3. Experimental
The 1H and 13C-NMR spectra were recorded on a Bruker Avance DPX300 with tetramethylsilane as an internal standard, all NMR spectra were obtained using CDCl3 as the solvent unless otherwise noted. GC-Mass was carried out on a 6890N GC-Agilent 5973N. Melting points were measured on an Cole-Parmer melting point apparatus and are not corrected. Commercially available compounds were used in this work without further purification. The solvent tetrahydrofuran (THF) was dried by distillation from sodium and benzophenone. Substrates 4 were synthesized by our lab, while the others were commercially available. Column chromatography was performed using silica gel (200–300 mesh). TLC was performed on GF254 silica gel plates (Qingdao Haiyang Co., Ltd, Qingdao, China). NMR spectra were recorded on a Bruker Avance DPX300 spectrometer (300 MHz for 1H and 75 MHz for 13C) with tetramethylsilane as the internal standard.
(Z)-4-Hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-2,6,6-trimethylcyclohex-2-enone (3a): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 3a (1.08 g, 87%) as a red-brown oil. 1H-NMR (CDCl3, 25 °C): δ 5.94–5.91 (m, 1H), 5.90–5.84 (m, 1H), 4.41 (s, 1H), 4.26–4.24 (d, J = 6.7, 2H), 3.49 (s, 1H), 2.53–2.39 (m, 2H), 2.14 (s, 3H), 1.87 (s, 3H), 1.23 (s, 3H), 1.11 (s, 3H). 13C-NMR (CDCl3, 25 °C) : δ 199.0, 136.7, 125.5, 119.8, 92.7, 85.1, 74.4, 60.6, 41.7, 25.0, 22.7, 19.7.
4-Hydroxy-2,6,6-trimethyl-4-((trimethylsilyl)ethynyl)cyclohex-2-enone (3b): n-BuLi (2.1 mL, 5 mmol, 2.4 mol/L) was added dropwise to a solution of ethynyltrimethylsilane (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 5:1) to afford 3b (1.14 g, 91%) as a white solid, mp: 76–78 °C. 1H-NMR (CDCl3, 25 °C) δ 5.77 (d, J = 1.1 Hz, 1H), 2.71 (s, 1H), 2.38 (q, J1 = 16.4 Hz, J2 = 37.2 Hz, 2H), 2.04 (d, J = 1.3 Hz, 3H), 1.12 (s, 3H), 1.06 (s, 3H), 0.10 (s, 9H). 13C-NMR (CDCl3, 25 °C) δ 198.4, 126.0, 103.9, 92.2, 74.6, 58.2, 49.0, 41.5, 24.9, 19.5, 18.2, -0.3.MS:273.1 (M + Na+).
Ethyl 3-(1′-hydroxy-3′,5′,5′-trimethyl-4′-oxocyclohex-2′-en-1′-yl)propiolate (3c): LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of ethylpropiolate (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 10:1) to afford 3c (1.19 g, 95%) as a white solid, 72–73 °C. 1H-NMR (CDCl3, 25 °C) δ 5.91 (d, J = 1.3 Hz, 1H), 4.25 (q, J1 = 7.1 Hz, J2 = 14.2 Hz, 2H), 3.52 (s, 1H), 2.47 (t, J = 12.0 Hz, 2H), 2.14 (d, J = 1.4 Hz, 3H), 1.32 (t, J = 7.2 Hz, 3H), 1.24 (s, 3H), 1.12 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ. 197.7, 158.2, 152.8, 126.8, 85.0, 78.4, 74.4, 62.3, 48.7, 42.0, 24.9, 21.7, 19.6, 13.8. MS:273.0 (M + Na+).
Methyl 3-(1′-hydroxy-3′,5′,5′-trimethyl-4′-oxocyclohex-2′-en-1′-yl)propiolate (3d): LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of methyl propiolate (0.42 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 9:1) to afford 3d (1.08 g, 92%) as a red-brown oil. 1H-NMR (CDCl3, 25 °C) δ 5.92 (d, J = 1.3 Hz, 1H), 4.22 (s, 1H), 3.79 (s, 3H), 2.49 (q, J = 6.6 Hz, 2H), 2.15 (d, J = 1.3 Hz, 3H), 1.25 (s, 3H), 1.13 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 198.0, 153.2, 126.6, 85.7, 77.4, 74.2, 52.8, 41.9, 24.8, 19.5. MS:258.9 (M + Na+).
