Probing Early Misfolding Events in Prion Protein Mutants by NMR Spectroscopy


Abstract
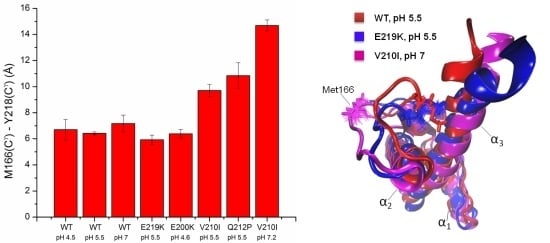
:
{kind=link}
{kind=link}
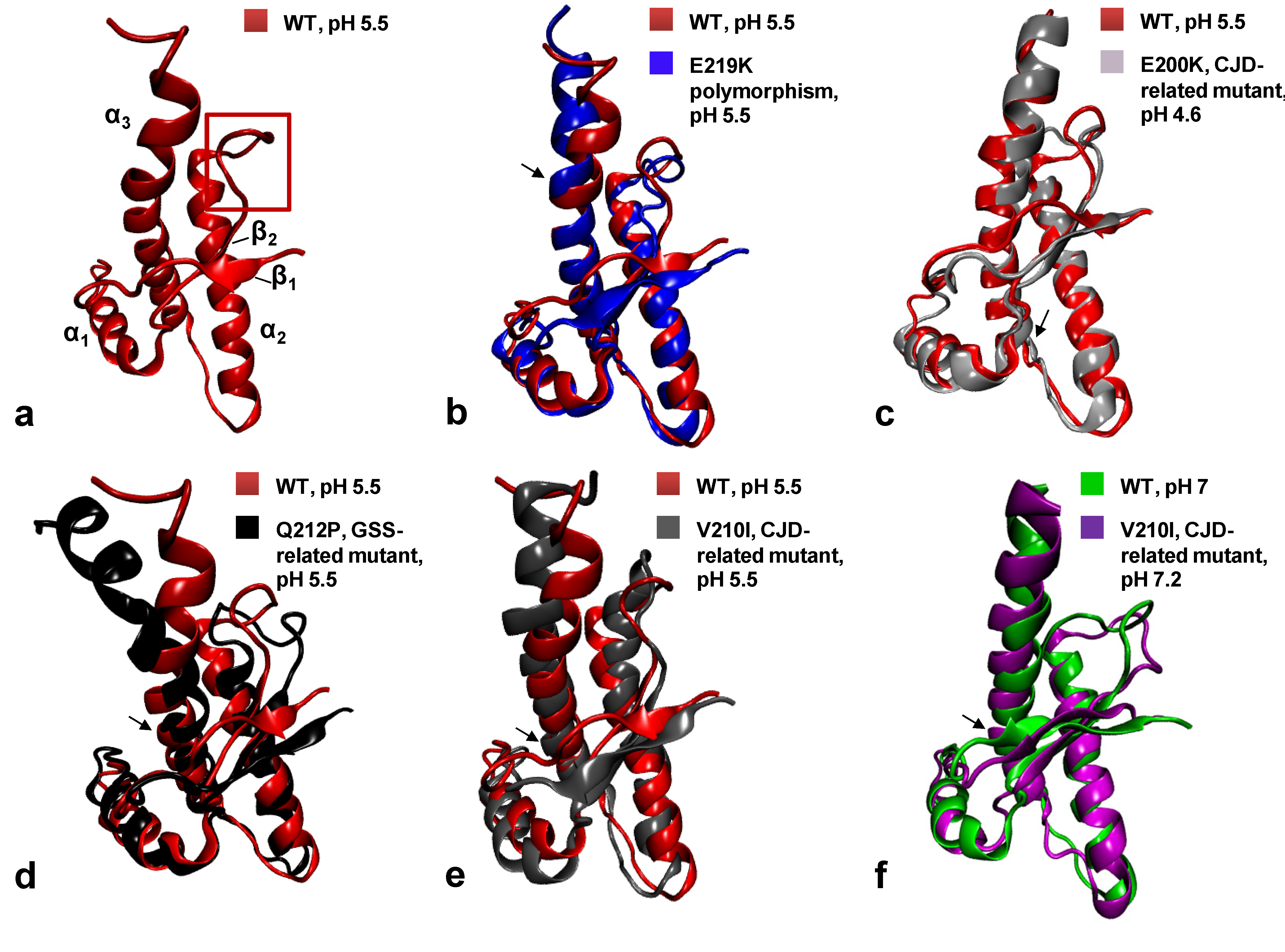{kind=link}
{kind=link}
1. Introduction
2. Structural Features of PrPC

3. Defining PrPC Functions
4. Prion Conversion and PrPSc Molecular Models
5. Genetic Human Prion Diseases: An Overview
5.1. Clinico-Pathological Features of Genetic Human Prion Diseases
5.2. Influential Polymorphisms in PRNP
6. Structural Biology of HuPrP Pathological Mutants

6.1. The Q212P Mutant NMR Structure
6.2. NMR Structures of V210I at Two Physiological pH Values

6.3. NMR Structures of WT HuPrP and of HuPrP Containing E219K Protective Polymorphism
6.4. Common Structural Traits in HuPrP Pathological Mutants
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Prusiner, S.B. Novel proteinaceous infectious particles cause scrapie. Science 1982, 216, 136–144. [Google Scholar]
- Head, M.W.; Ironside, J.W. Review: Creutzfeldt-jakob disease: Prion protein type, disease phenotype and agent strain. Neuropathol. Appl. Neurobiol. 2012, 38, 296–310. [Google Scholar] [CrossRef]
- Imran, M.; Mahmood, S. An overview of animal prion diseases. Virol. J. 2011, 8, 493. [Google Scholar] [CrossRef]
- Ladogana, A.; Puopolo, M.; Croes, E.A.; Budka, H.; Jarius, C.; Collins, S.; Klug, G.M.; Sutcliffe, T.; Giulivi, A.; Alperovitch, A.; et al. Mortality from creutzfeldt-jakob disease and related disorders in europe, australia, and canada. In Neurology; 2005; Volume 64, pp. 1586–1591. [Google Scholar]
- Mead, S. Prion disease genetics. Eur. J. Hum. Genet. 2006, 14, 273–281. [Google Scholar] [CrossRef]
- Aguzzi, A.; Falsig, J. Prion propagation, toxicity and degradation. Nat. Neurosci. 2012, 15, 936–939. [Google Scholar] [CrossRef]
- Legname, G.; Giachin, G.; Benetti, F. Structural studies of prion proteins and prions. In Non-Fibrillar Amyloidogenic Protein Assemblies-Common Cytotoxins Underlying Degenerative Diseases; Rahimi, F., Bitan, G., Eds.; Springer: Dordrecht, The Netherlands, 2012; pp. 289–317. [Google Scholar]
- Legname, G.; Tran, T.N.L.; Giachin, G. Synthetic prions. In Prions and Prion Diseases: New Developments; Verdier, J., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2012; pp. 61–83. [Google Scholar]
- Mallucci, G.R. Prion neurodegeneration: Starts and stops at the synapse. Prion 2009, 3, 195–201. [Google Scholar]
- Solomon, I.H.; Schepker, J.A.; Harris, D.A. Prion neurotoxicity: Insights from prion protein mutants. Curr. Issues Mol. Biol. 2010, 12, 51–61. [Google Scholar]
- Basler, K.; Oesch, B.; Scott, M.; Westaway, D.; Walchli, M.; Groth, D.F.; McKinley, M.P.; Prusiner, S.B.; Weissmann, C. Scrapie and cellular prp isoforms are encoded by the same chromosomal gene. Cell 1986, 46, 417–428. [Google Scholar] [CrossRef] [Green Version]
- Linden, R.; Martins, V.R.; Prado, M.A.; Cammarota, M.; Izquierdo, I.; Brentani, R.R. Physiology of the prion protein. Physiol. Rev. 2008, 88, 673–728. [Google Scholar] [CrossRef]
- Benvegnu, S.; Poggiolini, I.; Legname, G. Neurodevelopmental expression and localization of the cellular prion protein in the central nervous system of the mouse. J. Comp. Neurol. 2010, 518, 1879–1891. [Google Scholar] [CrossRef]
- Manson, J.; West, J.D.; Thomson, V.; McBride, P.; Kaufman, M.H.; Hope, J. The prion protein gene: A role in mouse embryogenesis? Development 1992, 115, 117–122. [Google Scholar]
- McKinley, M.P.; Hay, B.; Lingappa, V.R.; Lieberburg, I.; Prusiner, S.B. Developmental expression of prion protein gene in brain. Dev. Biol. 1987, 121, 105–110. [Google Scholar] [CrossRef]
- Poggiolini, I.; Legname, G. Mapping the prion protein distribution in marsupials: Insights from comparing opossum with mouse cns. PLoS One 2012, 7, e50370. [Google Scholar] [CrossRef]
- Sales, N.; Hassig, R.; Rodolfo, K.; Di Giamberardino, L.; Traiffort, E.; Ruat, M.; Fretier, P.; Moya, K.L. Developmental expression of the cellular prion protein in elongating axons. Eur. J. Neurosci. 2002, 15, 1163–1177. [Google Scholar] [CrossRef]
- Stahl, N.; Borchelt, D.R.; Prusiner, S.B. Differential release of cellular and scrapie prion proteins from cellular membranes by phosphatidylinositol-specific phospholipase C. Biochemistry 1990, 29, 5405–5412. [Google Scholar] [CrossRef]
- Taylor, D.R.; Hooper, N.M. The prion protein and lipid rafts. Mol. Membr. Biol. 2006, 23, 89–99. [Google Scholar] [CrossRef]
- Hornemann, S.; Schorn, C.; Wuthrich, K. Nmr structure of the bovine prion protein isolated from healthy calf brains. EMBO Rep. 2004, 5, 1159–1164. [Google Scholar] [CrossRef]
- Wopfner, F.; Weidenhofer, G.; Schneider, R.; von Brunn, A.; Gilch, S.; Schwarz, T.F.; Werner, T.; Schatzl, H.M. Analysis of 27 mammalian and 9 avian prps reveals high conservation of flexible regions of the prion protein. J. Mol. Biol. 1999, 289, 1163–1178. [Google Scholar] [CrossRef]
- Millhauser, G.L. Copper and the prion protein: Methods, structures, function, and disease. Annu Rev. Phys. Chem. 2007, 58, 299–320. [Google Scholar] [CrossRef]
