Protein Structure Validation and Identification from Unassigned Residual Dipolar Coupling Data Using 2D-PDPA

Abstract
:
1. Introduction

2. Results and Discussion
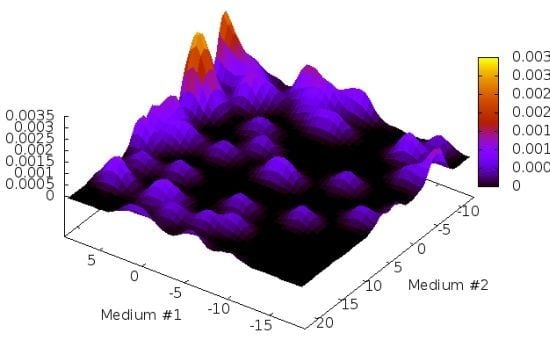
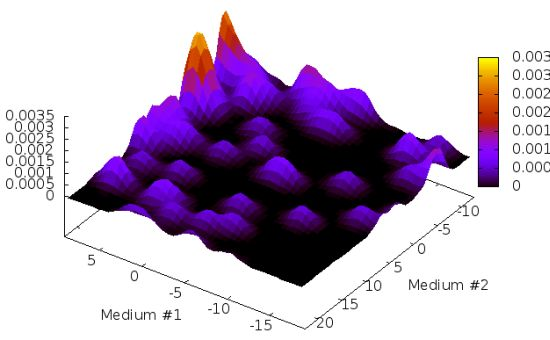
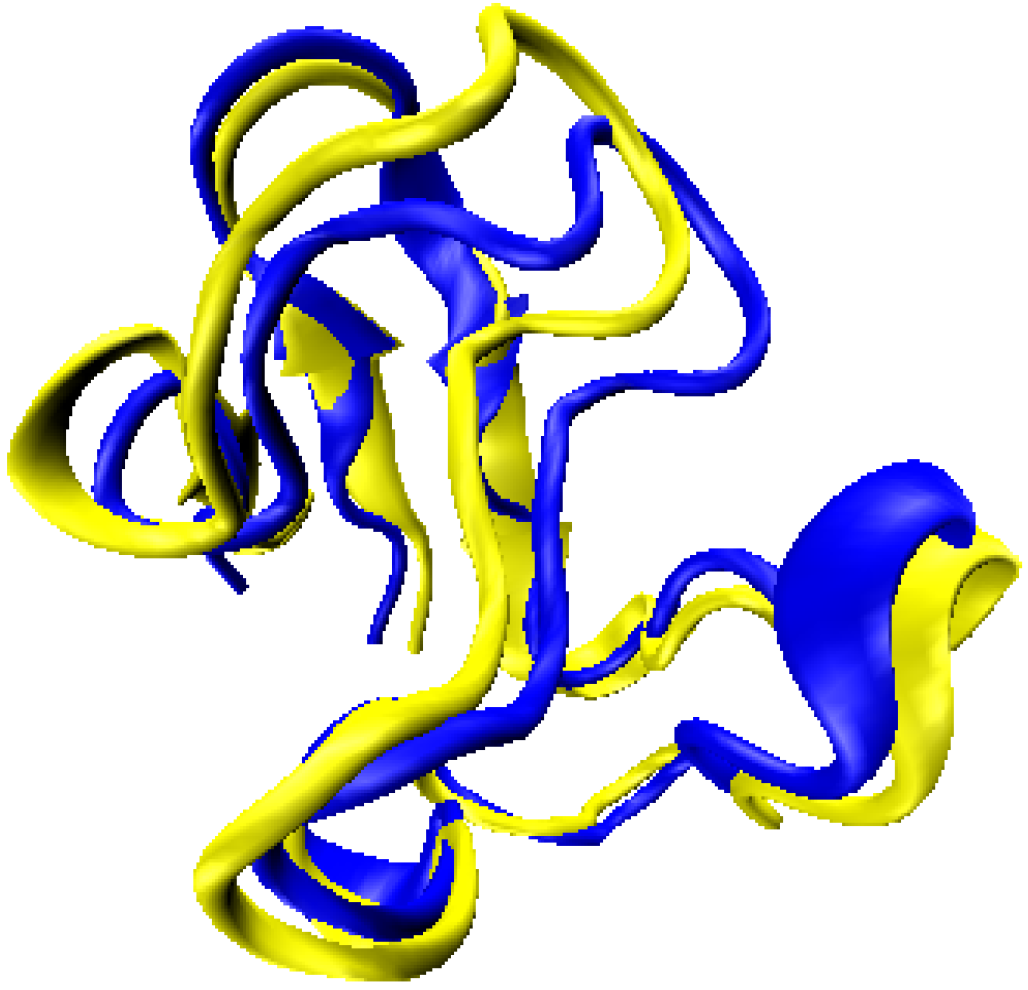
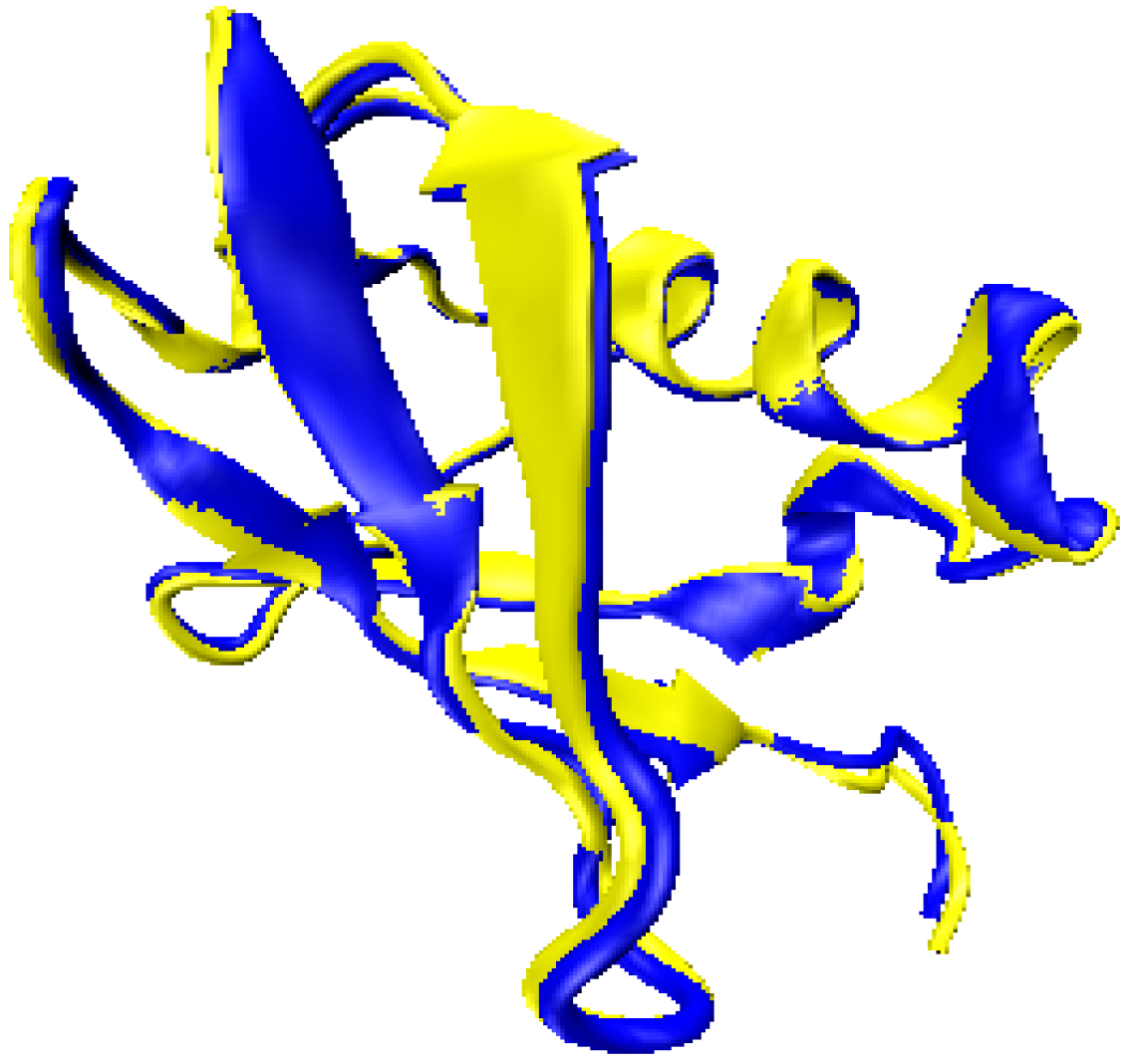
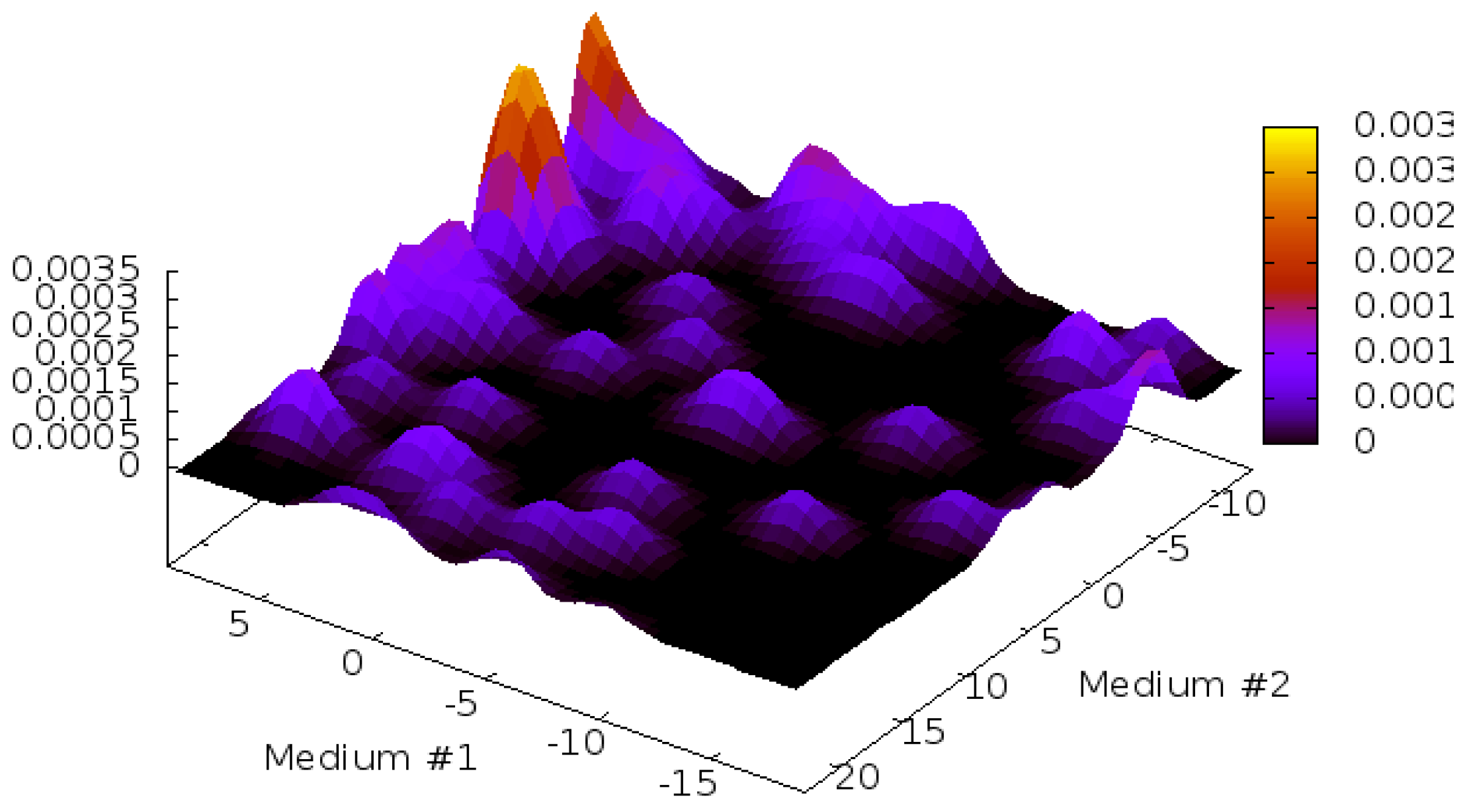
2.1. Structure Identification from Simulated RDC Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
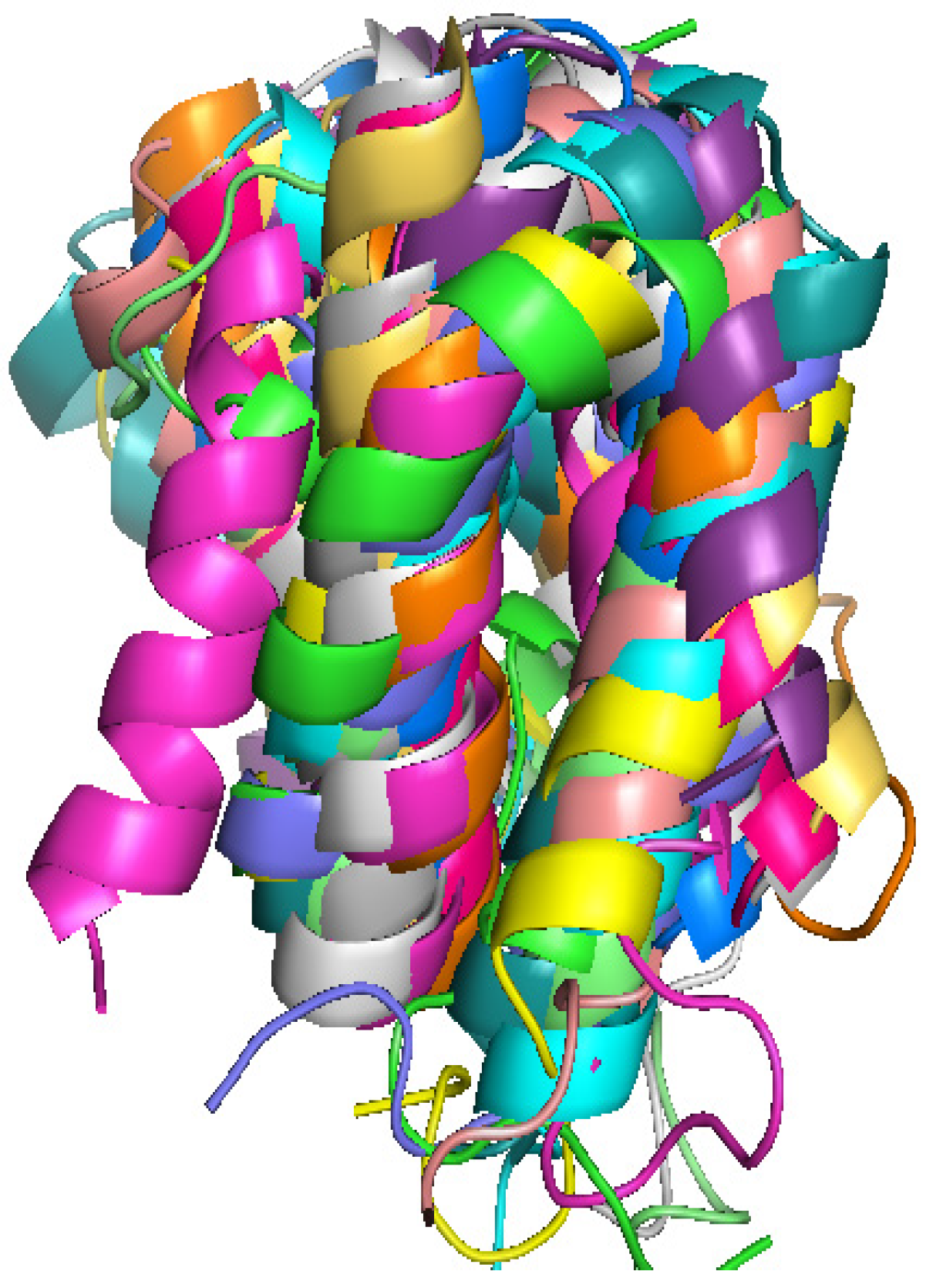{kind=link}
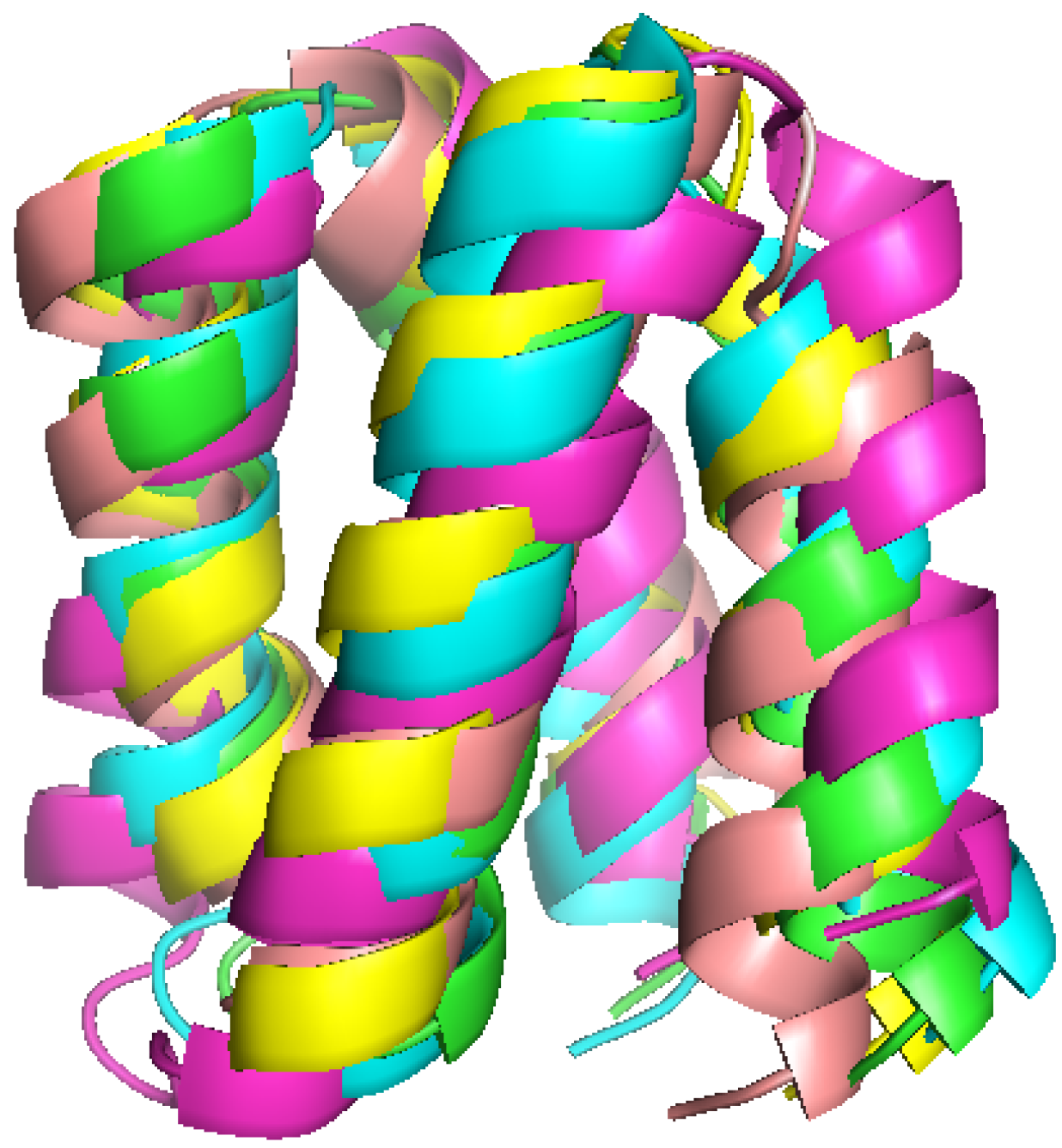{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Target Structure | Size (# of NH Vectors) | Error Added | Rank |
|---|---|---|---|
| 1BRF | 46 | ±1 hz | 1 |
| 1P7E | 55 | ±1 hz | 1 |
| 1SF0 | 67 | ±1 hz | 1 |
| 1BQZ | 75 | ±1 hz | 1 |
| 110M | 149 | ±1 hz | 1 |
| 1NCX | 160 | ±1 hz | 1 |
| 1QHS_A | 172 | ±1 hz | 1 |
| 3FIB | 241 | ±1 hz | 1 |
| 16VP_A | 289 | ±1 hz | 1 |
| 1VSG_A | 353 | ±1 hz | 1 |
| 1A4Y_A | 445 | ±1 hz | 1 |
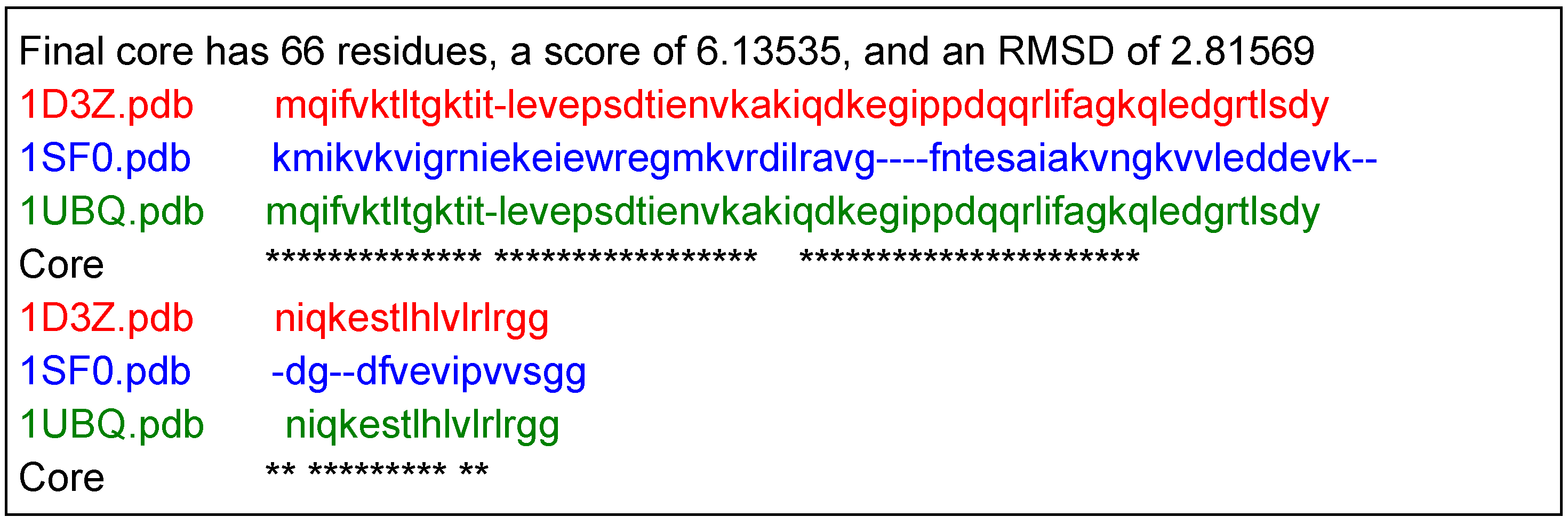
2.2. Structure Identification Using Experimental RDC Data
| Library Structure | Size (# of NH vectors) | Rank | Raw Score |
|---|---|---|---|
| 1P7E | 55 | 1 | 0.45 |
| 1IGD | 59 | 2 | 0.47 |
| 1P7F | 55 | 3 | 0.48 |

| Library Structure | Size (# of NH vectors) | Rank | Raw Score |
|---|---|---|---|
| 1BRF | 46 | 1 | 0.661 |
| 1RWD | 43 | 2 | 0.667 |

| Library Structure | Size (# of NH vectors) | Rank | Raw Score |
|---|---|---|---|
| 1D3Z | 72 | 1 | 0.378 |
| 1UBQ | 72 | 2 | 0.450 |
| 1SF0 | 67 | 3 | 0.452 |



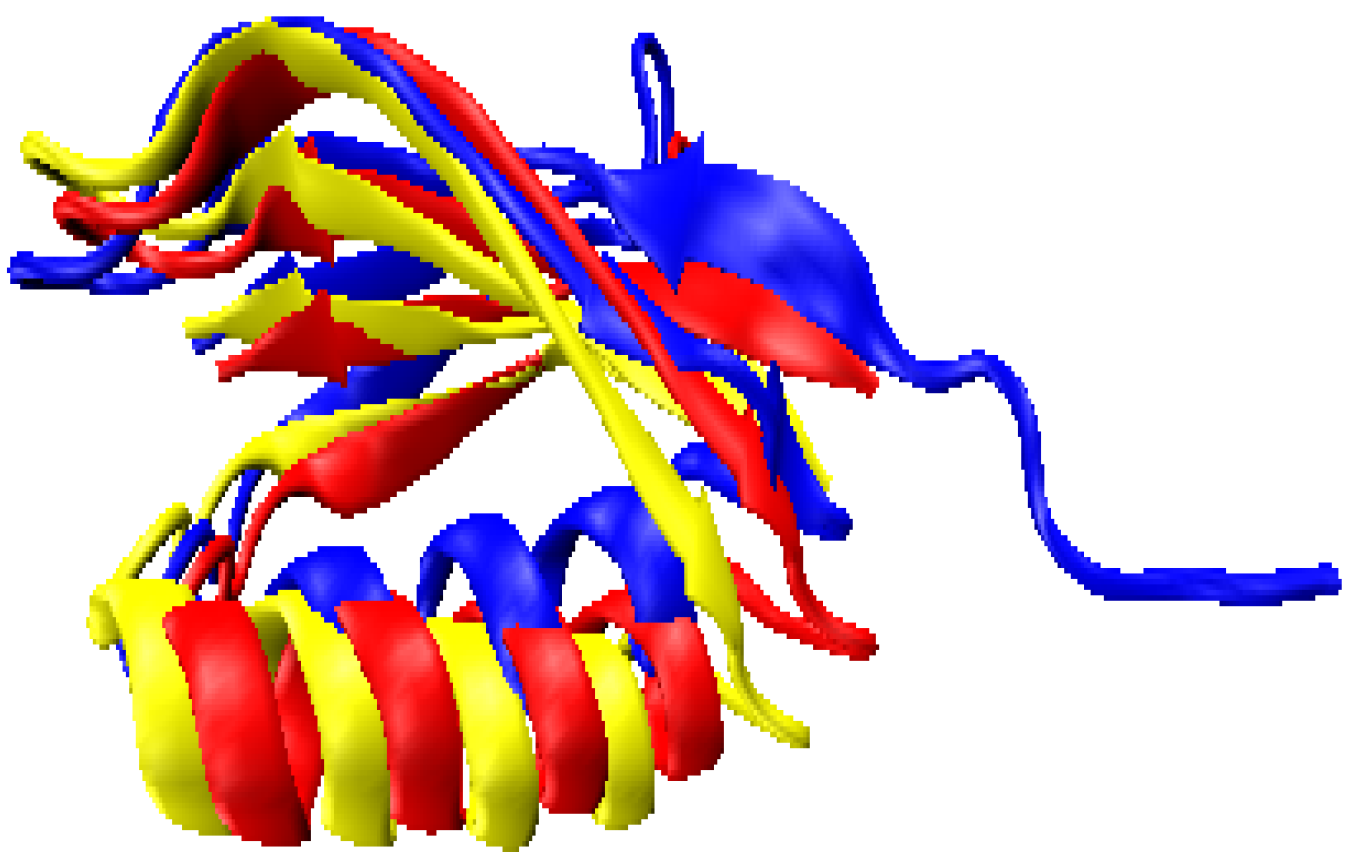
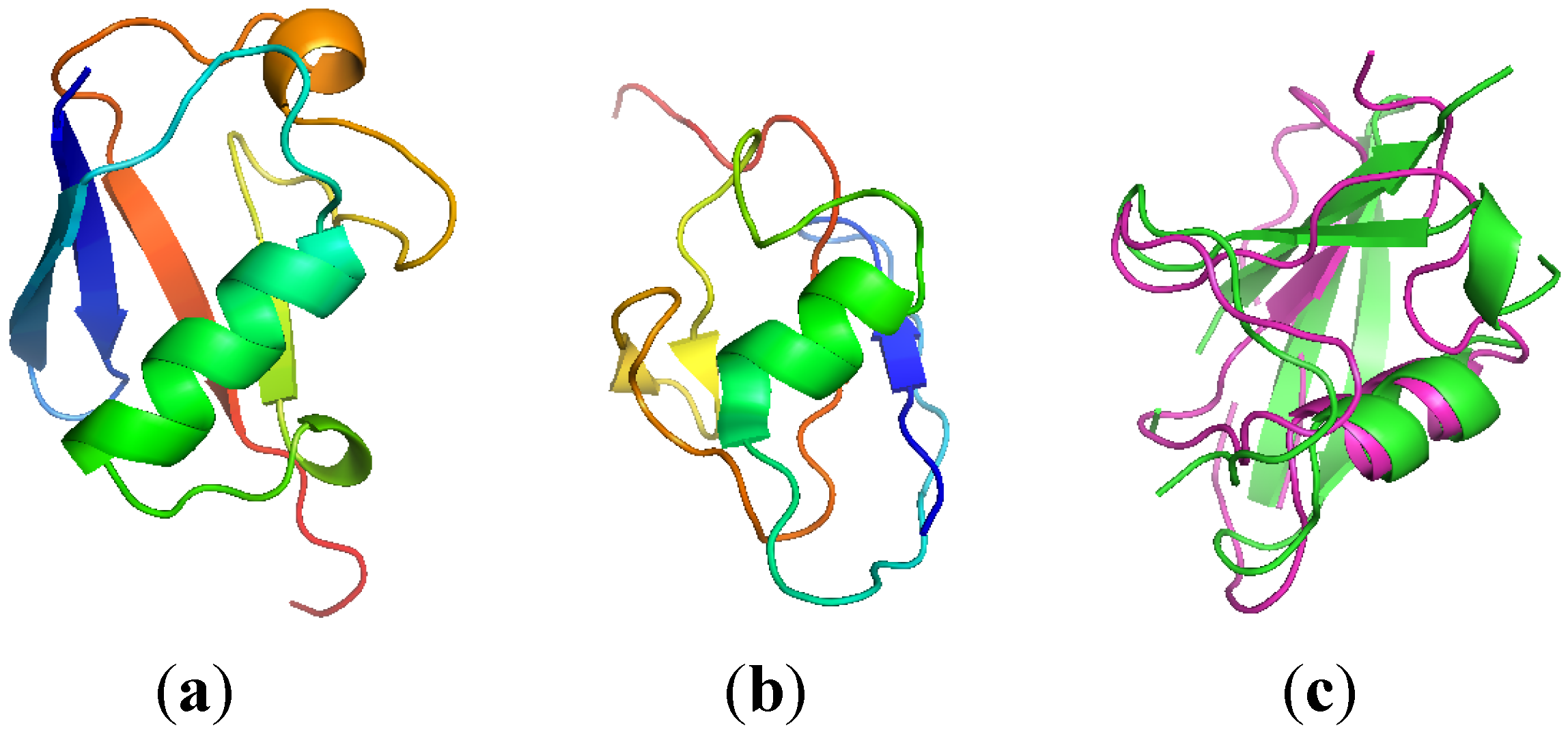
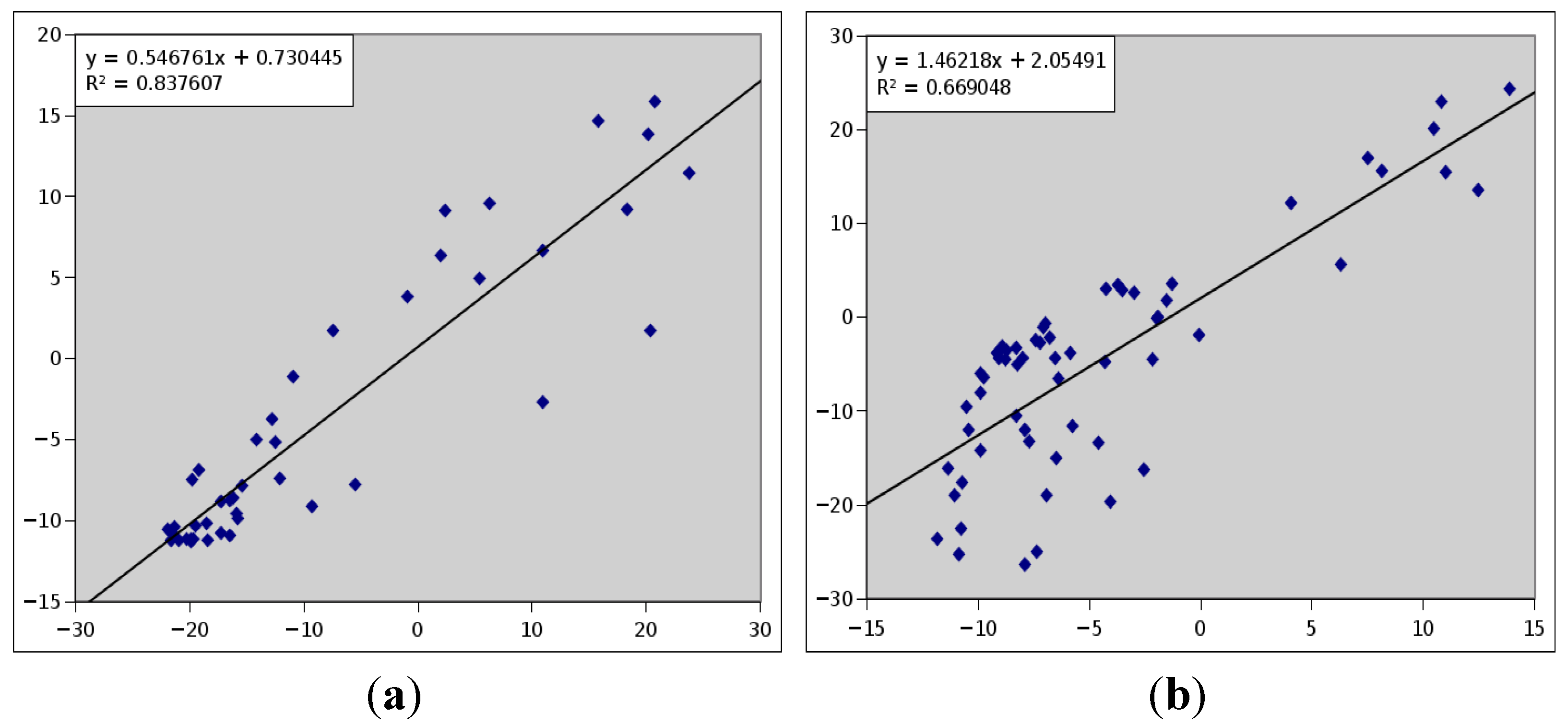
2.3. Computationally Modeled Structures of PF2048.1

| R1 | R2 | R3 | R4 | R5 | R6 | R7 | R8 | R9 | R10 | I1 | I2 | I3 | I4 | I5 | |
| R1 | 0 | 6.51 | 2.93 | 3.01 | 2.95 | 3.39 | 4.33 | 3.37 | 2.73 | 5.42 | 4.43 | 5.14 | 4.82 | 4.25 | 3.48 |
| R2 | 6.51 | 0 | 7.32 | 7.52 | 6.62 | 6.44 | 7.04 | 8.05 | 7.83 | 6.29 | 7.49 | 7.51 | 7.93 | 7.69 | 7.71 |
| R3 | 2.93 | 7.32 | 0 | 4.8 | 5.17 | 3.08 | 6.19 | 4.06 | 3.69 | 4.11 | 6.45 | 7.21 | 7.06 | 6.21 | 5.29 |
| R4 | 3.01 | 7.52 | 4.8 | 0 | 3.34 | 5.26 | 2.91 | 3.75 | 2.68 | 7.31 | 3.08 | 3.78 | 3.85 | 3.23 | 2.74 |
| R5 | 2.95 | 6.62 | 5.17 | 3.34 | 0 | 4.72 | 3.1 | 4.04 | 3.94 | 6.81 | 3.32 | 3.78 | 3.66 | 2.96 | 2.83 |
| R6 | 3.39 | 6.44 | 3.08 | 5.26 | 4.72 | 0 | 5.75 | 5.69 | 3.75 | 3.49 | 6.52 | 7 | 6.84 | 6.18 | 5.56 |