4-(3′,3′-Dimethylbut-1′-yn-1′-yl)-4-hydroxy-2,6,6-trimethylcyclohex-2-enone (3e): n-BuLi (2.1 mL, 5 mmol, 2.4 mol/L) was added dropwise to a solution of 3,3-dimethyl-1-butyne (0.41 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at –78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether /EtOAc, 10:1) to afford 3e (1.09 g, 93%) as a red-brown oil. 1H-NMR (CDCl3, 25 °C) δ 5.81 (d, J = 1.0 Hz, 1H), 3.0 (s, 1H), 2.43 (d, J = 8.5 Hz, 2H), 2.11 (d, J = 1.2 Hz, 3H), 1.22 (s, 9H), 1.17 (s, 3H), 1.09 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 198.6, 125.4, 95.9, 74.0, 41.5, 30.6, 27.4, 24.9, 19.6. MS:257.0 (M + Na+).
4-Hydroxy-2,6,6-trimethyl-4-(phenylethynyl)cyclohex-2-enone (3f): n-BuLi (2.1 mL, 5 mmol, 2.4 mol/L) was added dropwise to a solution of phenylacetylene (0.51 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4-ketoisophorone 1 (0.76 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 10:1) to afford 3f (1.19 g, 94%) as a red-brown oil. 1H-NMR (CDCl3, 25 °C) δ 7.42–7.38 (m, 2H), 7.32–7.26 (m, 3H), 5.89 (d, J = 1.3 Hz, 1H), 3.88 (s, 1H), 2.50 (q, J1 = 19.2 Hz, J2 = 23.2 Hz, 2H), 2.16 (d, J = 1.4 Hz, 3H), 1.22 (s, 3H), 1.14 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 198.7, 131.5, 128.6, 128.1, 125.6, 121.8, 87.7, 86.7, 76.5, 74.4, 49.1, 41.8, 24.9, 21.7, 19.6. MS:277.0 (M + Na+).
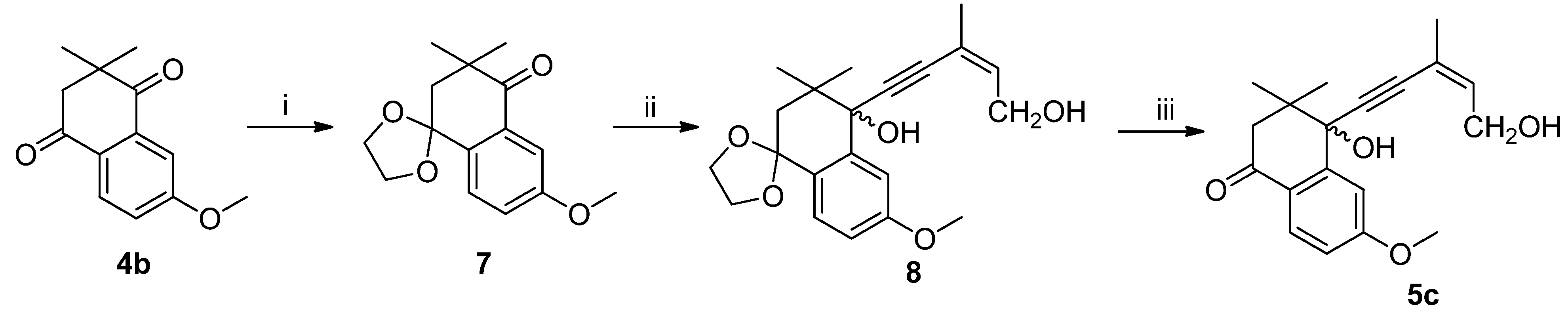
(Z)-4-hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-6-methoxy-3,3-dimethyl-3,4-dihydronaphthalen-1(2H)-one (5c): To a stirred solution of 4b (2.0 g, 9.2 mmol), ethylene glycol (1.14 g, 18.4 mmol), methylorthoformate (1.46 g, 13.8 mmol) and p-TSA (0.15g, 0.87 mmol) in 150 mL diethyl ether under an atmosphere of argon at r.t for 16h. The reaction was quenched by slow addition of aqueous NaHCO3 solution (5 mL) and extracted with diethyl ether (3 × 50 mL). The organic phase was washed with water (2 × 50 mL) and dried over anhydrous Na2SO4 and filtered, filtrate concentrated under reduced pressure. The residue was subjected to silica gel chromatography using PE and EtOAc (6:1) as eluant to afford ketal 7 as a yellow oil (2.05 g, yield 85%). 7: 1H-NMR (CDCl3, 25 °C) δ 7.49–7.46 (m, 2H), 7.14 (dd, J1 = 2.8 Hz, J2 = 8.5 Hz, 1H), 4.22–4.08 (m, 4H), 3.84 (s, 3H), 2.20 (s, 2H), 1.30 (s, 6H). 13C-NMR (CDCl3, 25 °C) δ 201.9, 160.2, 135.2, 132.3, 127.3, 121.5, 109.2, 105.0, 64.9, 55.3, 45.3, 42.4, 26.6.