- Walter, E.D.; Stevens, D.J.; Visconte, M.P.; Millhauser, G.L. The prion protein is a combined zinc and copper binding protein: Zn2+ alters the distribution of Cu2+ coordination modes. J. Am. Chem. Soc. 2007, 129, 15440–15441. [Google Scholar] [CrossRef]
- D’Angelo, P.; Della Longa, S.; Arcovito, A.; Mancini, G.; Zitolo, A.; Chillemi, G.; Giachin, G.; Legname, G.; Benetti, F. Effects of the pathological q212p mutation on human prion protein non-octarepeat copper-binding site. Biochemistry 2012, 51, 6068–6079. [Google Scholar] [CrossRef]
- Hasnain, S.S.; Murphy, L.M.; Strange, R.W.; Grossmann, J.G.; Clarke, A.R.; Jackson, G.S.; Collinge, J. Xafs study of the high-affinity copper-binding site of human prp(91–231) and its low-resolution structure in solution. J. Mol. Biol. 2001, 311, 467–473. [Google Scholar] [CrossRef]
- Chakrabarti, O.; Ashok, A.; Hegde, R.S. Prion protein biosynthesis and its emerging role in neurodegeneration. Trends. Biochem. Sci. 2009, 34, 287–295. [Google Scholar] [CrossRef]
- Li, A.; Christensen, H.M.; Stewart, L.R.; Roth, K.A.; Chiesa, R.; Harris, D.A. Neonatal lethality in transgenic mice expressing prion protein with a deletion of residues 105–125. EMBO J. 2007, 26, 548–558. [Google Scholar] [CrossRef]
- Biljan, I.; Giachin, G.; Ilc, G.; Zhukov, I.; Plavec, J.; Legname, G. Structural basis for the protective effect of the human prion protein carrying the dominant-negative e219k polymorphism. Biochem. J. 2012, 446, 243–251. [Google Scholar] [CrossRef]
- Calzolai, L.; Zahn, R. Influence of ph on nmr structure and stability of the human prion protein globular domain. J. Biol. Chem. 2003, 278, 35592–35596. [Google Scholar] [CrossRef]
- Zahn, R.; Liu, A.; Luhrs, T.; Riek, R.; von Schroetter, C.; Lopez Garcia, F.; Billeter, M.; Calzolai, L.; Wider, G.; Wuthrich, K. Nmr solution structure of the human prion protein. Proc. Natl. Acad. Sci. USA 2000, 97, 145–150. [Google Scholar] [CrossRef]
- Adrover, M.; Pauwels, K.; Prigent, S.; De Chiara, C.; Xu, Z.; Chapuis, C.; Pastore, A.; Rezaei, H. Prion fibrillization is mediated by a native structural element that comprises helices h2 and h3. J. Biol. Chem. 2010, 285, 21004–21012. [Google Scholar]
- Rossetti, G.; Giachin, G.; Legname, G.; Carloni, P. Structural facets of disease-linked human prion protein mutants: A molecular dynamic study. Proteins-Struct. Funct. Bioinform 2010, 78, 3270–3280. [Google Scholar] [CrossRef]
- van der Kamp, M.W.; Daggett, V. Pathogenic mutations in the hydrophobic core of the human prion protein can promote structural instability and misfolding. J. Mol. Biol. 2010, 404, 732–748. [Google Scholar] [CrossRef]
- Shen, L.; Ji, H.F. Mutation directional selection sheds light on prion pathogenesis. Biochem. Biophys. Res. Commun. 2011, 410, 159–163. [Google Scholar] [CrossRef]
- Donne, D.G.; Viles, J.H.; Groth, D.; Mehlhorn, I.; James, T.L.; Cohen, F.E.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Structure of the recombinant full-length hamster prion protein prp(29–231): The n terminus is highly flexible. Proc. Natl. Acad. Sci. USA 1997, 94, 13452–13457. [Google Scholar] [CrossRef]
- Gossert, A.D.; Bonjour, S.; Lysek, D.A.; Fiorito, F.; Wuthrich, K. Prion protein nmr structures of elk and of mouse/elk hybrids. Proc. Natl. Acad. Sci. USA 2005, 102, 646–650. [Google Scholar] [CrossRef]
- Christen, B.; Perez, D.R.; Hornemann, S.; Wuthrich, K. Nmr structure of the bank vole prion protein at 20 degrees c contains a structured loop of residues 165–171. J. Mol. Biol. 2008, 383, 306–312. [Google Scholar] [CrossRef]
- Christen, B.; Hornemann, S.; Damberger, F.F.; Wuthrich, K. Prion protein nmr structure from tammar wallaby (macropus eugenii) shows that the beta2-alpha2 loop is modulated by long-range sequence effects. J. Mol. Biol. 2009, 389, 833–845. [Google Scholar] [CrossRef]
- Wen, Y.; Li, J.; Yao, W.; Xiong, M.; Hong, J.; Peng, Y.; Xiao, G.; Lin, D. Unique structural characteristics of the rabbit prion protein. J. Biol. Chem. 2010, 285, 31682–31693. [Google Scholar]
- Perez, D.R.; Damberger, F.F.; Wuthrich, K. Horse prion protein nmr structure and comparisons with related variants of the mouse prion protein. J. Mol. Biol. 2010, 400, 121–128. [Google Scholar] [CrossRef]
- Scouras, A.D.; Daggett, V. Disruption of the x-loop turn of the prion protein linked to scrapie resistance. Protein Eng. Des. Sel. 2012, 25, 243–249. [Google Scholar] [CrossRef]
- Colby, D.W.; Prusiner, S.B. Prions. Cold Spring Harb Perspect. Biol. 2011, 3, a006833. [Google Scholar] [CrossRef]
- Prusiner, S.B. Prions. Proc. Natl. Acad. Sci. USA 1998, 95, 13363–13383. [Google Scholar] [CrossRef]
- Legname, G.; Baskakov, I.V.; Nguyen, H.O.; Riesner, D.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Synthetic mammalian prions. Science 2004, 305, 673–676. [Google Scholar] [CrossRef]
