18F-Labeled Peptides: The Future Is Bright


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. General 18F Radiochemistry Concepts for Peptide Labeling
2.1. Concept 1: The Prosthetic Group Approach for 18F-Radiolabeling of Peptides

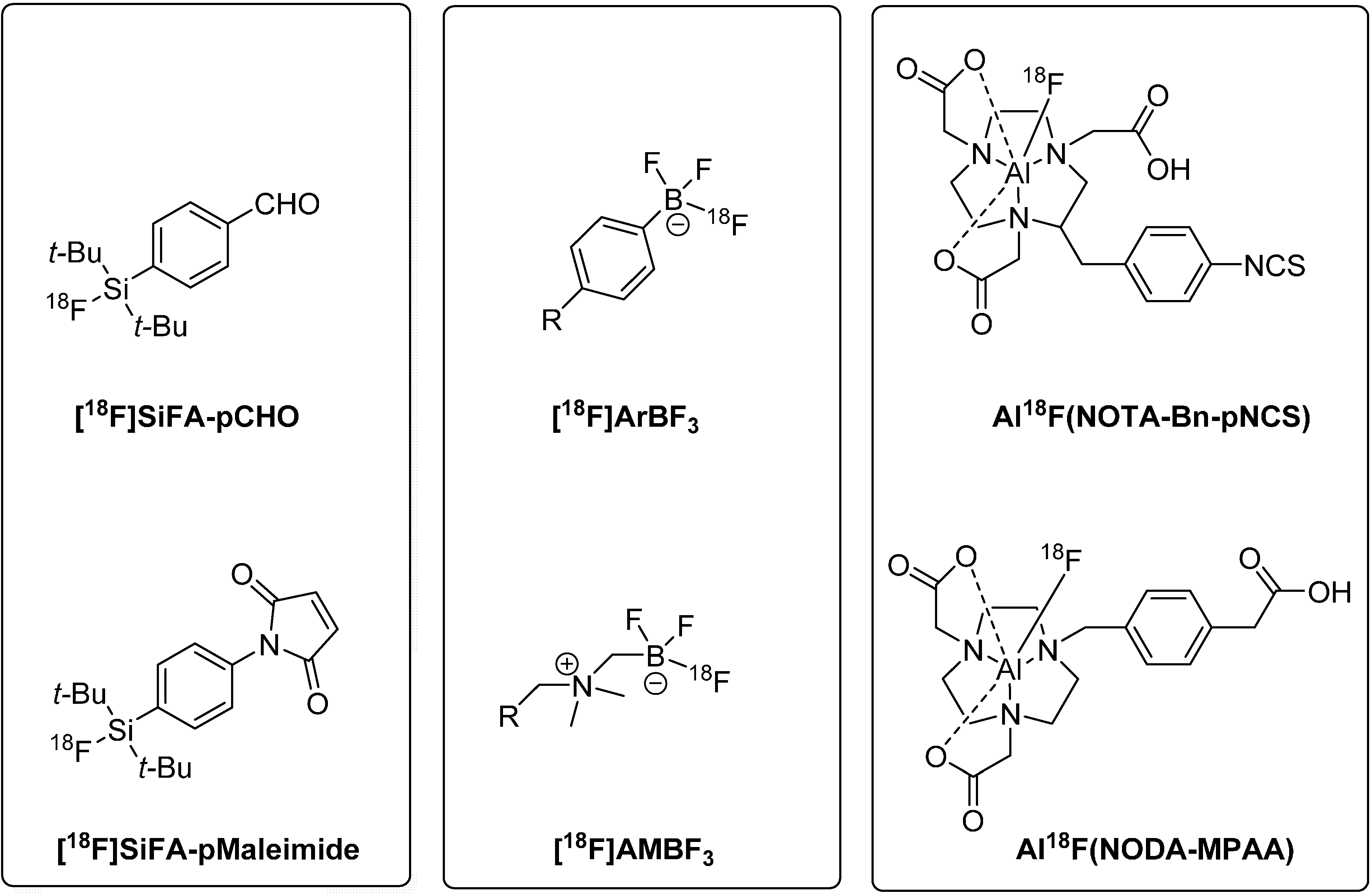
2.2. Concept 2: 18F-Radiolabeling of Peptides via [18F]Fluoride Acceptor Chemistry
2.3. Concept 3: Click Chemistry for Radiolabeling of Peptides with Fluorine-18

3. Recent Technology Advances in Peptide Labeling with 18F
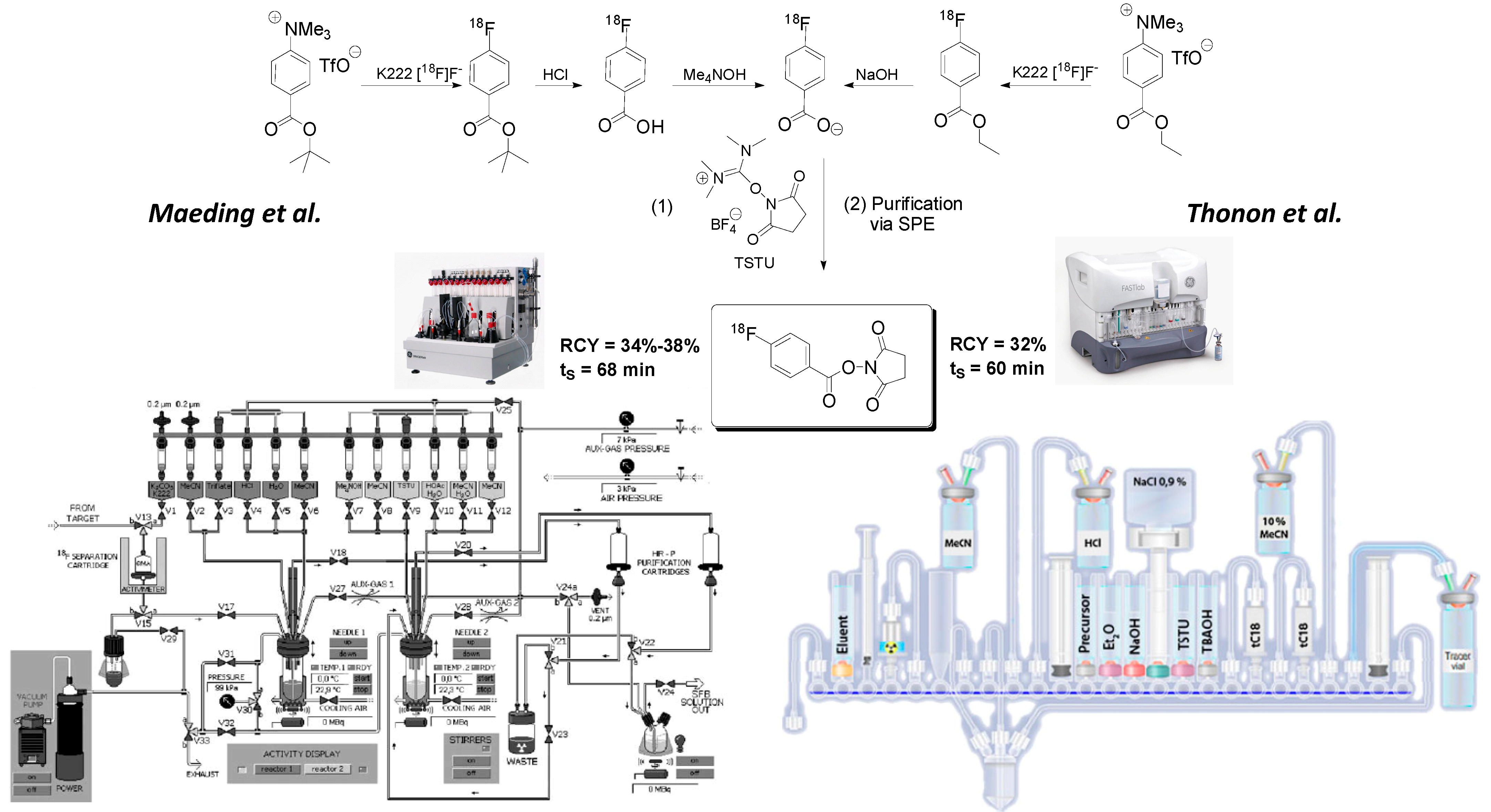
3.1. Automated Synthesis of 18F-Labeled Peptides

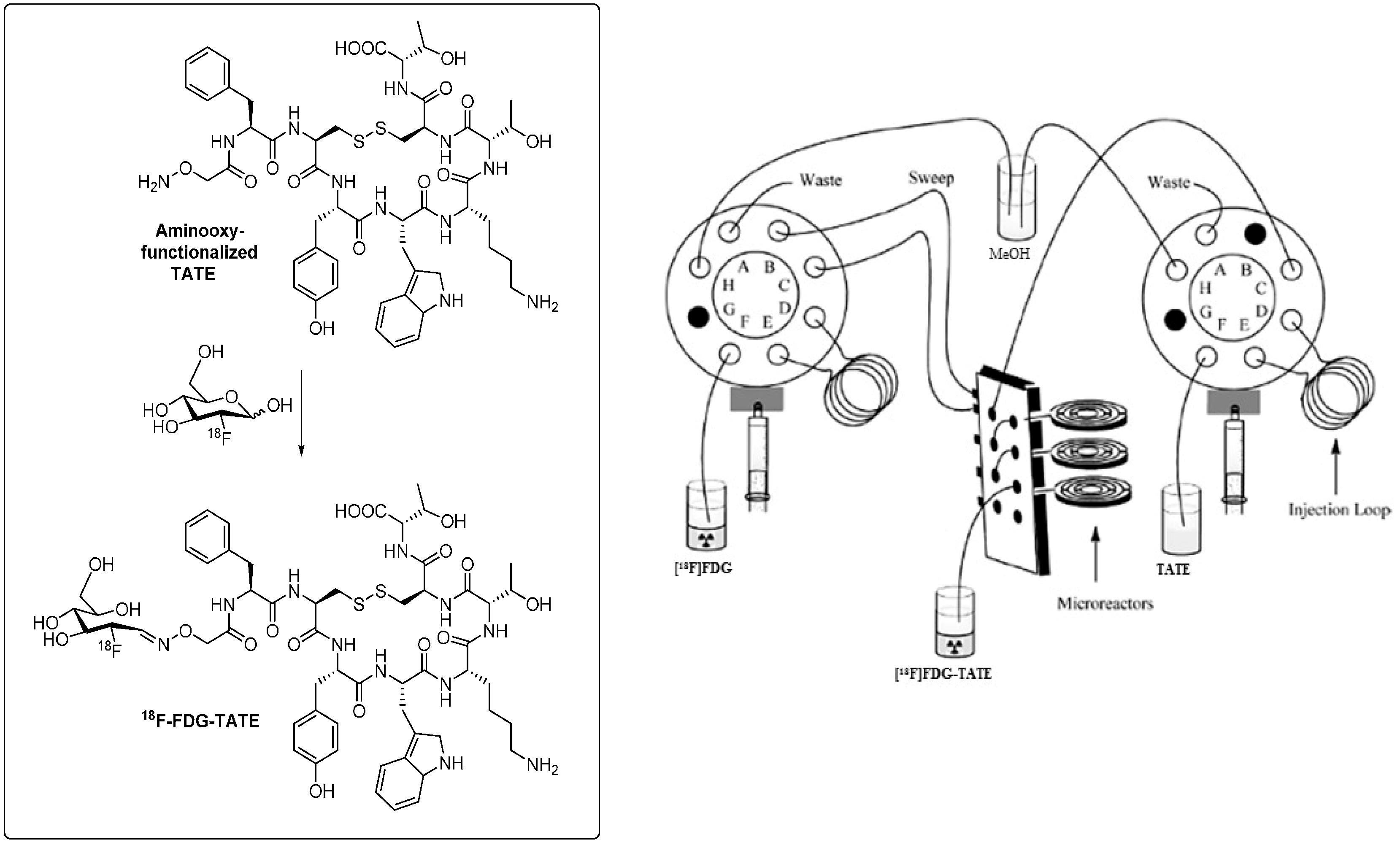
3.2. Application of Microfluidic Technology for 18F-Labeling of Peptides

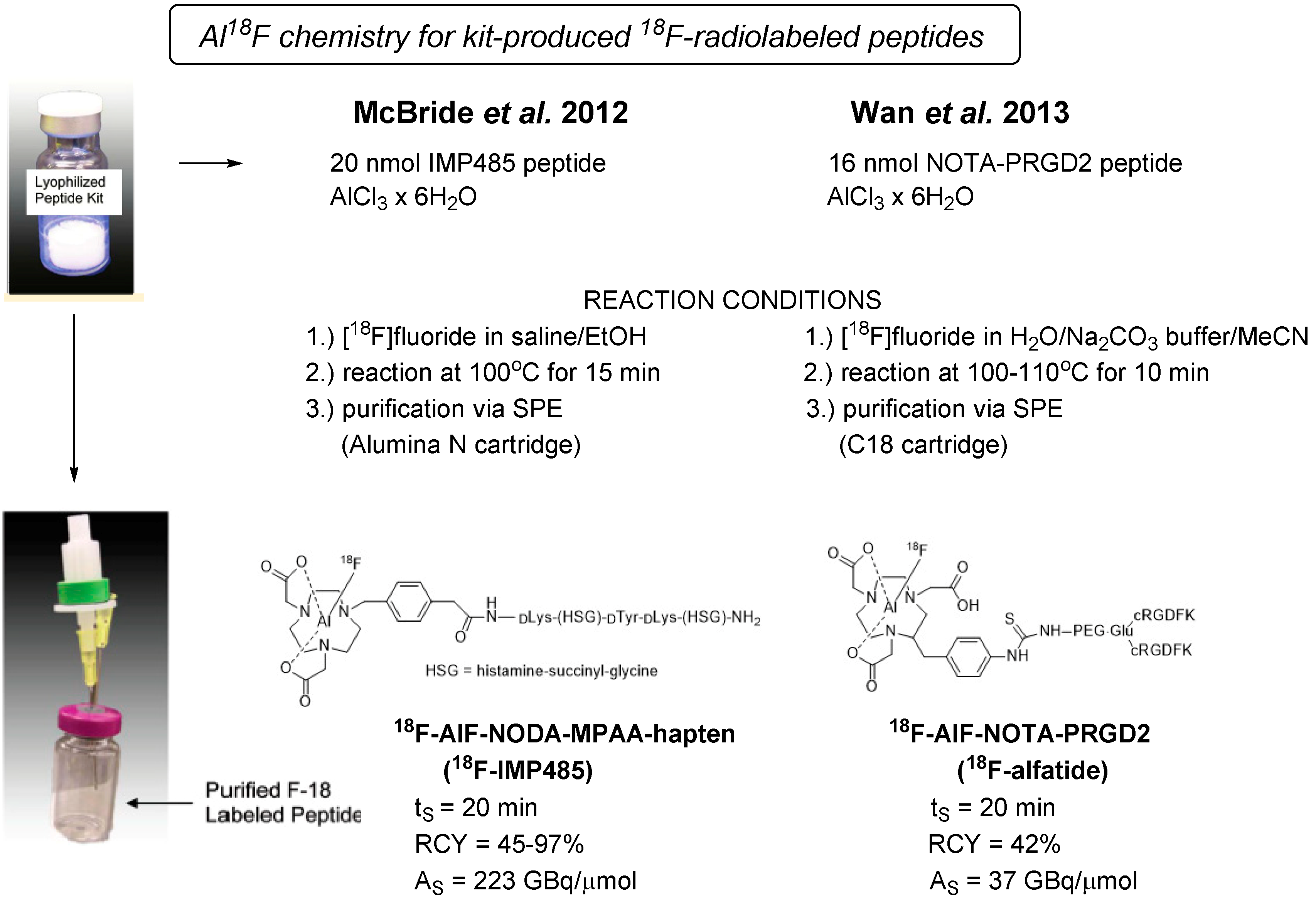
3.3. Kit-Like Preparation of 18F-Labeled Peptides

4. Challenges and Trends in Peptide Receptor-Targeted Molecular Imaging
5. Summary and Conclusions
Acknowledgments
Conflicts of Interest
References
- Hopkins, A.L.; Groom, C.R. The druggable genome. Nat. Rev. Drug Discov. 2002, 1, 727–730. [Google Scholar] [CrossRef] [PubMed]
- Rask-Andersen, M.; Masuram, S.; Schiöth, H.B. The druggable genome: Evaluation of drug targets in clinical trials suggests major shifts in molecular class and indication. Annu. Rev. Pharmacol. Toxicol. 2014, 54, 9–26. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C. Peptide receptors as molecular targets for cancer diagnosis and therapy. Endocr. Rev. 2003, 24, 389–427. [Google Scholar] [CrossRef] [PubMed]
- Olberg, D.E.; Hjuelsten, O.K. Labeling strategies of peptides with 18F for positron emission tomography. Curr. Top. Med. Chem. 2010, 10, 1669–1679. [Google Scholar] [CrossRef] [PubMed]
- Reubi, J.C.; Landolt, A.M. High density of somatostatin receptors in pituitary tumors from acromegalic patients. J. Clin. Endocrinol. Metab. 1984, 59, 1148–1151. [Google Scholar] [CrossRef] [PubMed]
- Krenning, E.P.; Bakker, W.H.; Breeman, W.A.; Koper, J.W.; Kooij, P.P.; Ausema, L.; Lameris, J.S.; Reubi, J.C.; Lamberts, S.W. Localisation of endocrine-related tumours with radioiodinated analogue of somatostatin. Lancet 1989, 1, 242–244. [Google Scholar] [CrossRef] [PubMed]
- Lasne, M.-C.; Perrio, C.; Rouden, J.; Barré, L.; Roeda, D.; Dolle, F.; Crouzel, C. Chemistry of β+-Emitting Compounds Based on Fluorine-18. Top. Curr. Chem. 2002, 222, 201–258. [Google Scholar]
