, V.C.; Mynar, J.L.; Fréchet, J.M.J.; Raymond, K.N. Conjugation effects of various linkers on Gd(III) MRI contrast agents with dendrimers: Optimizing the hydroxypyridinonate (HOPO) ligands with nontoxic, Degradable Esteramide (EA) dendrimers for high relaxivity. J. Am. Chem. Soc. 2011, 133, 2390–2393. [Google Scholar] [CrossRef]
- De León-Rodríguez, L.M.; Lubag, A.; Udugamasooriya, D.G.; Proneth, B.; Brekken, R.A.; Sun, X.; Kodadek, T.; Sherry, A.D. MRI detection of VEGFR2 in vivo using a low molecular weight peptoid-(Gd)8-dendron for targeting. J. Am. Chem. Soc. 2010, 132, 12829–12831. [Google Scholar] [CrossRef]
- Bryson, J.M.; Chu, W.; Lee, J.; Reineke, T.M. A β-cyclodextrin “click cluster” decorated with seven paramagnetic chelates containing two water exchange sites. Bioconjugate Chem. 2008, 19, 1505–1509. [Google Scholar] [CrossRef]
- Song, Y.; Kohlmeir, E.K.; Meade, T.J. Synthesis of multimeric mr contrast agents for cellular imaging. J. Am. Chem. Soc. 2008, 130, 6662–6663. [Google Scholar] [CrossRef]
- Livramento, J.B.; Tóth, É.; Sour, A.; Borel, A.; Merbach, A.E.; Ruloff, R. High relaxivity confined to a small molecular space: A metallostar-based, Potential mri contrast agent. Angew. Chem. Intl. Ed. 2005, 14, 1480–1484. [Google Scholar]
- Bayer, M.J.; Hawthorne, M.F. An improved method for the synthesis of [closo-B12(OH)12]−2. Inorg. Chem. 2004, 43, 2018–2020. [Google Scholar] [CrossRef]
- Farha, O.K.; Julius, R.L.; Lee, M.W.; Huertas, R.E.; Knobler, C.B.; Hawthorne, M.F. Synthesis of stable Dodecaalkoxy derivatives of hypercloso-B12H12. J. Am. Chem. Soc. 2005, 127, 18243–18251. [Google Scholar]
- Jalisatgi, S.S.; Kulkarni, V.S.; Tang, B.; Houston, Z.H.; Lee, M.W.; Hawthorne, M.F. A convenient route to diversely substituted icosahedral closomer nanoscaffolds. J. Am. Chem. Soc. 2011, 133, 12382–12385. [Google Scholar] [CrossRef]
- Goswami, L.N.; Chakravarty, S.; Lee, M.W.; Jalisatgi, S.S.; Hawthorne, M.F. Extensions of the icosahedral closomer structure by using azide–alkyne click reactions. Angew. Chem. Int. Ed. 2011, 50, 4689–4691. [Google Scholar] [CrossRef]
- Pushechnikov, A.; Jalisatgi, S.S.; Hawthorne, M.F. Dendritic closomers: Novel spherical hybrid dendrimers. Chem. Comm. 2013, 49, 3579–3581. [Google Scholar] [CrossRef]
- Goswami, L.N.; Ma, L.; Cai, Q.; Sarma, S.J.; Jalisatgi, S.S.; Hawthorne, M.F. cRGD Peptide-conjugated Icosahedral closo-B122− core carrying multiple Gd3+-DOTA chelates for αvβ3 Integrin-Targeted Tumor Imaging (MRI). Inorg. Chem. 2013, 52, 1701–1709. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “Ligation” of Azides and Terminal Alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Lutz, J.-F. 1,3-dipolar cycloadditions of azides and alkynes: A universal ligation tool in polymer and materials science. Angew. Chem. Int. Ed. 2007, 46, 1018–1025. [Google Scholar] [CrossRef]
- Goswami, L.N.; Ma, L.; Chakravarty, S.; Cai, Q.; Jalisatgi, S.S.; Hawthorne, M.F. Discrete Nanomolecular Polyhedral Borane Scaffold Supporting Multiple Gadolinium(III) Complexes as a High Performance MRI Contrast Agent. Inorg. Chem. 2013, 52, 1694–1700. [Google Scholar] [CrossRef]