- Turnbaugh, J.A.; Unterberger, U.; Saa, P.; Massignan, T.; Fluharty, B.R.; Bowman, F.P.; Miller, M.B.; Supattapone, S.; Biasini, E.; Harris, D.A. The n-terminal, polybasic region of prp(c) dictates the efficiency of prion propagation by binding to prp(sc). J. Neurosci. 2012, 32, 8817–8830. [Google Scholar] [CrossRef]
- Thakur, A.K.; Srivastava, A.K.; Srinivas, V.; Chary, K.V.; Rao, C.M. Copper alters aggregation behavior of prion protein and induces novel interactions between its n- and c-terminal regions. J. Biol. Chem. 2011, 286, 38533–38545. [Google Scholar]
- Spevacek, A.R.; Evans, E.G.; Miller, J.L.; Meyer, H.C.; Pelton, J.G.; Millhauser, G.L. Zinc drives a tertiary fold in the prion protein with familial disease mutation sites at the interface. Structure 2013, 21, 236–246. [Google Scholar] [CrossRef]
- Bueler, H.; Fischer, M.; Lang, Y.; Bluethmann, H.; Lipp, H.P.; DeArmond, S.J.; Prusiner, S.B.; Aguet, M.; Weissmann, C. Normal development and behaviour of mice lacking the neuronal cell-surface prp protein. Nature 1992, 356, 577–582. [Google Scholar] [CrossRef]
- Criado, J.R.; Sanchez-Alavez, M.; Conti, B.; Giacchino, J.L.; Wills, D.N.; Henriksen, S.J.; Race, R.; Manson, J.C.; Chesebro, B.; Oldstone, M.B. Mice devoid of prion protein have cognitive deficits that are rescued by reconstitution of prp in neurons. Neurobiol. Dis. 2005, 19, 255–265. [Google Scholar] [CrossRef]
- Maglio, L.E.; Martins, V.R.; Izquierdo, I.; Ramirez, O.A. Role of cellular prion protein on ltp expression in aged mice. Brain Res. 2006, 1097, 11–18. [Google Scholar] [CrossRef]
- Maglio, L.E.; Perez, M.F.; Martins, V.R.; Brentani, R.R.; Ramirez, O.A. Hippocampal synaptic plasticity in mice devoid of cellular prion protein. Brain Res. Mol. Brain Res. 2004, 131, 58–64. [Google Scholar] [CrossRef]
- Caiati, M.D.; Safiulina, V.F.; Fattorini, G.; Sivakumaran, S.; Legname, G.; Cherubini, E. Prpc controls via protein kinase a the direction of synaptic plasticity in the immature hippocampus. J. Neurosci. 2013, 33, 2973–2983. [Google Scholar] [CrossRef]
- Colling, S.B.; Collinge, J.; Jefferys, J.G. Hippocampal slices from prion protein null mice: Disrupted Ca2+-activated K+ currents. Neurosci. Lett. 1996, 209, 49–52. [Google Scholar] [CrossRef]
- Fuhrmann, M.; Bittner, T.; Mitteregger, G.; Haider, N.; Moosmang, S.; Kretzschmar, H.; Herms, J. Loss of the cellular prion protein affects the Ca2+ homeostasis in hippocampal ca1 neurons. J. Neurochem. 2006, 98, 1876–1885. [Google Scholar] [CrossRef]
- Mallucci, G.R.; Ratte, S.; Asante, E.A.; Linehan, J.; Gowland, I.; Jefferys, J.G.; Collinge, J. Post-natal knockout of prion protein alters hippocampal ca1 properties, but does not result in neurodegeneration. EMBO J. 2002, 21, 202–210. [Google Scholar] [CrossRef]
- McLennan, N.F.; Brennan, P.M.; McNeill, A.; Davies, I.; Fotheringham, A.; Rennison, K.A.; Ritchie, D.; Brannan, F.; Head, M.W.; Ironside, J.W.; et al. Prion protein accumulation and neuroprotection in hypoxic brain damage. Am. J. Pathol. 2004, 165, 227–235. [Google Scholar] [CrossRef]
- Spudich, A.; Frigg, R.; Kilic, E.; Kilic, U.; Oesch, B.; Raeber, A.; Bassetti, C.L.; Hermann, D.M. Aggravation of ischemic brain injury by prion protein deficiency: Role of erk-1/-2 and stat-1. Neurobiol. Dis. 2005, 20, 442–449. [Google Scholar] [CrossRef]
- Walz, R.; Amaral, O.B.; Rockenbach, I.C.; Roesler, R.; Izquierdo, I.; Cavalheiro, E.A.; Martins, V.R.; Brentani, R.R. Increased sensitivity to seizures in mice lacking cellular prion protein. Epilepsia 1999, 40, 1679–1682. [Google Scholar] [CrossRef]
- Rangel, A.; Burgaya, F.; Gavin, R.; Soriano, E.; Aguzzi, A.; Del Rio, J.A. Enhanced susceptibility of prnp-deficient mice to kainate-induced seizures, neuronal apoptosis, and death: Role of ampa/kainate receptors. J. Neurosci. Res. 2007, 85, 2741–2755. [Google Scholar] [CrossRef] [Green Version]
- Rangel, A.; Madronal, N.; Gruart, A.; Gavin, R.; Llorens, F.; Sumoy, L.; Torres, J.M.; Delgado-Garcia, J.M.; Del Rio, J.A. Regulation of gaba(a) and glutamate receptor expression, synaptic facilitation and long-term potentiation in the hippocampus of prion mutant mice. PLoS One 2009, 4, e7592. [Google Scholar] [CrossRef]
- Brown, D.R.; Nicholas, R.S.; Canevari, L. Lack of prion protein expression results in a neuronal phenotype sensitive to stress. J. Neurosci. Res. 2002, 67, 211–224. [Google Scholar] [CrossRef]
- Khosravani, H.; Zhang, Y.; Tsutsui, S.; Hameed, S.; Altier, C.; Hamid, J.; Chen, L.; Villemaire, M.; Ali, Z.; Jirik, F.R.; et al. Prion protein attenuates excitotoxicity by inhibiting nmda receptors. J. Cell. Biol. 2008, 181, 551–565. [Google Scholar] [CrossRef]
- Viles, J.H.; Klewpatinond, M.; Nadal, R.C. Copper and the structural biology of the prion protein. Biochem. Soc. Trans. 2008, 36, 1288–1292. [Google Scholar] [CrossRef]
- Kanaani, J.; Prusiner, S.B.; Diacovo, J.; Baekkeskov, S.; Legname, G. Recombinant prion protein induces rapid polarization and development of synapses in embryonic rat hippocampal neurons in vitro. J. Neurochem. 2005, 95, 1373–1386. [Google Scholar] [CrossRef]