- Sánchez-Crespo, A.; Andreo, P.; Larsson, S.A. Positron flight in human tissues and its influence on PET image spatial resolution. Eur. J. Nucl. Med. Mol. Imaging 2004, 31, 44–51. [Google Scholar] [CrossRef] [PubMed]
- Shively, J.E. 18F-labeling for immuno-PET: Where speed and contrast meet. J. Nucl. Med. 2007, 48, 170–172. [Google Scholar] [PubMed]
- Wester, H.J.; Schottelius, M.; Scheidhauer, K.; Meisetschläger, G.; Herz, M.; Rau, F.C.; Reubi, J.C.; Schwaiger, M. PET imaging of somatostatin receptors: Design, synthesis and preclinical evaluation of a novel 18F-labelled, carbohydrated analogue of octreotide. Eur. J. Nucl. Med. Mol. Imaging 2003, 30, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Meisetschläger, G.; Poethko, T.; Stahl, A.; Wolf, I.; Scheidhauer, K.; Schottelius, M.; Herz, M.; Wester, H.J.; Schwaiger, M. Gluc-Lys([18F]FP)-TOCA PET in patients with SSTR-positive tumors: Biodistribution and diagnostic evaluation compared with [111In]DTPA-octreotide. J. Nucl. Med. 2006, 47, 566–573. [Google Scholar] [PubMed]
- Schottelius, M.; Wester, H.J. Molecular imaging targeting peptide receptors. Methods 2009, 48, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Koopmans, K.P.; Glaudemans, A.W. Rationale for the use of radiolabelled peptides in diagnosis and therapy. Eur. J. Nucl. Med. Mol. Imaging 2012, 39 (Suppl. 1), S4–S10. [Google Scholar] [CrossRef]
- Li, X.G.; Helariutta, K.; Roivainen, A.; Jalkanen, S.; Knuuti, J.; Airaksinen, A.J. Using 5-deoxy-5-[18F]fluororibose to glycosylate peptides for positron emission tomography. Nat. Protoc. 2014, 9, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Haubner, R.; Weber, W.A.; Beer, A.J.; Vabuliene, E.; Reim, D.; Sarbia, M.; Becker, K.F.; Goebel, M.; Hein, R.; Wester, H.J.; et al. Noninvasive visualization of the activated alphavbeta3 integrin in cancer patients by positron emission tomography and [18F]Galacto-RGD. PLoS Med. 2005, 2, e70. [Google Scholar] [CrossRef] [Green Version]
- Vallabhajosula, S. Molecular Imaging: Radiopharmaceuticals for PET and SPECT, 1st ed.; Springer: Berlin/Heidelberg, Germany, 2009; Chapter 15; p. 234. [Google Scholar]
- Kuhnast, B.; Dollé, F. The challenge of labeling macromolecules with fluorine-18: Three decades of research. Curr. Radiopharm. 2010, 3, 174–201. [Google Scholar] [CrossRef]