n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 7 (1.31 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 8 (1.54 g, 86%) as a yellow oil. 8: 1H-NMR (300 MHz, CDCl3, 25 °C) δ 7.39 (d, J = 7.5 Hz, 1H), 7.31 (d, J = 2.6 Hz, 1H), 6.89 (dd, J1 = 2.6 Hz, J2 = 8.6 Hz, 1H), 5.85 (t, J = 1.4 Hz, 1H), 4.22 (d, J = 6.8 Hz, 2H), 4.17-4.05 (m, 4H), 3.81 (s, 3H), 3.08 (s, 1H), 2.22 (d, J = 14.2 Hz, 1H), 2.03 (d, J = 14.2 Hz, 1H), 1.87 (d, J = 0.9 Hz, 3H), 1.15 (s, 3H), 1.12 (s, 3H). 13C-NMR (75 MHz, CDCl3, 25 °C) δ 160.1, 141.4, 136.1, 128.7, 127.6, 120.3, 120.3, 114.9, 111.0, 105.8, 95.2, 85.0, 75.1, 64.6, 60.9, 55.2, 42.9, 39.2, 25.2, 23.5, 22.9.
To a stirred solution of 8 (1.50 g, 4.2 mmol) in acetone (30 mL), p-TSA (60 mg) was added, and the mixture was stirred for 1 h at room temperature. A saturated NaHCO3 solution (5 mL) was added, and it was extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (3 × 20 mL), dried over anhydrous Na2SO4 and concentrated to a residue, which was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 5c (1.17 g, 89%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 7.99 (d, J = 8.6 Hz, 1H), 7.39 (d, J = 2.5 Hz, 1H), 6.92 (dd, J1 = 2.5 Hz, J2 = 8.7 Hz, 1H), 5.94-5.89 (m, 1H), 4.29 (d, J = 6.7 Hz, 2H), 3.90 (s, 3H), 2.86-2.63 (m, 3H), 1.90 (s, 3H), 1.17 (s, 6H). 13C-NMR (CDCl3, 25 °C) δ 195.7, 164.4, 136.6, 130.8, 129.5, 128.8, 123.6, 120.0, 114.2, 94.2, 85.9, 74.8, 65.5, 61.2, 55.5, 41.5, 30.5, 24.9, 22.9.
(Z)-4-Hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-2,2-dimethyl-3,4-dihydronaphthalen-1(2H)-one (6a): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4a (0.94 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at –78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 6a (1.24 g, 87%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.02 (d, J = 9.2 Hz, 1H), 7.90 (d, J = 8.9 Hz, 1H), 7.63 (t, J = 7.4 Hz, 1H), 7.44 (t, J = 7.6 Hz, 1H), 5.94–5.89 (m, 1H), 4.28 (d, J = 6.6 Hz, 2H), 2.94 (d, J = 17.4 Hz, 2H), 2.64 (d, J = 17.4 Hz, 2H), 1.91 (d, J = 1.1 Hz, 3H), 1.77 (s, 2H), 1.21 (s, 3H), 1.17 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 197.1, 144.0, 136.5, 134.3, 130.0, 128.6, 127.0, 126.9, 120.0, 94.3, 85.9, 74.7, 61.25, 48.5, 41.4, 25.0, 23.1, 22.9.