- Aguzzi, A.; Baumann, F.; Bremer, J. The prion’s elusive reason for being. Annu Rev. Neurosci 2008, 31, 439–477. [Google Scholar] [CrossRef]
- Didonna, A. Prion protein and its role in signal transduction. Cell. Mol. Biol. Lett. 2013, 18, 209–230. [Google Scholar] [CrossRef]
- Mouillet-Richard, S.; Ermonval, M.; Chebassier, C.; Laplanche, J.L.; Lehmann, S.; Launay, J.M.; Kellermann, O. Signal transduction through prion protein. Science 2000, 289, 1925–1928. [Google Scholar] [CrossRef]
- Santuccione, A.; Sytnyk, V.; Leshchyns'ka, I.; Schachner, M. Prion protein recruits its neuronal receptor ncam to lipid rafts to activate p59(fyn) and to enhance neurite outgrowth. J. Cell Biol. 2005, 169, 341–354. [Google Scholar] [CrossRef] [Green Version]
- Schmitt-Ulms, G.; Legname, G.; Baldwin, M.A.; Ball, H.L.; Bradon, N.; Bosque, P.J.; Crossin, K.L.; Edelman, G.M.; DeArmond, S.J.; Cohen, F.E.; et al. Binding of neural cell adhesion molecules (n-cams) to the cellular prion protein. J. Mol. Biol. 2001, 314, 1209–1225. [Google Scholar] [CrossRef]
- Roffe, M.; Beraldo, F.H.; Bester, R.; Nunziante, M.; Bach, C.; Mancini, G.; Gilch, S.; Vorberg, I.; Castilho, B.A.; Martins, V.R.; et al. Prion protein interaction with stress-inducible protein 1 enhances neuronal protein synthesis via mtor. Proc. Natl. Acad. Sci. USA 2010, 107, 13147–13152. [Google Scholar] [CrossRef]
- Devanathan, V.; Jakovcevski, I.; Santuccione, A.; Li, S.; Lee, H.J.; Peles, E.; Leshchyns'ka, I.; Sytnyk, V.; Schachner, M. Cellular form of prion protein inhibits reelin-mediated shedding of caspr from the neuronal cell surface to potentiate caspr-mediated inhibition of neurite outgrowth. J. Neurosci. 2010, 30, 9292–9305. [Google Scholar] [CrossRef]
- Senatore, A.; Colleoni, S.; Verderio, C.; Restelli, E.; Morini, R.; Condliffe, S.B.; Bertani, I.; Mantovani, S.; Canovi, M.; Micotti, E.; et al. Mutant prp suppresses glutamatergic neurotransmission in cerebellar granule neurons by impairing membrane delivery of vgcc alpha(2)delta-1 subunit. Neuron 2012, 74, 300–313. [Google Scholar] [CrossRef]
- Prusiner, S.B. Molecular biology of prion diseases. Science 1991, 252, 1515–1522. [Google Scholar] [CrossRef]
- Jarrett, J.T.; Lansbury, P.T., Jr. Seeding “one-dimensional crystallization” of amyloid: A pathogenic mechanism in alzheimer's disease and scrapie? Cell 1993, 73, 1055–1058. [Google Scholar] [CrossRef]
- Caughey, B.; Raymond, G.J.; Bessen, R.A. Strain-dependent differences in beta-sheet conformations of abnormal prion protein. J. Biol. Chem. 1998, 273, 32230–32235. [Google Scholar] [CrossRef]
- Caughey, B.W.; Dong, A.; Bhat, K.S.; Ernst, D.; Hayes, S.F.; Caughey, W.S. Secondary structure analysis of the scrapie-associated protein prp 27–30 in water by infrared spectroscopy. Biochemistry 1991, 30, 7672–7680. [Google Scholar] [CrossRef]
- Gasset, M.; Baldwin, M.A.; Fletterick, R.J.; Prusiner, S.B. Perturbation of the secondary structure of the scrapie prion protein under conditions that alter infectivity. Proc. Natl. Acad. Sci. USA 1993, 90, 1–5. [Google Scholar] [CrossRef]
- Goormaghtigh, E.; Cabiaux, V.; Ruysschaert, J.M. Secondary structure and dosage of soluble and membrane proteins by attenuated total reflection fourier-transform infrared spectroscopy on hydrated films. Eur. J. Biochem. 1990, 193, 409–420. [Google Scholar] [CrossRef]
- Pan, K.M.; Baldwin, M.; Nguyen, J.; Gasset, M.; Serban, A.; Groth, D.; Mehlhorn, I.; Huang, Z.; Fletterick, R.J.; Cohen, F.E.; et al. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. USA 1993, 90, 10962–10966. [Google Scholar] [CrossRef]
- Spassov, S.; Beekes, M.; Naumann, D. Structural differences between tses strains investigated by ft-ir spectroscopy. Biochim. Biophys. Acta 2006, 1760, 1138–1149. [Google Scholar] [CrossRef]
- Surewicz, W.K.; Apostol, M.I. Prion protein and its conformational conversion: A structural perspective. Top. Curr. Chem. 2011, 305, 135–167. [Google Scholar] [CrossRef]
- Wille, H.; Bian, W.; McDonald, M.; Kendall, A.; Colby, D.W.; Bloch, L.; Ollesch, J.; Borovinskiy, A.L.; Cohen, F.E.; Prusiner, S.B.; et al. Natural and synthetic prion structure from X-ray fiber diffraction. Proc. Natl. Acad. Sci. USA 2009, 106, 16990–16995. [Google Scholar] [CrossRef]
- Smirnovas, V.; Baron, G.S.; Offerdahl, D.K.; Raymond, G.J.; Caughey, B.; Surewicz, W.K. Structural organization of brain-derived mammalian prions examined by hydrogen-deuterium exchange. Nat. Struct. Mol. Biol. 2011, 18, 504–506. [Google Scholar] [CrossRef]
- Croes, E.A.; Theuns, J.; Houwing-Duistermaat, J.J.; Dermaut, B.; Sleegers, K.; Roks, G.; Van den Broeck, M.; van Harten, B.; van Swieten, J.C.; Cruts, M.; et al. Octapeptide repeat insertions in the prion protein gene and early onset dementia. J. Neurol. Neurosurg. Psychiatr. 2004, 75, 1166–1170. [Google Scholar] [CrossRef]
- Kovacs, G.G.; Puopolo, M.; Ladogana, A.; Pocchiari, M.; Budka, H.; Van Duijn, C.; Collins, S.J.; Boyd, A.; Giulivi, A.; Coulthart, M.; et al. Genetic prion disease: The eurocjd experience. Hum. Genet. 2005, 118, 166–174. [Google Scholar] [CrossRef]