- Liu, S.; Shen, B.; Chin, F.T.; Cheng, Z. Recent progress in radiofluorination of peptides for PET imaging. Curr.Org. Synth. 2011, 8, 584–592. [Google Scholar] [CrossRef]
- Haubner, R.; Wester, H.J.; Burkhart, F.; Senekowitsch-Schmidtke, R.; Weber, W.; Goodman, S.L.; Kessler, H.; Schwaiger, M. Glycosylated RGD-containing peptides: Tracer for tumor targeting and angiogenesis imaging with improved biokinetics. J. Nucl. Med. 2001, 42, 326–336. [Google Scholar] [PubMed]
- Haubner, R.; Wester, H.J.; Weber, W.A.; Mang, C.; Ziegler, S.I.; Goodman, S.L.; Senekowitsch-Schmidtke, R.; Kessler, H.; Schwaiger, M. Noninvasive imaging of alpha(v)beta3 integrin expression using 18F-labeled RGD-containing glycopeptide and positron emission tomography. Cancer Res. 2001, 61, 1781–1785. [Google Scholar] [PubMed]
- Vaidyanathan, G.; Zalutsky, M.R. Labeling proteins with fluorine-18 using N-succinimidyl 4-[18F]fluorobenzoate. Int. J. Rad. Appl. Instrum. B 1992, 19, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Wuest, F.; Hultsch, C.; Bergmann, R.; Johannsen, B.; Henle, T. Radiolabelling of isopeptide N epsilon-(gamma-glutamyl)-l-lysine by conjugation with N-succinimidyl-4-[18F]fluorobenzoate. Appl. Radiat. Isot. 2003, 59, 43–48. [Google Scholar] [CrossRef] [PubMed]
- Mäding, P.; Füchtner, F.; Wüst, F. Module-assisted synthesis of the bifunctional labelling agent N-succinimidyl 4-[(18)F]fluorobenzoate ([(18)F]SFB). Appl. Radiat. Isot. 2005, 63, 329–332. [Google Scholar] [CrossRef] [PubMed]
- Tang, G.; Zeng, W.B.; Yu, M.X.; Kabalka, G. Facile synthesis of N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) for protein labeling. J. Label. Compd. Radiopharm. 2008, 51, 68–71. [Google Scholar] [CrossRef]
- Thonon, D.; Goblet, D.; Goukens, E.; Kaisin, G.; Paris, J.; Aerts, J.; Lignon, S.; Franci, X.; Hustinx, R.; Luxen, A. Fully automated preparation and conjugation of N-succinimidyl 4-[18F]fluorobenzoate ([18F]SFB) with RGD peptide using a GE FASTlab™ synthesizer. Mol. Imaging Biol. 2011, 13, 1088–1095. [Google Scholar] [CrossRef] [PubMed]
- Marik, J.; Sutcliffe, J. Fully automated preparation of n.c.a. 4-[18F]fluorobenzoic acid and N-succinimidyl 4-[18F]fluorobenzoate using a Siemens/CTI chemistry process control unit (CPCU). Appl. Radiat. Isot. 2007, 65, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Sutcliffe-Goulden, J.L.; O’Doherty, M.J.; Marsden, P.K.; Hart, I.R.; Marshall, J.F.; Bansal, S.S. Rapid solid phase synthesis and biodistribution of 18F-labelled linear peptides. Eur. J. Nucl. Med. Mol. Imaging 2002, 29, 754–759. [Google Scholar] [CrossRef] [PubMed]