| R7 | 4.33 | 7.04 | 6.19 | 2.91 | 3.1 | 5.75 | 0 | 5.45 | 4.2 | 7.73 | 2.89 | 3.02 | 3.56 | 3.22 | 3.43 |
| R8 | 3.37 | 8.05 | 4.06 | 3.75 | 4.04 | 5.69 | 5.45 | 0 | 4.36 | 7.06 | 4.87 | 5.77 | 5.74 | 4.61 | 3.73 |
| R9 | 2.73 | 7.83 | 3.69 | 2.68 | 3.94 | 3.75 | 4.2 | 4.36 | 0 | 6 | 4.92 | 5.57 | 5.25 | 4.77 | 4.04 |
| R10 | 5.42 | 6.29 | 4.11 | 7.31 | 6.81 | 3.49 | 7.73 | 7.06 | 6 | 0 | 8.48 | 8.85 | 8.86 | 8.23 | 7.6 |
| I1 | 4.43 | 7.49 | 6.45 | 3.08 | 3.32 | 6.52 | 2.89 | 4.87 | 4.92 | 8.48 | 0 | 1.21 | 2.75 | 1.31 | 1.91 |
| I2 | 5.14 | 7.51 | 7.21 | 3.78 | 3.78 | 7 | 3.02 | 5.77 | 5.57 | 8.85 | 1.21 | 0 | 2.55 | 1.89 | 2.89 |
| I3 | 4.82 | 7.93 | 7.06 | 3.85 | 3.66 | 6.84 | 3.56 | 5.74 | 5.25 | 8.86 | 2.75 | 2.55 | 0 | 3.07 | 3.62 |
| I4 | 4.25 | 7.69 | 6.21 | 3.23 | 2.96 | 6.18 | 3.22 | 4.61 | 4.77 | 8.23 | 1.31 | 1.89 | 3.07 | 0 | 1.44 |
| I5 | 3.48 | 7.71 | 5.29 | 2.74 | 2.83 | 5.56 | 3.43 | 3.73 | 4.04 | 7.6 | 1.91 | 2.89 | 3.62 | 1.44 | 0 |
2.4. 2D-PDPA Ranking of the Modeled Structures
| Modeled structure | 2D-PDPA raw score | 2D-PDPA corrected score |
|---|---|---|
| R5 | 0.74 | 0.36 |
| R1 | 0.79 | 0.41 |
| R8 | 0.81 | 0.43 |
| R4 | 0.81 | 0.43 |
| R7 | 0.82 | 0.44 |
| R2 | 0.82 | 0.44 |
| R6 | 0.83 | 0.45 |
| R10 | 0.84 | 0.46 |
| R9 | 0.85 | 0.47 |
| R3 | 0.87 | 0.49 |
| Modeled structure | 2D-PDPA raw score | 2D-PDPA corrected score |
|---|---|---|
| I5 | 0.73 | 0.35 |
| I4 | 0.76 | 0.38 |
| I2 | 0.78 | 0.40 |
| I3 | 0.81 | 0.43 |
| I1 | 0.82 | 0.44 |

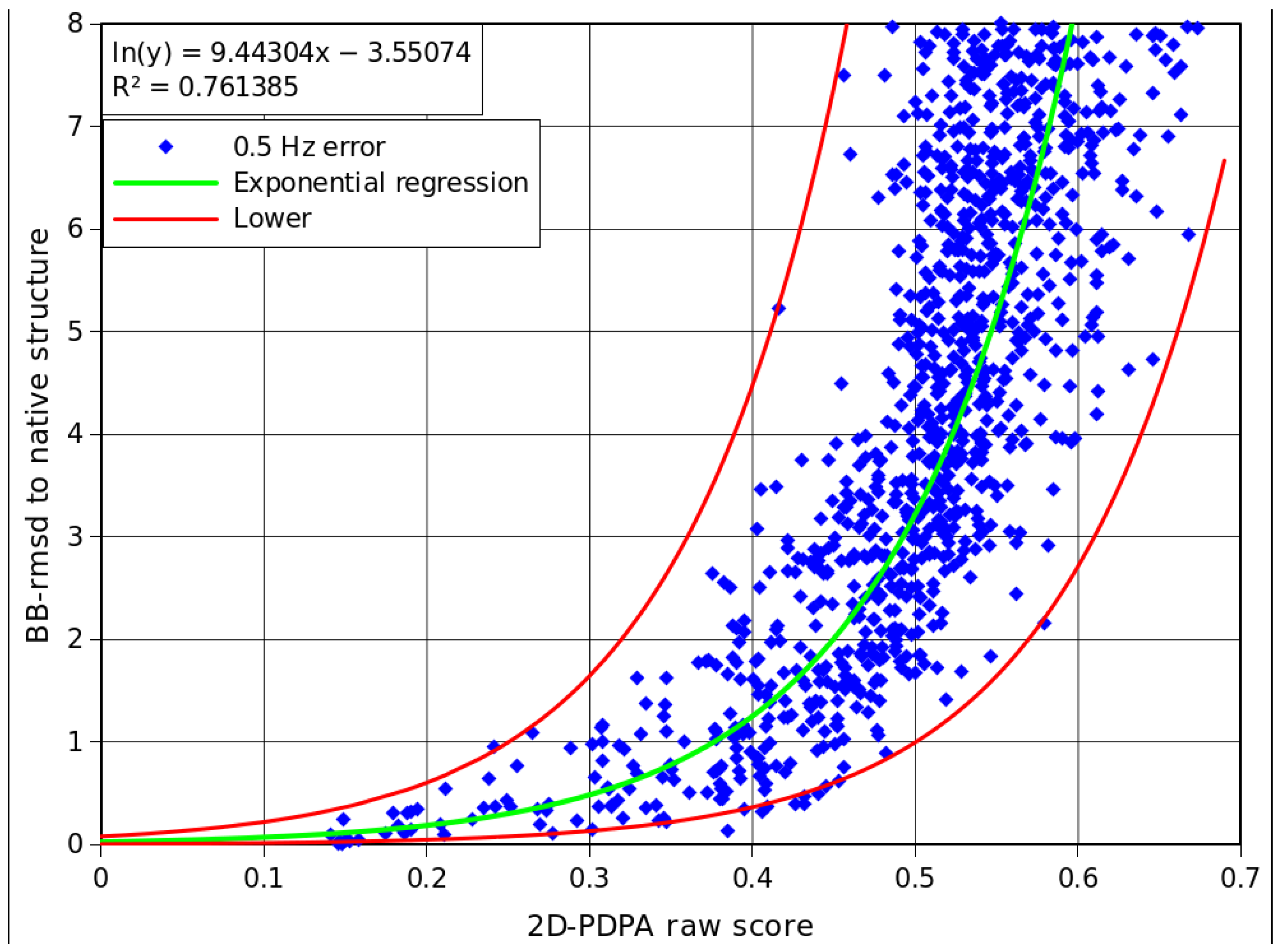
2.5. Interpretation of 2D-PDPA Results for Modeled Structures of Pf2048

| Modeled structure | 2D-PDPA corrected score | Lower bb-rmsd(Å) | Upper bb-rmsd(Å) |
|---|---|---|---|
| I5 | 0.35 | 0.22 | 2.72 |
| R5 | 0.36 | 0.25 | 3.00 |
| I4 | 0.38 | 0.30 | 3.67 |
| I2 | 0.40 | 0.37 | 4.48 |
| R1 | 0.41 | 0.41 | 4.95 |
3. Experimental
3.1. General Experimental Approach and Targeted Protein Structures
| Structure | X-ray/NMR | # of Residues | CATH Classification | Data Source |
|---|---|---|---|---|
| 1BRF | X-Ray | 53 | 2.20.28.10 | Synthetic |
| 1RWD | NMR | 53 | 2.20.28.10 | Experimental |
| 1P7E | NMR | 56 | 3.10.20.10 | Experimental & Synthetic |
| 1D3Z | NMR | 76 | 3.10.20.90 | Experimental |
| 1SF0 | NMR | 77 | 3.10.20.30 | Synthetic |
| 1BQZ | NMR | 77 | 1.10.287.110 | Synthetic |
| 110M | X-Ray | 154 | 1.10.490.10 | Synthetic |