- Legname, G.; Zanusso, G. Molecular pathogenesis of prion diseases. In Miscellanea on Encephalopathies; Tanasescu, R., Ed.; InTech: New York, NY, USA, 2012; pp. 95–112. [Google Scholar]
- Gambetti, P.; Kong, Q.; Zou, W.; Parchi, P.; Chen, S.G. Sporadic and familial cjd: Classification and characterisation. Br. Med. Bull. 2003, 66, 213–239. [Google Scholar] [CrossRef]
- Schelzke, G.; Eigenbrod, S.; Romero, C.; Varges, D.; Breithaupt, M.; Taratuto, A.L.; Kretzschmar, H.A.; Zerr, I. Genetic prion disease with codon 196 prnp mutation: Clinical and pathological findings. Neurobiol. Aging. 2011, 32, 756.e1–756.e9. [Google Scholar] [CrossRef]
- Gambetti, P.; Parchi, P.; Petersen, R.B.; Chen, S.G.; Lugaresi, E. Fatal familial insomnia and familial creutzfeldt-jakob disease: Clinical, pathological and molecular features. Brain Pathol. 1995, 5, 43–51. [Google Scholar] [CrossRef]
- Goldfarb, L.G.; Petersen, R.B.; Tabaton, M.; Brown, P.; LeBlanc, A.C.; Montagna, P.; Cortelli, P.; Julien, J.; Vital, C.; Pendelbury, W.W.; et al. Fatal familial insomnia and familial creutzfeldt-jakob disease: Disease phenotype determined by a DNA polymorphism. Science 1992, 258, 806–808. [Google Scholar]
- McLean, C.A.; Storey, E.; Gardner, R.J.; Tannenberg, A.E.; Cervenakova, L.; Brown, P. The d178n (cis-129m) “fatal familial insomnia” mutation associated with diverse clinicopathologic phenotypes in an australian kindred. Neurology 1997, 49, 552–558. [Google Scholar] [CrossRef]
- Pocchiari, M.; Ladogana, A.; Petraroli, R.; Cardone, F.; D’Alessandro, M. Recent italian ffi cases. Brain Pathol. 1998, 8, 564–566. [Google Scholar]
- Revesz, T.; Holton, J.L.; Lashley, T.; Plant, G.; Frangione, B.; Rostagno, A.; Ghiso, J. Genetics and molecular pathogenesis of sporadic and hereditary cerebral amyloid angiopathies. Acta Neuropathol. 2009, 118, 115–130. [Google Scholar] [CrossRef]
- Jansen, C.; Parchi, P.; Capellari, S.; Vermeij, A.J.; Corrado, P.; Baas, F.; Strammiello, R.; van Gool, W.A.; van Swieten, J.C.; Rozemuller, A.J. Prion protein amyloidosis with divergent phenotype associated with two novel nonsense mutations in prnp. Acta neuropathol. 2010, 119, 189–197. [Google Scholar] [CrossRef]
- Paquet, C.; Privat, N.; Kaci, R.; Polivka, M.; Dupont, O.; Haik, S.; Laplanche, J.L.; Hauw, J.J.; Gray, F. Cerebral amyloid angiopathy with co-localization of prion protein and beta-amyloid in an 85-year-old patient with sporadic creutzfeldt-jakob disease. Acta neuropathol. 2008, 116, 567–573. [Google Scholar] [CrossRef]
- Mahal, S.P.; Jablonski, J.; Suponitsky-Kroyter, I.; Oelschlegel, A.M.; Herva, M.E.; Oldstone, M.; Weissmann, C. Propagation of rml prions in mice expressing prp devoid of gpi anchor leads to formation of a novel, stable prion strain. PLoS Pathog. 2012, 8, e1002746. [Google Scholar] [CrossRef]
- Rangel, A.; Race, B.; Striebel, J.; Chesebro, B. Non-amyloid and amyloid prion protein deposits in prion-infected mice differ in blockage of interstitial brain fluid. Neuropathol. Appl. Neurobiol. 2013, 39, 217–230. [Google Scholar] [CrossRef]
- Raymond, G.J.; Race, B.; Hollister, J.R.; Offerdahl, D.K.; Moore, R.A.; Kodali, R.; Raymond, L.D.; Hughson, A.G.; Rosenke, R.; Long, D.; et al. Isolation of novel synthetic prion strains by amplification in transgenic mice coexpressing wild-type and anchorless prion proteins. J. Virol. 2012, 86, 11763–11778. [Google Scholar] [CrossRef]
- Stohr, J.; Watts, J.C.; Legname, G.; Oehler, A.; Lemus, A.; Nguyen, H.O.; Sussman, J.; Wille, H.; DeArmond, S.J.; Prusiner, S.B.; et al. Spontaneous generation of anchorless prions in transgenic mice. Proc. Natl. Acad. Sci. USA 2011, 108, 21223–21228. [Google Scholar] [CrossRef]
- Beck, J.A.; Poulter, M.; Campbell, T.A.; Adamson, G.; Uphill, J.B.; Guerreiro, R.; Jackson, G.S.; Stevens, J.C.; Manji, H.; Collinge, J.; et al. Prnp allelic series from 19 years of prion protein gene sequencing at the mrc prion unit. Human mutat. 2010, 31, E1551–E1563. [Google Scholar] [CrossRef]
- Schelzke, G.; Stoeck, K.; Eigenbrod, S.; Grasbon-Frodl, E.; Raddatz, L.M.; Ponto, C.; Kretzschmar, H.A.; Zerr, I. Report about four novel mutations in the prion protein gene. Dement. Geriatr. Cogn. Disord. 2013, 35, 229–237. [Google Scholar] [CrossRef]
- Alperovitch, A.; Zerr, I.; Pocchiari, M.; Mitrova, E.; de Pedro Cuesta, J.; Hegyi, I.; Collins, S.; Kretzschmar, H.; van Duijn, C.; Will, R.G. Codon 129 prion protein genotype and sporadic creutzfeldt-jakob disease. Lancet 1999, 353, 1673–1674. [Google Scholar] [CrossRef]
- Brandel, J.P.; Preece, M.; Brown, P.; Croes, E.; Laplanche, J.L.; Agid, Y.; Will, R.; Alperovitch, A. Distribution of codon 129 genotype in human growth hormone-treated cjd patients in france and the uk. Lancet 2003, 362, 128–130. [Google Scholar] [CrossRef]
- Cervenakova, L.; Goldfarb, L.G.; Garruto, R.; Lee, H.S.; Gajdusek, D.C.; Brown, P. Phenotype-genotype studies in kuru: Implications for new variant creutzfeldt-jakob disease. Proc. Natl. Acad. Sci. USA 1998, 95, 13239–13241. [Google Scholar] [CrossRef]