- Kuchar, M.; Pretze, M.; Kniess, T.; Steinbach, J.; Pietzsch, J.; Löser, R. Site-selective radiolabeling of peptides by (18)F-fluorobenzoylation with [(18F)]SFB in solution and on solid phase: A comparative study. Amino Acids 2012, 43, 1431–1443. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Wuest, M.; Krieger, S.S.; Rogers, B.E.; Friebe, M.; Bergmann, R.; Wuest, F. Synthesis and radiopharmacological evaluation of a high-affinity and metabolically stabilized 18F-labeled bombesin analogue for molecular imaging of gastrin-releasing peptide receptor-expressing prostate cancer. Nucl. Med. Biol. 2013, 40, 1025–1034. [Google Scholar] [CrossRef] [PubMed]
- White, J.B.; Hausner, S.H.; Carpenter, R.D.; Sutcliffe, J.L. Optimization of the solid-phase synthesis of [18F] radiolabeled peptides for positron emission tomography. Appl. Radiat. Isot. 2012, 70, 2720–2729. [Google Scholar] [CrossRef] [PubMed]
- Marik, J.; Hausner, S.H.; Fix, L.A.; Gagnon, M.K.J.; Sutcliffe, J.L. Solid-phase synthesis of 2-[18F]fluoropropionyl peptides. Bioconjugate Chem. 2006, 17, 1017–1021. [Google Scholar] [CrossRef]
- Hausner, S.H.; Marik, J.; Gagnon, M.K.J.; Sutcliffe, J.L. In vivo positron emission tomography (PET) imaging with an αvβ6 specific peptide radiolabeled using 18F-“click” chemistry: Evaluation and comparison with the corresponding 4-[18F]fluorobenzoyl- and 2-[18F]fluoropropionyl-peptides. J. Med. Chem. 2008, 51, 5901–5904. [Google Scholar] [CrossRef] [PubMed]
- Berndt, M.; Pietzsch, J.; Wuest, F. Labeling of low-density lipoproteins using the 18F-labeled thiol-reactive reagent N-[6-(4-[18F]fluorobenzylidene)aminooxyhexyl]maleimide. Nucl. Med. Biol. 2007, 34, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Kniess, T.; Kuchar, M.; Pietzsch, J. Automated radiosynthesis of the thiol-reactive labeling agent N-[6-(4-[18F]fluorobenzylidene)aminooxyhexyl]maleimide ([18F]FBAM). Appl. Radiat. Isot. 2011, 69, 1226–1230. [Google Scholar] [CrossRef] [PubMed]
- Kiesewetter, D.O.; Jacobson, O.; Lang, L.; Chen, X. Automated radiochemical synthesis of [18F]FBEM: A thiol reactive synthon for radiofluorination of peptides and proteins. Appl. Radiat. Isot. 2011, 69, 410–414. [Google Scholar] [CrossRef] [PubMed]