| 1NCX | X-Ray | 162 | 1.10.238.10 | Synthetic |
| 1.10.238.10 | ||||
| 1QHS_A | X-Ray | 178 | 3.40.50.300 | Synthetic |
| 3FIB | X-Ray | 249 | 3.90.215.10 | Synthetic |
| 4.10.530.10 | ||||
| 16VP_A | X-Ray | 366 | 1.10.1290.10 | Synthetic |
| 1VSG_A | X-Ray | 364 | 3.90.150.10 | Synthetic |
| 1.10.470.10 | ||||
| 1A4Y_A | X-Ray | 460 | 3.80.10.10 | Synthetic |
3.2. Simulated and Experimental RDC Data of Target Proteins
| Sxx | Sxy | Sxz | Syy | Syz | Da (NH Hz) | R | |
|---|---|---|---|---|---|---|---|
| M1 | 3.000E−4 | 0.0 | 0.0 | 5.000E-4 | 0.0 | −9.45 | 0.25 |
| M2 | 1.066E−04 | 2.367E−04 | 3.603E−04 | −1.464E−04 | 4.323E−04 | 8.27 | 0.28 |

3.3. Library of Structures Representing Protein Fold Families
3.4. NMR Sample Preparation, Data Acquisition and Data
3.5. Computational Modeling of Pf2048.1
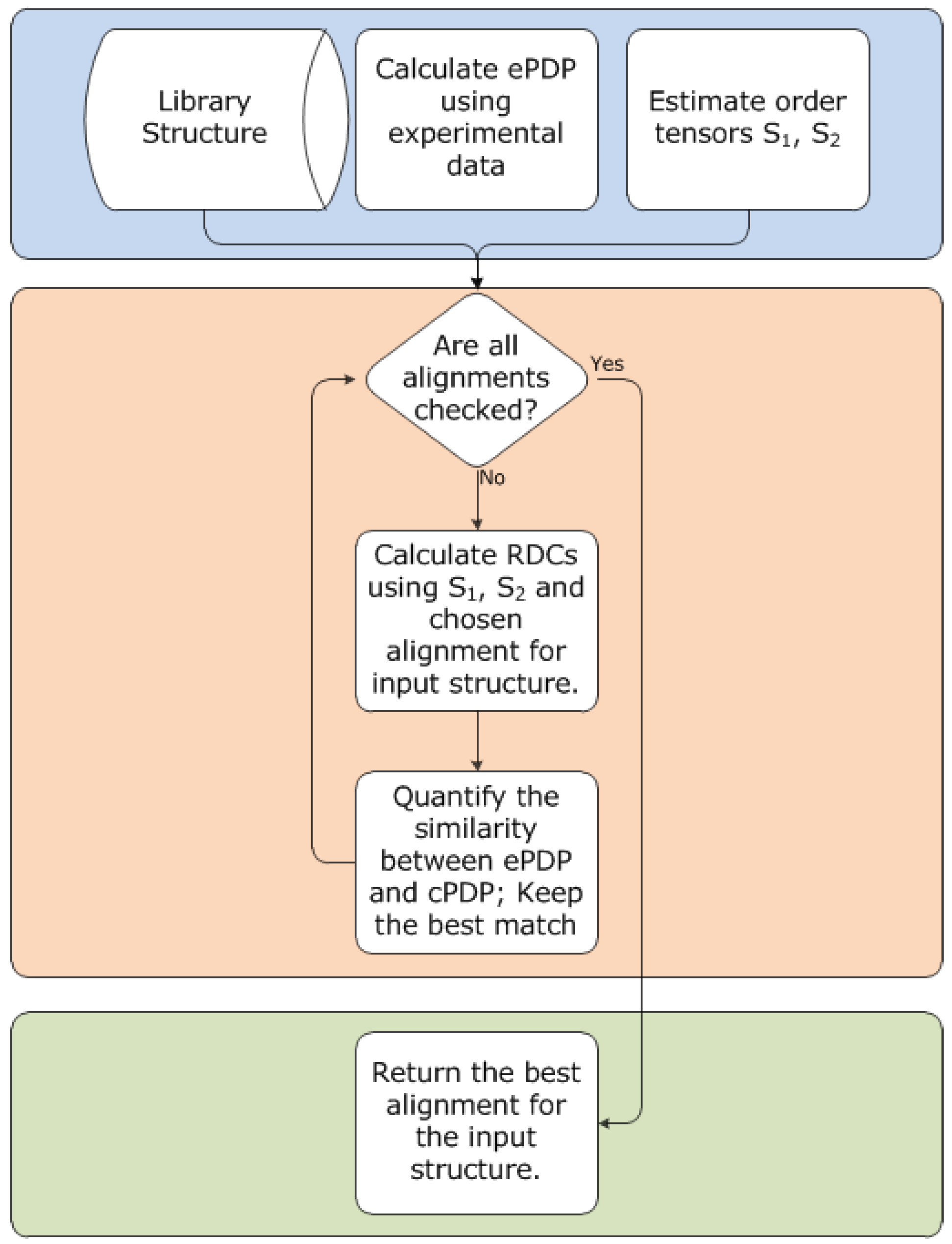
3.6. Outline of 2D-PDPA Method


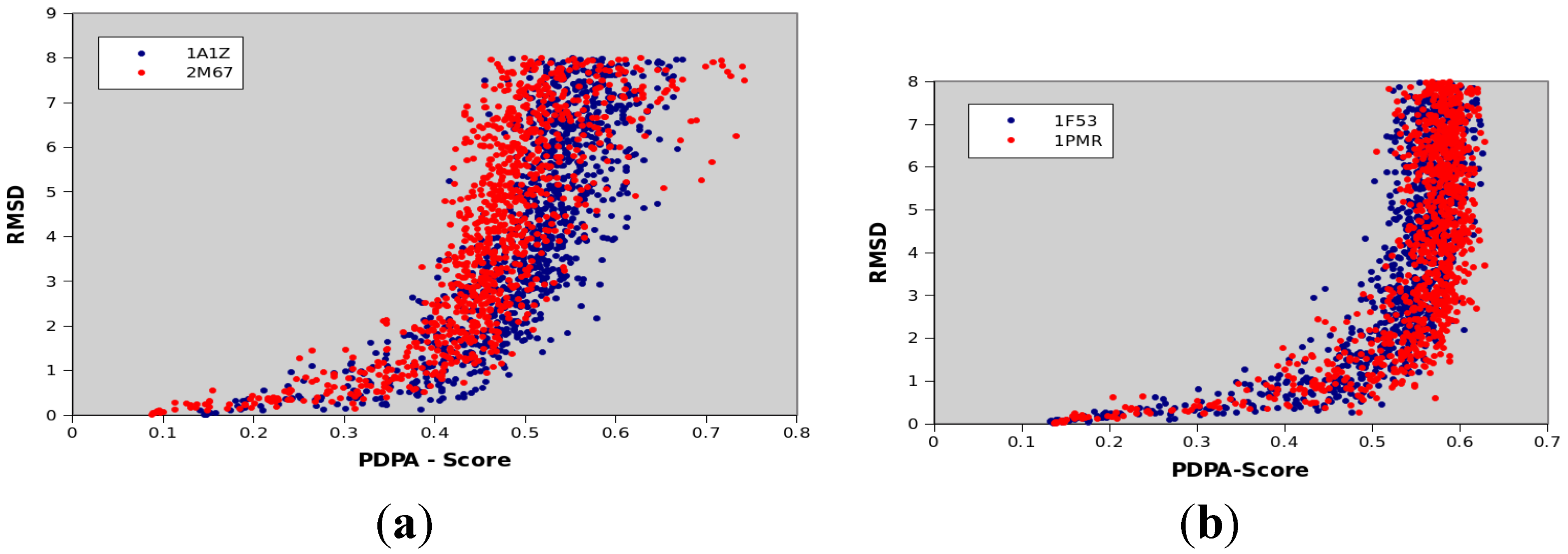
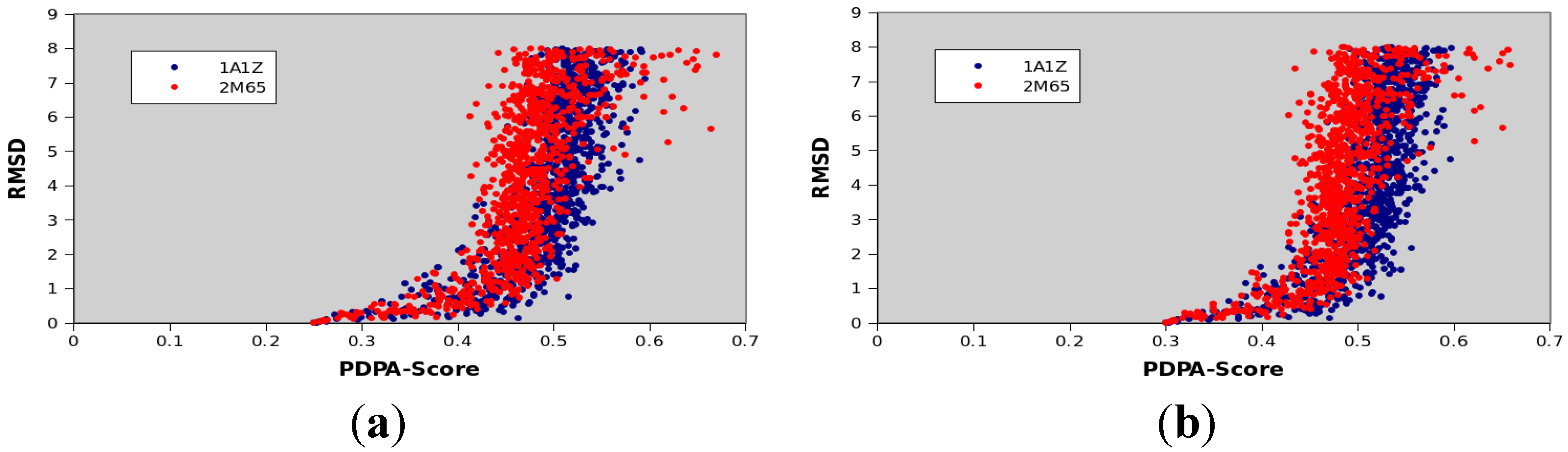
3.7. Scoring and Interpretation of 2D-PDPA Raw Scores

- Interpretations patterns are primarily a function of class of protein structure (α of β protein) and protein size
- Interpretation patterns depend on completeness of data
- Interpretation patterns exhibit a dependency on quality of experimental data, and more directly on the quality of the two estimated order tensors
| Protein PDBID | Protein Size | CATH Classification | Number of Secondary Structural Elements |
|---|---|---|---|
| 1A1Z | 83 | 1.10.533.10 | 11 α-helices |
| 2M67 | 81 | Not available yet | 6 α-helices |
| 1F53 | 84 | 2.60.20.30 | 6 β-strands |
| 1PMR | 80 | 2.40.50.100 | 6 β-strands |


3.8. Computational Facilities
4. Theoretical Background
4.1. Residual Dipolar Coupling





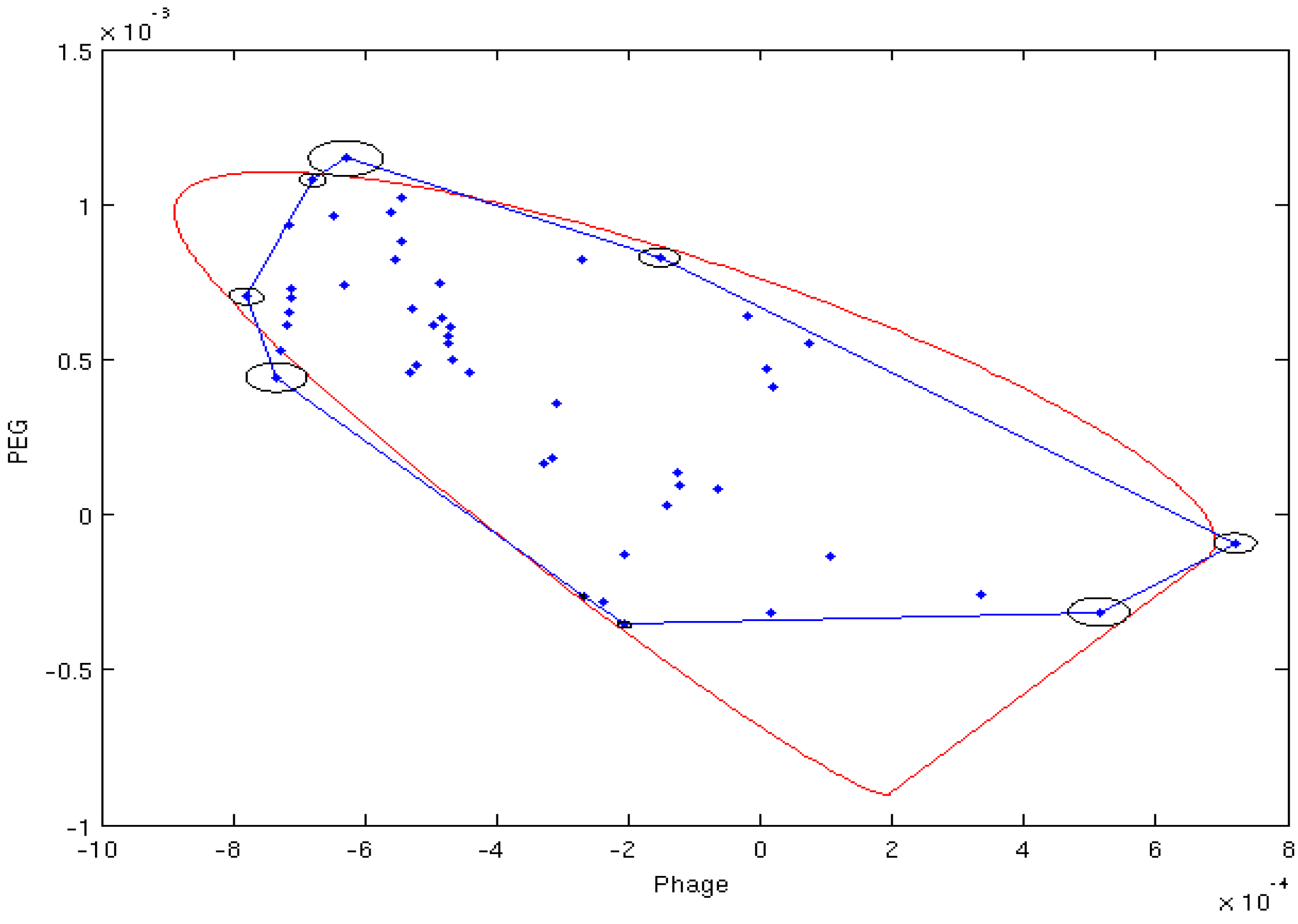
4.2. Estimation of Order Tensor and Calculation of RDCs
| Sxx | Sxy | Sxz | Syy | Syz | Da | R | |
|---|---|---|---|---|---|---|---|
| M1(Phage) | 2.04E−04 | 0.00E+00 | 0.00E+00 | 7.11E−04 | 0.00E+00 | −10.8 | 0.55 |
| M2 (PEG) | −9.14E−04 | 1.71E−05 | 1.61E−04 | −8.55E−05 | 3.89E−04 | −13.03 | 0.65 |
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- Grant, A.; Lee, D.; Orengo, C. Progress towards mapping the universe of protein folds. Genome Biol. 2004, 5, 107. [Google Scholar] [CrossRef]
- Fukunaga, K. Introduction to Statistical Pattern Recognition, 1st ed; Academic Press Inc.: San Diego, CA, USA, 1990; p. 591. [Google Scholar]