- Bishop, M.T.; Pennington, C.; Heath, C.A.; Will, R.G.; Knight, R.S. Prnp variation in uk sporadic and variant creutzfeldt jakob disease highlights genetic risk factors and a novel non-synonymous polymorphism. BMC Med. Genet. 2009, 10, 146. [Google Scholar] [CrossRef]
- Kitamoto, T.; Tateishi, J. Human prion diseases with variant prion protein. Philos. Trans. R. Soc. Lond., B, Biol. Sci. 1994, 343, 391–398. [Google Scholar] [CrossRef]
- Shibuya, S.; Higuchi, J.; Shin, R.W.; Tateishi, J.; Kitamoto, T. Codon 219 lys allele of prnp is not found in sporadic creutzfeldt-jakob disease. Ann. Neurol. 1998, 43, 826–828. [Google Scholar] [CrossRef]
- Mead, S.; Stumpf, M.P.; Whitfield, J.; Beck, J.A.; Poulter, M.; Campbell, T.; Uphill, J.B.; Goldstein, D.; Alpers, M.; Fisher, E.M.; et al. Balancing selection at the prion protein gene consistent with prehistoric kurulike epidemics. Science 2003, 300, 640–643. [Google Scholar] [CrossRef]
- Soldevila, M.; Calafell, F.; Andres, A.M.; Yague, J.; Helgason, A.; Stefansson, K.; Bertranpetit, J. Prion susceptibility and protective alleles exhibit marked geographic differences. Human Mutat. 2003, 22, 104–105. [Google Scholar]
- Nozaki, I.; Hamaguchi, T.; Sanjo, N.; Noguchi-Shinohara, M.; Sakai, K.; Nakamura, Y.; Sato, T.; Kitamoto, T.; Mizusawa, H.; Moriwaka, F.; et al. Prospective 10-year surveillance of human prion diseases in Japan. Brain 2010, 133, 3043–3057. [Google Scholar] [CrossRef]
- Nishida, Y.; Sodeyama, N.; Toru, Y.; Toru, S.; Kitamoto, T.; Mizusawa, H. Creutzfeldt-jakob disease with a novel insertion and codon 219 lys/lys polymorphism in prnp. Neurology 2004, 63, 1978–1979. [Google Scholar] [CrossRef]
- Seno, H.; Tashiro, H.; Ishino, H.; Inagaki, T.; Nagasaki, M.; Morikawa, S. New haplotype of familial creutzfeldt-jakob disease with a codon 200 mutation and a codon 219 polymorphism of the prion protein gene in a japanese family. Acta neuropathol. 2000, 99, 125–130. [Google Scholar] [CrossRef]
- Tanaka, Y.; Minematsu, K.; Moriyasu, H.; Yamaguchi, T.; Yutani, C.; Kitamoto, T.; Furukawa, H. A japanese family with a variant of gerstmann-straussler-scheinker disease. J. Neurol. Neurosurg. Psychiatr. 1997, 62, 454–457. [Google Scholar] [CrossRef]
- Hoshi, K.; Yoshino, H.; Urata, J.; Nakamura, Y.; Yanagawa, H.; Sato, T. Creutzfeldt-jakob disease associated with cadaveric dura mater grafts in Japan. Neurology 2000, 55, 718–721. [Google Scholar] [CrossRef]
- Ikawa, M.; Yoneda, M.; Matsunaga, A.; Nakagawa, H.; Kazama-Suzuki, A.; Miyashita, N.; Naiki, H.; Kitamoto, T.; Kuriyama, M. Unique clinicopathological features and prp profiles in the first autopsied case of dura mater graft-associated creutzfeldt-jakob disease with codon 219 lysine allele observed in japanese population. J. Neurol. Sci. 2009, 285, 265–267. [Google Scholar] [CrossRef]
- Lukic, A.; Beck, J.; Joiner, S.; Fearnley, J.; Sturman, S.; Brandner, S.; Wadsworth, J.D.; Collinge, J.; Mead, S. Heterozygosity at polymorphic codon 219 in variant creutzfeldt-jakob disease. Arch. Neurol. 2010, 67, 1021–1023. [Google Scholar] [CrossRef]
- Yamada, M. The first japanese case of variant creutzfeldt-jakob disease showing periodic electroencephalogram. Lancet 2006, 367, 874. [Google Scholar] [CrossRef]
- Mead, S.; Whitfield, J.; Poulter, M.; Shah, P.; Uphill, J.; Campbell, T.; Al-Dujaily, H.; Hummerich, H.; Beck, J.; Mein, C.A.; et al. A novel protective prion protein variant that colocalizes with kuru exposure. N. Engl. J. Med. 2009, 361, 2056–2065. [Google Scholar] [CrossRef]
- Palmer, M.S.; Mahal, S.P.; Campbell, T.A.; Hill, A.F.; Sidle, K.C.; Laplanche, J.L.; Collinge, J. Deletions in the prion protein gene are not associated with cjd. Hum. Mol. Genet. 1993, 2, 541–544. [Google Scholar] [CrossRef]
- Apetri, A.C.; Surewicz, K.; Surewicz, W.K. The effect of disease-associated mutations on the folding pathway of human prion protein. J. Biol. Chem. 2004, 279, 18008–18014. [Google Scholar] [CrossRef]
- Liemann, S.; Glockshuber, R. Influence of amino acid substitutions related to inherited human prion diseases on the thermodynamic stability of the cellular prion protein. Biochemistry 1999, 38, 3258–3267. [Google Scholar] [CrossRef]
- Swietnicki, W.; Petersen, R.B.; Gambetti, P.; Surewicz, W.K. Familial mutations and the thermodynamic stability of the recombinant human prion protein. J. Biol. Chem. 1998, 273, 31048–31052. [Google Scholar]
- Kaneko, K.; Zulianello, L.; Scott, M.; Cooper, C.M.; Wallace, A.C.; James, T.L.; Cohen, F.E.; Prusiner, S.B. Evidence for protein x binding to a discontinuous epitope on the cellular prion protein during scrapie prion propagation. Proc. Natl. Acad. Sci. USA 1997, 94, 10069–10074. [Google Scholar] [CrossRef]
- Telling, G.C.; Scott, M.; Mastrianni, J.; Gabizon, R.; Torchia, M.; Cohen, F.E.; DeArmond, S.J.; Prusiner, S.B. Prion propagation in mice expressing human and chimeric prp transgenes implicates the interaction of cellular prp with another protein. Cell 1995, 83, 79–90. [Google Scholar] [CrossRef]