- Wuest, F.; Berndt, M.; Bergmann, R.; van den Hoff, J.; Pietzsch, J. Synthesis and application of [18F]FDG-maleimidehexyloxime ([18F]FDG-MHO): A [18F]FDG-based prosthetic group for the chemoselective18F-labeling of peptides and proteins. Bioconjugate Chem. 2008, 19, 1202–1210. [Google Scholar] [CrossRef]
- Schirrmacher, R.; Bradtmöller, G.; Schirrmacher, E.; Thews, O.; Tillmanns, J.; Siessmeier, T.; Buchholz, H.G.; Bartenstein, P.; Wängler, B.; Niemeyer, C.M.; et al. 18F-labeling of peptides by means of an organosilicon-based fluoride acceptor. Angew. Chem. Int. Ed. 2006, 45, 6047–6050. [Google Scholar] [CrossRef]
- Höhne, A.; Yu, L.; Mu, L.; Reiher, M.; Voigtmann, U.; Klar, U.; Graham, K.; Schubiger, P.A.; Ametamey, S.M. Organofluorosilanes as model compounds for 18F-labeled silicon-based PET tracers and their hydrolytic stability: Experimental data and theoretical calculations (PET = positron emission tomography). Chemistry 2009, 15, 3736–3743. [Google Scholar] [CrossRef] [PubMed]
- Höhne, A.; Mu, L.; Honer, M.; Schubiger, P.A.; Ametamey, S.M.; Graham, K.; Stellfeld, T.; Borkowski, S.; Berndorff, D.; Klar, U.; et al. Synthesis, 18F-labeling, and in vitro and in vivo studies of bombesin peptides modified with silicon-based building blocks. Bioconjugate Chem. 2008, 19, 1871–1879. [Google Scholar] [CrossRef]
- Lindner, S.; Michler, C.; Leidner, S.; Rensch, C.; Wängler, C.; Schirrmacher, R.; Bartenstein, P.; Wängler, B. Synthesis and in vitro and in vivo evaluation of SiFA-tagged bombesin and RGD peptides as tumor imaging probes for positron emission tomography. Bioconjugate Chem. 2014, 25, 738–749. [Google Scholar] [CrossRef]
- Bernard-Gauthier, V.; Wängler, C.; Schirrmacher, E.; Kostikov, A.; Jurkschat, K.; Wängler, B.; Schirrmacher, R. 18F-Labeled Silicon-Based Fluoride Acceptors: Potential Opportunities for Novel Positron Emitting Radiopharmaceuticals. Biomed. Res. Int. 2014, 2014. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Ting, R.; Harwig, C.W.; Keller, U.; Bellac, C.L.; Lange, P.F.; Inkster, J.A.H.; Schaffer, P.; Adam, M.J.; Ruth, T.J.; et al. Towards kit-like 18F-labeling of marimastat, a noncovalent inhibitor drug for in vivo PET imaging cancer associated matrix metalloproteases. Med. Chem. Commun. 2011, 2, 942–949. [Google Scholar] [CrossRef]