- Greshenfeld, N.A. The Nature of Mathematical Modeling; Cambridge University Press: New York, NY, USA, 1998. [Google Scholar]
- Quinn, M.J.; Flomenhoft, M.L.; Jones, E.A. Parallel Programming in C with MPI and OpenMP; Flomenhoft, M.L., Ed.; McGraw-Hill Higher Education: New York, NY, USA, 2003. [Google Scholar]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinforma. 2008, 9, 40. [Google Scholar] [CrossRef]
- Dunbrack, R.L. Sequence comparison and protein structure prediction. Curr. Opin. Struct. Biol. 2006, 16, 374–384. [Google Scholar] [CrossRef]
- Zhang, Y. Template-based modeling and free modeling by I-TASSER in CASP7. Proteins 2007, 69 (Suppl. 8), 108–117. [Google Scholar] [CrossRef]
- Adams, M.W.W.; Dailey, H.A.; Delucas, L.J.; Luo, M.; Prestegard, J.H.; Rose, J.P.; Wang, B.C. The southeast collaboratory for structural genomics: A high-throughput gene to structure factory. Acc. Chem. Res. 2003, 36, 191–198. [Google Scholar] [CrossRef]
- Brenner, S.E.; Levitt, M. Expectations from structural genomics. Protein Sci. 2000, 9, 197–200. [Google Scholar] [CrossRef]
- Service, R. Structural biology - Structural genomics, round 2. Science 2005, 307, 1554–1558. [Google Scholar] [CrossRef]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The protein data bank. Nucleic Acids Res. 2000, 28, 235–242. [Google Scholar] [CrossRef]
- Wu, S.; Skolnick, J.; Zhang, Y. Ab initio modeling of small proteins by iterative TASSER simulations. BMC Biol. 2007, 5, 17. [Google Scholar] [CrossRef]
- Kim, D.E.; Chivian, D.; Baker, D. Protein structure prediction and analysis using the Robetta server. Nucleic Acids Res. 2004, 32, W526–W531. [Google Scholar] [CrossRef]
- Vassar, R.; Citron, M. AÎ2-generating enzymes: Recentadvances in Î2- and Î3-secretase research. Neuron 2000, 27, 419–422. [Google Scholar] [CrossRef]
- Murzin, A.G.; Brenner, S.E.; Hubbard, T.; Chothia, C. SCOP - a structural classification of proteins database for the investigation of sequences and structures. J. Mol. Biol. 1995, 247, 536–540. [Google Scholar]
- Orengo, C.A.; Michie, A.D.; Jones, S.; Jones, D.T.; Swindells, M.B.; Thornton, J.M. CATH - a hierarchic classification of protein domain structures. Structure 1997, 5, 1093–1108. [Google Scholar] [CrossRef]
- Miao, X.; Waddell, P.J.; Valafar, H. TALI: Local alignment of protein structures using backbone torsion angles. J. Bioinforma. Comput. Boil. 2008, 6, 163–181. [Google Scholar] [CrossRef]