Ethyl 3-(1′-hydroxy-3′,3′-dimethyl-4′-oxo-1′,2′,3′,4′-tetrahydronaphthalen-1′-yl)propiolate 6b: LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of ethylpropiolate (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4a (0.94 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 10:1) to afford 6b (1.34 g, 94%) as a red-brown liquid. 1H-NMR (CDCl3, 25 °C) δ 8.00 (dd, J1 = 1.0 Hz, J2 = 7.7 Hz, 1H), 7.89 (d, J = 7.5 Hz, 1H), 7.62 (dt, J1 = 1.3 Hz, J2 = 7.5 Hz, 1H), 4.22 (q, J1 = 7.1 Hz, J2 = 14.2 Hz, 2H), 3.97 (s, 1H), 2.90 (d, J = 17.3 Hz, 1H), 2.63 (d, J = 17.3 Hz, 1H), 1.29 (t, J = 7.1 Hz, 3H), 1.22 (s, 3H), 1.18 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 196.7, 153.0, 142.2, 134.4, 129.8, 129.0, 127.3, 126.9, 86.4, 79.0, 74.1, 62.2, 41.4, 24.6, 13.8. MS:309.0 (M+Na+).
(Z)-4-Hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-7-methoxy-2,2-dimethyl-3,4-dihydronaphthalen-1(2H)-one (6c): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at –78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at –78 °C. A solution of 4b (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 6c (1.35 g, 86%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 7.96 (d, J = 8.7 Hz, 1H), 7.39 (d, J = 2.4 Hz, 1H), 6.89 (dd, J1 = 2.4 Hz, J2 = 8.7 Hz, 1H), 5.91–5.87 (m, 1H), 4.26 (d, J = 6.4 Hz, 2H), 3.87 (s, 3H), 2.78-2.61 (m, 3H), 1.87 (s, 3H), 1.15 (s, 6H). 13C-NMR (CDCl3, 25 °C) δ 196.2, 164.3, 146.8, 136.4, 129.3, 123.4, 119.9, 114.0, 111.8, 94.1, 85.6, 74.4, 60.8, 55.4, 48.2, 41.4, 30.7, 24.8, 22.8.
(Z)-4-Hydroxy-4-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-6-methoxy-2,2-dimethyl-3,4-dihydronaphthalen-1(2H)-one (6d): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at –78 °C. A solution of 4c (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at –78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether /EtOAc, 2:1) to afford 6d (1.32 g, 84%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 7.81 (d, J = 8.6 Hz, 1H), 7.46 (d, J = 2.8 Hz, 1H), 7.13 (dd, J1 = 2.8 Hz, J2 = 8.6 Hz, 1H), 5.90–5.85 (m, 1H), 4.27 (d, J = 6.7 Hz, 2H), 3.83 (s, 3H), 2.88 (d, J = 16.8 Hz, 1H), 2.58 (d, J = 16.8 Hz, 1H), 1.88 (d, J = 0.78 Hz, 3H), 1.20 (s, 3H), 1.13 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 197.4, 159.5, 136.7, 136.2, 131.0, 128.9, 121.5, 120.0, 109.3, 94.4, 85.4, 74.1, 60.9, 55.4, 48.2, 41.5, 24.9, 23.2, 22.9.
4-Hydroxy-6-methoxy-2,2-dimethyl-4-((trimethylsilyl)ethynyl)-3,4-dihydronaphthalen-1(2H)-one (6e): n-BuLi (2.1 mL, 5 mmol, 2.4 mol/L) was added dropwise to a solution of ethynyltrimethylsilane (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at –78 °C. A solution of 4c (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 6:1) to afford 6e (1.39 g, 88%) as a red-brown liquid. 1H-NMR (CDCl3, 25 °C) δ 7.84 (d, J = 8.5 Hz, 1H), 7.48 (d, J = 2.7 Hz, 1H), 7.16 (dd, J1 = 2.7 Hz, J2 = 8.6 Hz, 1H), 3.85 (s, 3H), 2.92 (d, J = 16.6 Hz, 1H), 2.56 (d, J = 16.6 Hz, 1H), 2.53 (s, 1H), 1.22 (s, 3H), 1.13 (s, 3H), 0.20 (s, 9H). 13C-NMR (CDCl3, 25 °C) δ 197.1, 159.7, 131.2, 129.0, 121.5, 109.3, 105.4, 74.2, 55.5, 41.3, 24.8, −0.2. MS: 339.0 (M+Na+).