- Liu, Z.; Pourghiasian, M.; Bénard, F.; Pan, J.; Lin, K.S.; Perrin, D.M. Preclinical evaluation of a high-affinity 18F-trifluoroborate octreotate derivative for somatostatin receptor imaging. J. Nucl. Med. 2014, 55, 1499–1505. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Pourghiasian, M.; Radtke, M.A.; Lau, J.; Pan, J.; Dias, G.M.; Yapp, D.; Lin, K.S.; Bénard, F.; Perrin, D.M. An organotrifluoroborate for broadly applicable one-step 18F-labeling. Angew. Chem. Int. Ed. 2014. [Google Scholar] [CrossRef]
- McBride, W.J.; Sharkey, R.M.; Karacay, H.; D’Souza, C.A.; Rossi, E.A.; Laverman, P.; Chang, C.H.; Boerman, O.C.; Goldenberg, D.M. A novel method of 18F-radiolabeling for PET. J. Nucl. Med. 2009, 50, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; McBride, W.J.; Sharkey, R.M.; Eek, A.; Joosten, L.; Oyen, W.J.; Goldenberg, D.M.; Boerman, O.C. A novel facile method of labeling octreotide with (18)F-fluorine. J. Nucl. Med. 2010, 51, 454–461. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Liu, H.; Jiang, H.; Xu, Y.; Zhang, H.; Cheng, Z. One-step radiosynthesis of 18F-AlF-NOTA-RGD2 for tumor angiogenesis PET imaging. Eur. J. Nucl. Med. Mol. Imaging 2011, 38, 1732–1741. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, I.; Terry, S.Y.; McBride, W.J.; Goldenberg, D.M.; Laverman, P.; Franssen, G.M.; Oyen, W.J.; Boerman, O.C. Imaging integrin alpha-v-beta-3 expression in tumors with an 18F-labeled dimeric RGD peptide. Contrast Media Mol. Imaging 2013, 8, 238–245. [Google Scholar] [CrossRef] [PubMed]
- Dijkgraaf, I.; Franssen, G.M.; McBride, W.J.; D’Souza, C.A.; Laverman, P.; Smith, C.J.; Goldenberg, D.M.; Oyen, W.J.; Boerman, O.C. PET of tumors expressing gastrin-releasing peptide receptor with an 18F-labeled bombesin analog. J. Nucl. Med. 2012, 53, 947–952. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Hu, X.; Liu, H.; Bu, L.; Ma, X.; Cheng, K.; Li, J.; Tian, M.; Zhang, H.; Cheng, Z. A comparative study of radiolabeled bombesin analogs for the PET imaging of prostate cancer. J. Nucl. Med. 2013, 54, 2132–2138. [Google Scholar] [CrossRef] [PubMed]
- Mamat, C.; Ramenda, T.; Wuest, F. Application of click chemistry for the synthesis of radiotracers for molecular imaging. Mini Rev. Org. Chem. 2009, 6, 21–34. [Google Scholar] [CrossRef]
- Wängler, C.; Schirrmacher, R.; Bartenstein, P.; Wängler, B. Click-chemistry reactions in radiopharmaceutical chemistry: Fast & easy introduction of radiolabels into biomolecules for in vivo imaging. Curr. Med. Chem. 2010, 17, 1092–1116. [Google Scholar] [CrossRef] [PubMed]