- Shealy, P.; Valafar, H. Multiple structure alignment with msTALI. BMC Bioinforma. 2012, 13, 105. [Google Scholar] [CrossRef]
- Shen, Y.; Vernon, R.; Baker, D.; Bax, A. De novo protein structure generation from incomplete chemical shift assignments. J. Biomol. NMR 2009, 43, 63–78. [Google Scholar] [CrossRef]
- Rohl, C.A.; Baker, D. De novo determination of protein backbone structure from residual dipolar couplings using Rosetta. J. Am. Chem. Soc. 2002, 124, 2723–2729. [Google Scholar] [CrossRef]
- Valafar, H.; Prestegard, J.H. Rapid classification of a protein fold family using a statistical analysis of dipolar couplings. Bioinformatics (Oxford, England) 2003, 19, 1549–1555. [Google Scholar] [CrossRef]
- Bansal, S.; Miao, X.; Adams, M.W.W.; Prestegard, J.H.; Valafar, H. Rapid classification of protein structure models using unassigned backbone RDCs and probability density profile analysis (PDPA). J. Magn. Reson. 2008, 192, 60–68. [Google Scholar] [CrossRef]
- Azurmendi, H.F.; Martin-Pastor, M.; Bush, C.A. Conformational studies of Lewis X and Lewis A trisaccharides using NMR residual dipolar couplings. Biopolymers 2002, 63, 89–98. [Google Scholar] [CrossRef]
- Adeyeye, J.; Azurmendi, H.F.; Stroop, C.J.M.; Sozhamannan, S.; Williams, A.L.; Adetumbi, A.M.; Johnson, J.A.; Bush, C.A. Conformation of the hexasaccharide repeating subunit from the Vibrio cholerae O139 capsular polysaccharide. Biochemistry 2003, 42, 3979–3988. [Google Scholar] [CrossRef]
- Tian, F.; Al-Hashimi, H.M.; Craighead, J.L.; Prestegard, J.H. Conformational analysis of a flexible oligosaccharide using residual dipolar couplings. J. Am. Chem. Soc. 2001, 123, 485–492. [Google Scholar] [CrossRef]
- Tjandra, N.; Tate, S.; Ono, A.; Kainosho, M.; Bax, A. The NMR structure of a DNA dodecamer in an aqueous dilute liquid crystalline phase. J. Am. Chem. Soc. 2000, 122, 6190–6200. [Google Scholar]
- Vermeulen, A.; Zhou, H.; Pardi, A. Determining DNA global structure and DNA bending by application of NMR residual dipolar couplings. J. Am. Chem. Soc. 2000, 122, 9638–9647. [Google Scholar] [CrossRef]
- Al-Hashimi, H.M.; Gorin, A.; Majumdar, A.; Gosser, Y.; Patel, D.J. Towards structural Genomics of RNA: Rapid NMR resonance assignment and simultaneous RNA tertiary structure determination using residual dipolar couplings. J. Mol. Biol. 2002, 318, 637–649. [Google Scholar] [CrossRef]