- Ashok, A.; Hegde, R.S. Selective processing and metabolism of disease-causing mutant prion proteins. PLoS Pathog. 2009, 5, e1000479. [Google Scholar] [CrossRef]
- Biljan, I.; Ilc, G.; Giachin, G.; Plavec, J.; Legname, G. Structural rearrangements at physiological ph: Nuclear magnetic resonance insights from the v210i human prion protein mutant. Biochemistry 2012, 51, 7465–7474. [Google Scholar] [CrossRef]
- Biljan, I.; Ilc, G.; Giachin, G.; Raspadori, A.; Zhukov, I.; Plavec, J.; Legname, G. Toward the molecular basis of inherited prion diseases: Nmr structure of the human prion protein with v210i mutation. J. Mol. Biol. 2011, 412, 660–673. [Google Scholar] [CrossRef]
- Ilc, G.; Giachin, G.; Jaremko, M.; Jaremko, L.; Benetti, F.; Plavec, J.; Zhukov, I.; Legname, G. Nmr structure of the human prion protein with the pathological q212p mutation reveals unique structural features. PLoS One 2010, 5, e11715. [Google Scholar]
- Zhang, Y.; Swietnicki, W.; Zagorski, M.G.; Surewicz, W.K.; Sonnichsen, F.D. Solution structure of the e200k variant of human prion protein. Implications for the mechanism of pathogenesis in familial prion diseases. J. Biol. Chem. 2000, 275, 33650–33654. [Google Scholar]
- Bae, S.H.; Legname, G.; Serban, A.; Prusiner, S.B.; Wright, P.E.; Dyson, H.J. Prion proteins with pathogenic and protective mutations show similar structure and dynamics. Biochemistry 2009, 48, 8120–8128. [Google Scholar] [CrossRef]
- Piccardo, P.; Kish, S.J.; Ang, L.C.; Young, K.; Bugiani, O.; Tagliavini, F.; Dlouhy, S.R.; Ghetti, B. Prion protein isoforms in the new variant of gerstmann-straussler-scheinker disease q212p. J. Neuropath. Exp. Neur. 1998, 57, 885–990. [Google Scholar] [CrossRef]
- Young, K.; Piccardo, P.; Kish, S.J.; Ang, L.C.; Dlouhy, S.; Ghetti, B. Gerstmann-straussler-scheinker disease (gss) with a mutation at prion protein (prp) residue 212. J. Neuropath. Exp. Neur. 1998, 57, 369–528. [Google Scholar] [CrossRef]
- Ladogana, A.; Puopolo, M.; Poleggi, A.; Almonti, S.; Mellina, V.; Equestre, M.; Pocchiari, M. High incidence of genetic human transmissible spongiform encephalopathies in italy. Neurology 2005, 64, 1592–1597. [Google Scholar] [CrossRef]
- Casey, J.R.; Grinstein, S.; Orlowski, J. Sensors and regulators of intracellular ph. Nat. Rev. Mol. Cell Biol. 2010, 11, 50–61. [Google Scholar] [CrossRef]
- Godsave, S.F.; Wille, H.; Kujala, P.; Latawiec, D.; DeArmond, S.J.; Serban, A.; Prusiner, S.B.; Peters, P.J. Cryo-immunogold electron microscopy for prions: Toward identification of a conversion site. J. Neurosci. 2008, 28, 12489–12499. [Google Scholar] [CrossRef]
- Marijanovic, Z.; Caputo, A.; Campana, V.; Zurzolo, C. Identification of an intracellular site of prion conversion. PLoS Pathog. 2009, 5, e1000426. [Google Scholar] [CrossRef]
- Pimpinelli, F.; Lehmann, S.; Maridonneau-Parini, I. The scrapie prion protein is present in flotillin-1-positive vesicles in central- but not peripheral-derived neuronal cell lines. Eur. J. Neurosci. 2005, 21, 2063–2072. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. Vmd: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Fraczkiewicz, R.; Braun, W. Exact and efficient analytical calculation of the accessible surface areas and their gradients for macromolecules. J. Comput. Chem. 1998, 19, 319–333. [Google Scholar] [CrossRef]
- Alonso, D.O.; DeArmond, S.J.; Cohen, F.E.; Daggett, V. Mapping the early steps in the ph-induced conformational conversion of the prion protein. Proc. Natl. Acad. Sci. USA 2001, 98, 2985–2989. [Google Scholar]
- DeMarco, M.L.; Daggett, V. Molecular mechanism for low ph triggered misfolding of the human prion protein. Biochemistry 2007, 46, 3045–3054. [Google Scholar] [CrossRef]
- van der Kamp, M.W.; Daggett, V. Influence of ph on the human prion protein: Insights into the early steps of misfolding. Biophys. J. 2010, 99, 2289–2298. [Google Scholar] [CrossRef]
- Crozet, C.; Lin, Y.L.; Mettling, C.; Mourton-Gilles, C.; Corbeau, P.; Lehmann, S.; Perrier, V. Inhibition of prpsc formation by lentiviral gene transfer of prp containing dominant negative mutations. J. Cell Sci. 2004, 117, 5591–5597. [Google Scholar] [CrossRef]
- Geoghegan, J.C.; Miller, M.B.; Kwak, A.H.; Harris, B.T.; Supattapone, S. Trans-dominant inhibition of prion propagation in vitro is not mediated by an accessory cofactor. PLoS Pathog. 2009, 5, e1000535. [Google Scholar] [CrossRef]
- Hizume, M.; Kobayashi, A.; Teruya, K.; Ohashi, H.; Ironside, J.W.; Mohri, S.; Kitamoto, T. Human prion protein (prp) 219k is converted to prpsc but shows heterozygous inhibition in variant creutzfeldt-jakob disease infection. J. Biol. Chem. 2009, 284, 3603–3609. [Google Scholar]
- Lee, C.I.; Yang, Q.; Perrier, V.; Baskakov, I.V. The dominant-negative effect of the q218k variant of the prion protein does not require protein x. Protein Sci. 2007, 16, 2166–2173. [Google Scholar] [CrossRef]