- Pretze, M.; Pietzsch, D.; Mamat, C. Recent trends in bioorthogonal click-radiolabeling reactions using fluorine-18. Molecules 2013, 18, 8618–8665. [Google Scholar] [CrossRef] [PubMed]
- Kolb, H.C.; Finn, M.G.; Sharpless, K.B. Click chemistry: Diverse chemical function from a few good reactions. Angew. Chem. Int. Ed. 2001, 40, 2004–2021. [Google Scholar] [CrossRef]
- Rostovtsev, V.V.; Green, L.G.; Fokin, V.V.; Sharpless, K.B. A stepwise huisgen cycloaddition process: Copper(I)-catalyzed regioselective “ligation” of azides and terminal alkynes. Angew. Chem. Int. Ed. 2002, 41, 2596–2599. [Google Scholar] [CrossRef]
- Marik, J.; Sutcliffe, J.L. Click for PET: Rapid preparation of [18F]fluoropeptides using CuI catalyzed 1,3-dipolar cycloaddition. Tetrahedron Lett. 2006, 47, 6681–6684. [Google Scholar] [CrossRef]
- Ramenda, T.; Bergmann, R.; Wuest, F. Synthesis of 18F-labeled neurotensin(8–13) via copper-mediated 1,3-dipolar [3+2] cycloaddition reaction. Lett. Drug Des. Discov. 2007, 4, 279–285. [Google Scholar] [CrossRef]
- Glaser, M.; Arstad, E. “Click labeling” with 2-[18F]fluoroethylazide for positron emission tomography. Bioconjugate Chem. 2007, 18, 989–993. [Google Scholar] [CrossRef]
- Thonon, D.; Kech, C.; Paris, J.; Lemaire, C.; Luxen, A. New strategy for the preparation of clickable peptides and labeling with 1-(azidomethyl)-4-[(18)F]-fluorobenzene for PET. Bioconjugate Chem. 2009, 20, 817–823. [Google Scholar] [CrossRef]
- Li, Z.B.; Wu, Z.; Chen, K.; Chin, F.T.; Chen, X. Click chemistry for (18)F-labeling of RGD peptides and microPET imaging of tumor integrin alphavbeta3 expression. Bioconjugate Chem. 2007, 18, 1987–1994. [Google Scholar] [CrossRef]
- Maschauer, S.; Einsiedel, J.; Haubner, R.; Hocke, C.; Ocker, M.; Hübner, H.; Kuwert, T.; Gmeiner, P.; Prante, O. Labeling and glycosylation of peptides using click chemistry: A general approach to (18)F-glycopeptides as effective imaging probes for positron emission tomography. Angew. Chem. Int. Ed. 2010, 49, 976–979. [Google Scholar] [CrossRef]
- Li, Y.; Liu, Z.; Harwig, C.W.; Pourghiasian, M.; Lau, J.; Lin, K.S.; Schaffer, P.; Benard, F.; Perrin, D.M. (18)F-click labeling of a bombesin antagonist with an alkyne-(18)F-ArBF(3) (−): In vivo PET imaging of tumors expressing the GRP-receptor. Am. J. Nucl. Med. Mol. Imaging 2013, 3, 57–70. [Google Scholar] [PubMed]