Ethyl 3-(1′-hydroxy-7′-methoxy-3′,3′-dimethyl-4′-oxo-1′,2′,3′,4′-tetrahydronaphthalen-1′-yl)propiolate (6f): LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of ethylpropiolate (0.49 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4c (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 8:1) to afford 6f (1.48 g, 94%) as a red-brown liquid. 1H-NMR (CDCl3, 25 °C) δ 7.79 (d, J = 8.6 Hz, 1H), 7.48 (d, J = 2.8 Hz, 1H), 7.15 (dd, J1 = 2.8 Hz, J2 = 8.6 Hz, 1H), 4.23 (q, J1 = 7.1 Hz, J2 = 14.2 Hz, 2H), 3.85 (s, 3H), 3.50 (d, J = 5.7 Hz, 1H), 2.92 (d, J = 17.1 Hz, 1H), 2.64 (d, J = 17.1 Hz, 1H), 1.31 (t, J = 7.1 Hz, 3H), 1.25 (s, 3H), 1.17 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 196.5, 160.0, 153.1, 134.7, 131.2, 129.0, 121.6, 109.7, 86.5, 78.9, 73.9, 62.2, 55.5, 41.6, 24.7, 23.3, 13.8. MS:339.0 (M+Na+).
Methyl 3-(1′-hydroxy-7′-methoxy-3′,3′-dimethyl-4′-oxo-1′,2′,3′,4′-tetrahydronaphthalen-1′-yl)propiolate (6g): LDA (2.5 mL, 5 mmol, 2.0 mol/L) was added dropwise to a solution of methyl propiolate (0.42 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4c (1.09 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 9:1) to afford 6g (1.37 g, 91%) as a red-brown liquid. 1H-NMR (CDCl3, 25 °C) δ 7.78 (d, J = 8.6 Hz, 1H), 7.50 (d, J = 2.8 Hz, 1H), 7.16 (dd, J1 = 2.8 Hz, J2 = 8.6 Hz, 1H), 3.86 (s, 3H), 3.79 (s, 3H), 3.03 (s, 1H), 2.93 (d, J = 17.1 Hz, 1H), 2.60 (d, J = 17.1 Hz, 1H), 1.23 (s, 3H), 1.16 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 196.3, 160.2, 153.5, 134.5, 131.2, 128.9, 121.7, 109.8, 86.9, 78.7, 74.1, 55.6, 52.8, 48.0, 41.6, 24.7, 23.1. MS:325.0 (M+Na+).
(Z)-7-Bromo-4-hydroxy-4-(5′-hydroxy-3-methylpent-3′-en-1′-yn-1′-yl)-2,2-dimethyl-3,4-dihydronaphthalen-1(2H)-one (6h): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4d (1.33 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 2:1) to afford 6h (1.61 g, 89%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.06 (d, J = 1.8 Hz, 1H), 7.85 (d, J = 8.3 Hz, 1H), 7.55 (dd, J1 = 1.9 Hz, J2 = 8.3 Hz, 1H), 5.93–5.89 (m, 1H), 4.26 (d, J = 6.7 Hz, 2H), 4.03 (s, 1H), 2.81 (d, J = 24.1 Hz, 1H), 2.64 (d, J = 24.1 Hz, 1H), 2.59 (s, 1H), 1.88 (s, 3H), 1.16 (s, 6H). 13C-NMR (CDCl3, 25 °C) δ 196.6, 146.1, 136.6, 131.7, 130.4, 129.4, 128.7, 128.5, 120.1, 93.6, 86.3, 74.0, 60.8, 48.5, 41.5, 29.5, 24.8, 22.8, 22.7.