- Piron, F.; Oprea, C.; Cismaş, C.; Terec, A.; Roncali, J.; Grosu, I. Synthesis of Podands with Cyanurate or Isocyanurate Cores and Terminal Triple Bonds. Synthesis 2010, 1639–1644. [Google Scholar]
- Barge, A.; Cravotto, G.; Gianolio, E.; Fedeli, F. How to determine free Gd and free ligand in solution of Gd chelates. A technical note. Contrast Med. Mol. Imaging 2006, 1, 184–188. [Google Scholar] [CrossRef]
- Doble, D.M.; Botta, M.; Wang, J.; Aime, S.; Barge, A.; Raymond, K.N. Optimization of the relaxivity of MRI contrast agents: Effect of poly(ethylene glycol) chains on the water-exchange rates of Gd(III) complexes. J. Am. Chem. Soc. 2001, 123, 10758–10759. [Google Scholar] [CrossRef]
- Djanashvili, K.; Peters, J.A. How to determine the number of inner-sphere water molecules in Lanthanide(III) complexes by 17O NMR spectroscopy. A technical note. Contrast Media Mol. Imaging 2007, 2, 67–71. [Google Scholar] [CrossRef]
- Caravan, P.; Farrar, C.T.; Frullano, L.; Uppal, R. Influence of molecular parameters and increasing magnetic field strength on relaxivity of gadolinium- and manganese-based T1 contrast agents. Contrast Media Mol. Imaging 2009, 4, 89–100. [Google Scholar] [CrossRef]
- Noebauer-Huhmann, I.M.; Kraff, O.; Juras, V.; Szomolanyi1, P.; Maderwald, S.; Mlynarik, V.; Thyesohn, J.M.; Ladd, S.C.; Ladd, M.E.; Trattnig, S. MR Contrast Media at 7 Tesla — Preliminary Study on Relaxivities. In Proceedings of 16th Annual Meeting of ISMRM, Toronto, Canada, 2008; p. 1457.
- Sample Availability: Samples of the compounds are not available from the authors.
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Goswami, L.N.; Ma, L.; Kueffer, P.J.; Jalisatgi, S.S.; Hawthorne, M.F. Synthesis and Relaxivity Studies of a DOTA-Based Nanomolecular Chelator Assembly Supported by an Icosahedral Closo-B122− -Core for MRI: A Click Chemistry Approach. Molecules 2013, 18, 9034-9048. https://doi.org/10.3390/molecules18089034
Goswami LN, Ma L, Kueffer PJ, Jalisatgi SS, Hawthorne MF. Synthesis and Relaxivity Studies of a DOTA-Based Nanomolecular Chelator Assembly Supported by an Icosahedral Closo-B122− -Core for MRI: A Click Chemistry Approach. Molecules. 2013; 18(8):9034-9048. https://doi.org/10.3390/molecules18089034
Chicago/Turabian StyleGoswami, Lalit N., Lixin Ma, Peter J. Kueffer, Satish S. Jalisatgi, and M. Frederick Hawthorne. 2013. "Synthesis and Relaxivity Studies of a DOTA-Based Nanomolecular Chelator Assembly Supported by an Icosahedral Closo-B122− -Core for MRI: A Click Chemistry Approach" Molecules 18, no. 8: 9034-9048. https://doi.org/10.3390/molecules18089034
APA StyleGoswami, L. N., Ma, L., Kueffer, P. J., Jalisatgi, S. S., & Hawthorne, M. F. (2013). Synthesis and Relaxivity Studies of a DOTA-Based Nanomolecular Chelator Assembly Supported by an Icosahedral Closo-B122− -Core for MRI: A Click Chemistry Approach. Molecules, 18(8), 9034-9048. https://doi.org/10.3390/molecules18089034