- Al-Hashimi, H.M.; Gosser, Y.; Gorin, A.; Hu, W.; Majumdar, A.; Patel, D.J. Concerted motions in HIV-1 TAR RNA may allow access to bound state conformations: RNA dynamics from NMR residual dipolar couplings. J. Mol. Biol. 2002, 315, 95–102. [Google Scholar] [CrossRef]
- Assfalg, M.; Bertini, I.; Turano, P.; Grant Mauk, A.; Winkler, J.R.; Gray, H.B. 15N-1H Residual dipolar coupling analysis of native and alkaline-K79A Saccharomyces cerevisiae cytochrome c. Biophys. J. 2003, 84, 3917–3923. [Google Scholar] [CrossRef]
- Andrec, M.; Du, P.; Levy, R.M. Protein backbone structure determination using only residual dipolar couplings from one ordering medium. J. Biomol. NMR 2001, 21, 335–347. [Google Scholar] [CrossRef]
- Delaglio, F.; Kontaxis, G.; Bax, A. Protein structure determination using molecular fragment replacement and NMR dipolar couplings. J. Am. Chem. Soc. 2000, 122, 2142–2143. [Google Scholar] [CrossRef]
- Prestegard, J.H.; Mayer, K.L.; Valafar, H.; Benison, G.C. Determination of protein backbone structures from residual dipolar couplings. Methods Enzymol. 2005, 394, 175–209. [Google Scholar] [CrossRef]
- Valafar, H.; Mayer, K.; Bougault, C.; LeBlond, P.; Jenney, F.E.; Brereton, P.S.; Adams, M.; Prestegard, J.H. Backbone solution structures of proteins using residual dipolar couplings: Application to a novel structural genomics target. J. Struct. Funct. Genomics 2005, 5, 241–254. [Google Scholar] [CrossRef]
- Shealy, P.; Liu, Y.; Simin, M.; Valafar, H. Backbone resonance assignment and order tensor estimation using residual dipolar couplings. J. Biomol. NMR 2011, 50, 357–369. [Google Scholar] [CrossRef]
- Jung, Y.-S.S.; Sharma, M.; Zweckstetter, M. Simultaneous assignment and structure determination of protein backbones by using NMR dipolar couplings. Angew. Chem. Int. Ed. Engl. 2004, 43, 3479–3481. [Google Scholar] [CrossRef]
- Marassi, F.M.; Opella, S.J. Simultaneous resonance assignment and structure determination in the solid-state NMR spectrum of a membrane protein in lipid bilayers. Biophys. J. 2002, 82, 467A. [Google Scholar]
- Langmead, C.J.; Donald, B.R. An expectation/maximization nuclear vector replacement algorithm for automated NMR resonance assignments. J. Biomol. NMR 2004, 29, 111–138. [Google Scholar] [CrossRef]
- Bernadó, P.; Blackledge, M. Local dynamic amplitudes on the protein backbone from dipolar couplings: Toward the elucidation of slower motions in biomolecules. J. Am. Chem. Soc. 2004, 126, 7760–7761. [Google Scholar] [CrossRef]