(Z)-5-Hydroxy-5-(5′-hydroxy-3′-methylpent-3′-en-1′-yn-1′-yl)-2,7,7-trimethyl-6,7-dihydroquinolin-8(5H)-one (6i): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4e (1.01 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 1:1) to afford 6i (1.40 g, 94%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.17 (d, J = 7.9 Hz, 1H), 7.28 (d, J = 7.9 Hz, 1H), 5.85–5.83 (m, 1H), 5.34 (s, 1H), 4.16 (d, J = 6.5 Hz, 2H), 2.99 (d, J = 17.5 Hz, 1H), 2.59 (d, J = 17.5 Hz, 1H), 2.65 (s, 3H), 1.79 (s, 3H), 1.39 (s, 3H), 0.96 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 195.8, 163.5, 160.8, 136.8, 135.4, 123.4, 122.5, 119.4, 94.3, 85.2, 73.6, 60.8, 50.2, 40.7, 24.6, 24.6, 22.6, 20.9.
(Z)-5-Hydroxy-5-(5′-hydroxy-3-methylpent-3′-en-1′-yn-1′-yl)-7,7-dimethyl-2-phenyl-6,7-dihydroquinolin-8(5H)-one (6j): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4f (1.32 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 1:1) to afford 6j (1.73 g, 96%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.35 (d, J = 8.1 Hz, 1H), 8.11 (dd, J1 = 2.7 Hz, J2 = 7.9 Hz, 2H), 7.87 (d, J = 8.2 Hz, 1H), 7.55–7.51 (m, 3H), 5.84–5.79 (m, 1H), 5.33 (s, 1H), 4.16 (s, 2H), 3.07 (d, J = 17.5 Hz, 1H), 2.62 (d, J = 17.5 Hz, 1H), 1.80 (d, J = 1.1 Hz, 3H), 1.45 (s, 3H), 0.99 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 195.7, 161.3, 160.5, 137.3, 136.6, 136.3, 130.6, 129.0, 127.5, 123.4, 120.2, 119.8, 94.6, 85.3, 73.8, 61.2, 50.6, 40.9, 24.8, 22.8, 21.0.
(Z)-5-Hydroxy-5-(5′-hydroxy-3-methylpent-3′-en-1′-yn-1′-yl)-7,7-dimethyl-6,7-dihydroquinolin-8(5H)-one (6k): n-BuLi (4.2 mL, 10 mmol, 2.4 mol/L) was added dropwise to a solution of (Z)-3-methylpent-2-en-4-yn-1-ol (0.48 g, 5 mmol) in THF (10 mL) at −78 °C under a N2 atmosphere, and then the mixture was stirred for 1 h at −78 °C. A solution of 4g (0.94 g, 5 mmol) in THF (5 mL) was added slowly, the mixture was stirred for 0.5 h at −78 °C, and then it was warmed to room temperature and stirred for 2 h. The reaction was quenched with saturated aqueous NH4Cl solution (10 mL) and extracted with EtOAc (3 × 30 mL). The combined organics were washed with brine (2 × 20 mL), dried over anhydrous Na2SO4 and concentrated. The residue was purified by silica gel column chromatography (petroleum ether/EtOAc, 1:2) to afford 6c (1.35 g, 95%) as a yellow oil. 1H-NMR (CDCl3, 25 °C) δ 8.77 (dd, J1 = 1.7 Hz, J2 = 4.8 Hz, 1H), 8.30 (dd, J1 = 1.7 Hz, J2 = 7.8 Hz, 1H), 7.45 (dd, J1 = 4.8 Hz, J2 = 7.8 Hz, 1H), 5.87–5.82 (m, 1H), 5.48 (s, 1H), 4.14 (d, J = 6.5 Hz, 2H), 3.00 (d, J = 17.4 Hz, 1H), 2.68 (d, J = 17.4 Hz, 1H), 3.02 (s, 1H), 1.78 (d, J = 1.2 Hz, 3H), 1.40 (d, J = 6.8 Hz, 3H), 1.01 (s, 3H). 13C-NMR (CDCl3, 25 °C) δ 196.0, 161.4, 153.1, 136.9, 135.2, 124.9, 123.6, 119.3, 93.9, 85.7, 73.9, 60.6, 50.0, 40.7, 24.6, 22.6, 21.1.













{kind=link}