- Perrier, V.; Kaneko, K.; Safar, J.; Vergara, J.; Tremblay, P.; DeArmond, S.J.; Cohen, F.E.; Prusiner, S.B.; Wallace, A.C. Dominant-negative inhibition of prion replication in transgenic mice. Proc. Natl. Acad. Sci. USA 2002, 99, 13079–13084. [Google Scholar] [CrossRef]
- Chen, W.; van der Kamp, M.W.; Daggett, V. Diverse effects on the native beta-sheet of the human prion protein due to disease-associated mutations. Biochemistry 2010, 49, 9874–9881. [Google Scholar] [CrossRef]
- Meli, M.; Gasset, M.; Colombo, G. Dynamic diagnosis of familial prion diseases supports the beta2-alpha2 loop as a universal interference target. PLoS One 2011, 6, e19093. [Google Scholar] [CrossRef]
- Rossetti, G.; Cong, X.J.; Caliandro, R.; Legname, G.; Carloni, P. Common structural traits across pathogenic mutants of the human prion protein and their implications for familial prion diseases. J. Mol. Biol. 2011, 411, 700–712. [Google Scholar] [CrossRef]
- Lin, M.C.; Mirzabekov, T.; Kagan, B.L. Channel formation by a neurotoxic prion protein fragment. J. Biol. Chem. 1997, 272, 44–47. [Google Scholar]
- Pillot, T.; Lins, L.; Goethals, M.; Vanloo, B.; Baert, J.; Vandekerckhove, J.; Rosseneu, M.; Brasseur, R. The 118–135 peptide of the human prion protein forms amyloid fibrils and induces liposome fusion. J. Mol. Biol. 1997, 274, 381–393. [Google Scholar] [CrossRef]
- Gabizon, R.; Mckinley, M.P.; Prusiner, S.B. Purified prion proteins and scrapie infectivity copartition into liposomes. P. Natl. Acad. Sci. USA 1987, 84, 4017–4021. [Google Scholar] [CrossRef]
- Lehmann, S.; Harris, D.A. A mutant prion protein displays an aberrant membrane association when expressed in cultured-cells. J. Biol. Chem. 1995, 270, 24589–24597. [Google Scholar] [CrossRef]
- Chiovitti, K.; Corsaro, A.; Thellung, S.; Villa, V.; Paludi, D.; D’Arrigo, C.; Russo, C.; Perico, A.; Ianieri, A.; Di Cola, D.; et al. Intracellular accumulation of a mild-denatured monomer of the human prp fragment 90–231, as possible mechanism of its neurotoxic effects. J. Neurochem. 2007, 103, 2597–2609. [Google Scholar]
- Thellung, S.; Corsaro, A.; Villa, V.; Simi, A.; Vella, S.; Pagano, A.; Florio, T. Human prp90–231-induced cell death is associated with intracellular accumulation of insoluble and protease-resistant macroaggregates and lysosomal dysfunction. Cell Death. Dis. 2011, 2, e138. [Google Scholar] [CrossRef]
- Corsaro, A.; Thellung, S.; Bucciarelli, T.; Scotti, L.; Chiovitti, K.; Villa, V.; D'Arrigo, C.; Aceto, A.; Florio, T. High hydrophobic amino acid exposure is responsible of the neurotoxic effects induced by e200k or d202n disease-related mutations of the human prion protein. Int. J. Biochem. Cell Biol. 2011, 43, 372–382. [Google Scholar] [CrossRef]
- Gerum, C.; Schlepckow, K.; Schwalbe, H. The unfolded state of the murine prion protein and properties of single-point mutants related to human prion diseases. J. Mol. Biol 2010, 401, 7–12. [Google Scholar] [CrossRef]
- Gerum, C.; Silvers, R.; Wirmer-Bartoschek, J.; Schwalbe, H. Unfolded-state structure and dynamics influence the fibril formation of human prion protein. Angew. Chem. Int. Ed. Engl. 2009, 48, 9452–9456. [Google Scholar] [CrossRef]
- Kumar, J.; Sreeramulu, S.; Schmidt, T.L.; Richter, C.; Vonck, J.; Heckel, A.; Glaubitz, C.; Schwalbe, H. Prion protein amyloid formation involves structural rearrangements in the c-terminal domain. Chembiochem 2010, 11, 1208–1213. [Google Scholar] [CrossRef]
- Morillas, M.; Swietnicki, W.; Gambetti, P.; Surewicz, W.K. Membrane environment alters the conformational structure of the recombinant human prion protein. J. Biol. Chem. 1999, 274, 36859–36865. [Google Scholar] [CrossRef]
- Kazlauskaite, J.; Sanghera, N.; Sylvester, I.; Venien-Bryan, C.; Pinheiro, T.J.T. Structural changes of the prion protein in lipid membranes leading to aggregation and fibrillization. Biochemistry 2003, 42, 3295–3304. [Google Scholar] [CrossRef]
- Sanghera, N.; Pinheiro, T.J.T. Binding of prion protein to lipid membranes and implications for prion conversion. J. Mol. Biol. 2002, 315, 1241–1256. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Giachin, G.; Biljan, I.; Ilc, G.; Plavec, J.; Legname, G. Probing Early Misfolding Events in Prion Protein Mutants by NMR Spectroscopy. Molecules 2013, 18, 9451-9476. https://doi.org/10.3390/molecules18089451
Giachin G, Biljan I, Ilc G, Plavec J, Legname G. Probing Early Misfolding Events in Prion Protein Mutants by NMR Spectroscopy. Molecules. 2013; 18(8):9451-9476. https://doi.org/10.3390/molecules18089451
Chicago/Turabian StyleGiachin, Gabriele, Ivana Biljan, Gregor Ilc, Janez Plavec, and Giuseppe Legname. 2013. "Probing Early Misfolding Events in Prion Protein Mutants by NMR Spectroscopy" Molecules 18, no. 8: 9451-9476. https://doi.org/10.3390/molecules18089451
APA StyleGiachin, G., Biljan, I., Ilc, G., Plavec, J., & Legname, G. (2013). Probing Early Misfolding Events in Prion Protein Mutants by NMR Spectroscopy. Molecules, 18(8), 9451-9476. https://doi.org/10.3390/molecules18089451