- Bouvignies, G.; Bernadó, P.; Meier, S.; Cho, K.; Grzesiek, S.; Brüschweiler, R.; Blackledge, M. Identification of slow correlated motions in proteins using residual dipolar and hydrogen-bond scalar couplings. Proc. Natl. Acad. Sci. USA 2005, 102, 13885–13890. [Google Scholar]
- Bryson, M.; Tian, F.; Prestegard, J.H.; Valafar, H. REDCRAFT: A tool for simultaneous characterization of protein backbone structure and motion from RDC data. J. Magn. Reson. 2008, 191, 322–334. [Google Scholar] [CrossRef]
- Andrec, M.; Harano, Y.; Jacobson, M.P.; Friesner, R.A.; Levy, R.M. Complete protein structure determination using backbone residual dipolar couplings and sidechain rotamer prediction. J. Struct. Funct. Genomics 2002, 2, 103–111. [Google Scholar] [CrossRef]
- Meiler, J.; Peti, W.; Griesinger, C. DipoCoup: A versatile program for 3D-structure homology comparison based on residual dipolar couplings and pseudocontact shifts. J. Biomol. NMR 2000, 17, 283–294. [Google Scholar] [CrossRef]
- Tian, F.; Valafar, H.; Prestegard, J.H. A dipolar coupling based strategy for simultaneous resonance assignment and structure determination of protein backbones. J. Am. Chem. Soc. 2001, 123, 11791–11796. [Google Scholar] [CrossRef]
- Marassi, F.M.; Opella, S.J. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 2003, 12, 403–411. [Google Scholar] [CrossRef]
- Prestegard, J.H.; Bougault, C.M.; Kishore, A.I. Residual dipolar couplings in structure determination of biomolecules. Chem. Rev. 2004, 104, 3519–3540. [Google Scholar] [CrossRef]
- Bertone, P. SPINE: an integrated tracking database and data mining approach for identifying feasible targets in high-throughput structural proteomics. Nucleic Acids Res. 2001, 29, 2884–2898. [Google Scholar] [CrossRef]
- Jones, E.Y.; Davis, S.J.; Williams, A.F.; Harlos, K.; Stuart, D.I. Crystal structure at 2.8 A resolution of a soluble form of the cell adhesion molecule CD2. Nature 1992, 360, 232–239. [Google Scholar] [CrossRef]
- Murray, A.J.; Head, J.G.; Barker, J.J.; Brady, R.L. Engineering an intertwined form of CD2 for stability and assembly. Nat. Struct. Biol. 1998, 5, 778–782. [Google Scholar]
- Valafar, H.; Bryson, M.; Miao, X.; Shealy, P.; Mukhopadhyay, R.; Yandle, R.; Simin, M.; Fahim, A.; Irausquin, S.J. ValafarLab web page. Available online: http://ifestos.cse.sc.edu (accessed on 10 August 2013).
- Ulrich, E.L.; Akutsu, H.; Doreleijers, J.F.; Harano, Y.; Ioannidis, Y.E.; Lin, J.; Livny, M.; Mading, S.; Maziuk, D.; Miller, Z.; et al. BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [Google Scholar]
- Doreleijers, J.F.; Mading, S.; Maziuk, D.; Sojourner, K.; Yin, L.; Zhu, J.; Markley, J.L.; Ulrich, E.L. BioMagResBank database with sets of experimental NMR constraints corresponding to the structures of over 1400 biomolecules deposited in the Protein Data Bank. J. Biomol. NMR 2003, 26, 139–146. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Ramirez, B.E.; Delaglio, F.; Bax, A. Evaluation of backbone proton positions and dynamics in a small protein by liquid crystal NMR spectroscopy. J. Am. Chem. Soc. 2003, 125, 9179–9191. [Google Scholar] [CrossRef]
- Cornilescu, G.; Marquardt, J.L.; Ottiger, M.; Bax, A. Validation of protein structure from anisotropic carbonyl chemical shifts in a dilute liquid crystalline phase. J. Am. Chem. Soc. 1998, 120, 6836–6837. [Google Scholar] [CrossRef]
- Mukhopadhyay, R.; Miao, X.; Shealy, P.; Valafar, H. Efficient and accurate estimation of relative order tensors from lambda-maps. J. Magn. Reson. 2009, 198, 236–247. [Google Scholar] [CrossRef]
- Doreleijers, J.F.; Raves, M.L.; Rullmann, T.; Kaptein, R. Completeness of NOEs in protein structure: A statistical analysis of NMR data. J. Biomol. NMR 1999, 14, 123–132. [Google Scholar] [CrossRef]
- Bau, R.; Rees, D.C.; Kurtz, D.M., Jr; Scott, R.A.; Huang, H.; Adams, M.W.W.; Eidsness, M.K. Crystal structure of rubredoxin from Pyrococcus furiosus at 0.95 Å resolution, and the structures of N-terminal methionine and formylmethionine variants of Pf Rd. Contributions of N-terminal interactions to thermostability. J. Biol. Inorg. Chem. 1998, 3, 484–493. [Google Scholar] [CrossRef]
- Valafar, H.; Prestegard, J.H. REDCAT: A residual dipolar coupling analysis tool. J. Magn. Reson. 2004, 167, 228–241. [Google Scholar] [CrossRef]
- Word, J.M.; Lovell, S.C.; Richardson, J.S.; Richardson, D.C. Asparagine and glutamine: Using hydrogen atom contacts in the choice of side-chain amide orientation. J. Mol. Biol. 1999, 285, 1735–1747. [Google Scholar] [CrossRef]
- Holm, L.; Sander, C. The FSSP database of structurally aligned protein fold families. Nucleic Acids Res. 1994, 22, 3600–3609. [Google Scholar]
- Prestegard, J.H.; Kishore, A.I. Partial alignment of biomolecules: An aid to NMR characterization. Curr. Opin. Chem. Biol. 2001, 5, 584–590. [Google Scholar] [CrossRef]
- Otting, G.; Rückert, M.; Levitt, M. H.; Moshref, A. NMR experiments for the sign determination of homonuclear scalar and residual dipolar couplings. J. Biomol. NMR 2000, 16, 343–346. [Google Scholar] [CrossRef]
- Ottiger, M.; Bax, A. Determination of relative N−H N, N−C‘, C α −C‘, and C α −H α effective bond lengths in a protein by NMR in a dilute liquid crystalline phase. J. Am. Chem. Soc. 1998, 120, 12334–12341. [Google Scholar] [CrossRef]
- Delaglio, F.; Grzesiek, S.; Vuister, G.W.; Zhu, G.; Pfeifer, J.; Bax, A. NMRPipe: A multi- dimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 1995, 6, 277–293. [Google Scholar]
- Miao, X.; Mukhopadhyay, R.; Valafar, H.; MR&VH, M.X. Estimation of relative order tensors, and reconstruction of vectors in space using unassigned RDC data and its application. J. Magn. Reson. 2008, 194, 202–211. [Google Scholar]
- Saupe, A.; Englert, G. High-resolution nuclear magnetic resonance spectra of orientated molecules. Phys. Rev. Lett. 1963, 11, 462–464. [Google Scholar] [CrossRef]
- Blackledge, M. Recent progress in the study of biomolecular structure and dynamics in solution from residual dipolar couplings. Prog. Nuclear Magn. Reson. Spectrosc. 2005, 46, 23–61. [Google Scholar] [CrossRef]
- Baran, M.C.; Huang, Y.J.; Moseley, H.N.B.; Montelione, G.T. Automated analysis of protein NMR assignments and structures. Chem. Rev. 2004, 104, 3541–3555. [Google Scholar] [CrossRef]
- Mayer, K.L.; Qu, Y.; Bansal, S.; LeBlond, P.D.; Jenney, F.E.; Brereton, P.S.; Adams, M.W.W.; Xu, Y.; Prestegard, J.H. Structure determination of a new protein from backbone-centered NMR data and NMR-assisted structure prediction. Proteins Struct. Funct. Bioinf. 2006, 65, 480–489. [Google Scholar] [CrossRef]
- Wang, X.; Bansal, S.; Jiang, M.; Prestegard, J.H. RDC-assisted modeling of symmetric protein homo-oligomers. Protein Sci. 2008, 17, 899–907. [Google Scholar] [CrossRef]
- Serfiotis-Mitsa, D.; Herbert, A.P.; Roberts, G.A.; Soares, D.C.; White, J.H.; Blakely, G.W.; Uhrín, D.; Dryden, D.T.F. The structure of the KlcA and ArdB proteins reveals a novel fold and antirestriction activity against Type I DNA restriction systems in vivo but not in vitro. Nucleic Acids Res. 2010, 38, 1723–1737. [Google Scholar] [CrossRef]
- Wang, J.; Walsh, J.D.; Kuszewski, J.; Wang, Y.-X. Periodicity, planarity, and pixel (3P): A program using the intrinsic residual dipolar coupling periodicity-to-peptide plane correlation and phi/psi angles to derive protein backbone structures. J. Magn. Reson. 2007, 189, 90–103. [Google Scholar] [CrossRef]
- Stelzer, A.C.; Frank, A.T.; Bailor, M.H.; Andricioaei, I.; Al-Hashimi, H.M. Constructing atomic-resolution RNA structural ensembles using MD and motionally decoupled NMR RDCs. Methods 2009, 49, 167–173. [Google Scholar] [CrossRef]
- Croy, J.E.; Wuttke, D.S. Insights into the dynamics of specific telomeric single-stranded DNA recognition by Pot1pN. J. Mol. Biol. 2009, 387, 935–948. [Google Scholar] [CrossRef]
- Latham, M.P.; Hanson, P.; Brown, D.J.; Pardi, A. Comparison of alignment tensors generated for native tRNA(Val) using magnetic fields and liquid crystalline media. J. Biomol. NMR 2008, 40, 83–94. [Google Scholar] [CrossRef]
- Bailor, M.H.; Musselman, C.; Hansen, A.L.; Gulati, K.; Patel, D.J.; Al-Hashimi, H.M. Characterizing the relative orientation and dynamics of RNA A-form helices using NMR residual dipolar couplings. Nat. Protoc. 2007, 2, 1536–1546. [Google Scholar] [CrossRef]
- Mackeen, M.M.; Almond, A.; Deschamps, M.; Cumpstey, I.; Fairbanks, A.J.; Tsang, C.; Rudd, P.M.; Butters, T.D.; Dwek, R.A.; Wormald, M.R. The conformational properties of the Glc3Man unit suggest conformational biasing within the chaperone-assisted glycoprotein folding pathway. J. Mol. Biol. 2009, 387, 335–347. [Google Scholar] [CrossRef]
- Zhuang, T.; Lee, H.-S.; Imperiali, B.; Prestegard, J.H. Structure determination of a Galectin-3-carbohydrate complex using paramagnetism-based NMR constraints. Protein Sci. 2008, 17, 1220–1231. [Google Scholar] [CrossRef]
- Zhuang, T.; Leffler, H.; Prestegard, J.H. Enhancement of bound-state residual dipolar couplings: Conformational analysis of lactose bound to Galectin-3. Protein Sci. 2006, 15, 1780–1790. [Google Scholar] [CrossRef]
- Prestegard, J.H.; Yi, X. NMR Spectroscopy and Computer Modeling of Carbohydrates; Vliegenthar, J.F.G., Woods, R.J., Eds.; American Chemical Society: Washington, DC, USA, 2006; Volume 930, pp. 40–59. [Google Scholar]
- Teriete, P.; Franzin, C.M.; Choi, J.; Marassi, F.M. Structure of the Na,K-ATPase regulatory protein FXYD1 in micelles. Biochemistry 2007, 46, 6774–6783. [Google Scholar] [CrossRef]
- Gong, X.-M.; Franzin, C.; Thai, K.; Yu, J.; Marassi, F.M. Nuclear magnetic resonance structural studies of membrane proteins in micelles and bilayers. Methods Mol. Biol. 2007, 400, 515–529. [Google Scholar] [CrossRef]
- Valentine, K.G.; Pometun, M.S.; Kielec, J.M.; Baigelman, R.E.; Staub, J.K.; Owens, K.L.; Wand, A.J. Magnetic susceptibility-induced alignment of proteins in reverse micelles. J. Am. Chem. Soc. 2006, 128, 15930–15931. [Google Scholar]
- Franzin, C.M.; Yu, J.; Thai, K.; Choi, J.; Marassi, F.M. Correlation of gene and protein structures in the FXYD family proteins. J. Mol. Boil. 2005, 354, 743–750. [Google Scholar] [CrossRef]
- Bhattacharya, A.; Kurochkin, A.V.; Yip, G.N.B.; Zhang, Y.; Bertelsen, E.B.; Zuiderweg, E.R.P. Allostery in Hsp70 chaperones is transduced by subdomain rotations. J. Mol. Biol. 2009, 388, 475–490. [Google Scholar] [CrossRef]
- Seidel, R.D.; Zhuang, T.; Prestegard, J.H. Bound-state residual dipolar couplings for rapidly exchanging ligands of His-tagged proteins. J. Am. Chem. Soc. 2007, 129, 4834–4839. [Google Scholar] [CrossRef]
- Prestegard, J.H.; Al-Hashimi, H.M.; Tolman, J.R. NMR structures of biomolecules using field oriented media and residual dipolar couplings. Q. Rev. Biophys. 2000, 33, 371–424. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fahim, A.; Mukhopadhyay, R.; Yandle, R.; Prestegard, J.H.; Valafar, H. Protein Structure Validation and Identification from Unassigned Residual Dipolar Coupling Data Using 2D-PDPA. Molecules 2013, 18, 10162-10188. https://doi.org/10.3390/molecules180910162
Fahim A, Mukhopadhyay R, Yandle R, Prestegard JH, Valafar H. Protein Structure Validation and Identification from Unassigned Residual Dipolar Coupling Data Using 2D-PDPA. Molecules. 2013; 18(9):10162-10188. https://doi.org/10.3390/molecules180910162
Chicago/Turabian StyleFahim, Arjang, Rishi Mukhopadhyay, Ryan Yandle, James H. Prestegard, and Homayoun Valafar. 2013. "Protein Structure Validation and Identification from Unassigned Residual Dipolar Coupling Data Using 2D-PDPA" Molecules 18, no. 9: 10162-10188. https://doi.org/10.3390/molecules180910162
APA StyleFahim, A., Mukhopadhyay, R., Yandle, R., Prestegard, J. H., & Valafar, H. (2013). Protein Structure Validation and Identification from Unassigned Residual Dipolar Coupling Data Using 2D-PDPA. Molecules, 18(9), 10162-10188. https://doi.org/10.3390/molecules180910162