- Arumugam, S.; Chin, J.; Schirrmacher, R.; Popik, V.V.; Kostikov, A.P. [18F]azadibenzocyclooctyne ([18F]ADIBO): A biocompatible radioactive labeling synthon for peptides using catalyst free [3+2] cycloaddition. Bioorg. Med. Chem. Lett. 2011, 21, 6987–6991. [Google Scholar] [CrossRef] [PubMed]
- Hausner, S.H.; Carpenter, R.D.; Bauer, N.; Sutcliffe, J.L. Evaluation of an integrin αvβ6-specific peptide labeled with [18F]fluorine by copper-free, strain-promoted click chemistry. Nucl. Med. Biol. 2013, 40, 233–239. [Google Scholar] [CrossRef] [PubMed]
- Campbell-Verduyn, L.S.; Mirfeizi, L.; Schoonen, A.K.; Dierckx, R.A.; Elsinga, P.H.; Feringa, B.L. Strain-promoted copper-free “click” chemistry for 18F radiolabeling of bombesin. Angew. Chem. Int. Ed. 2011, 50, 11117–11120. [Google Scholar] [CrossRef]
- Selvaraj, R.; Liu, S.; Hassink, M.; Huang, C.W.; Yap, L.P.; Fox, J.M.; Li, Z.; Conti, P.S. Tetrazine-trans-cyclooctene ligation for the rapid construction of integrin αvβ3 targeted PET tracer based on a cyclic RGD peptide. Bioorg. Med. Chem. Lett. 2011, 21, 5011–5014. [Google Scholar] [CrossRef] [PubMed]
- Knight, J.C.; Richter, S.; Wuest, M.; Way, J.D.; Wuest, F. Synthesis and evaluation of an 18F-labelled norbornene derivative for copper-free click chemistry reactions. Org. Biomol. Chem. 2013, 11, 3817–3825. [Google Scholar] [CrossRef] [PubMed]
- Poethko, T.; Schottelius, M.; Thumshirn, G.; Hersel, U.; Herz, M.; Henriksen, G.; Kessler, H.; Schwaiger, M.; Wester, H.J. Two-step methodology for high-yield routine radiohalogenation of peptides: (18)F-labeled RGD and octreotide analogs. J. Nucl. Med. 2004, 45, 892–902. [Google Scholar] [PubMed]
- Lee, Y.S.; Jeong, J.M.; Kim, H.W.; Chang, Y.S.; Kim, Y.J.; Hong, M.K.; Rai, G.B.; Chi, D.Y.; Kang, W.J.; Kang, J.H.; et al. An improved method of 18F peptide labeling: Hydrazone formation with HYNIC-conjugated c(RGDyK). Nucl. Med. Biol. 2006, 33, 677–683. [Google Scholar] [CrossRef] [PubMed]
- Wuest, F.; Hultsch, C.; Berndt, M.; Bergmann, R. Direct labelling of peptides with 2-[18F]fluoro-2-deoxy-d-glucose ([18F]FDG). Bioorg. Med. Chem. Lett 2009, 19, 5426–5428. [Google Scholar] [CrossRef] [PubMed]
- Hultsch, C.; Schottelius, M.; Auernheimer, J.; Alke, A.; Wester, H.J. (18)F-Fluoroglucosylation of peptides, exemplified on cyclo(RGDfK). Eur. J. Nucl. Med. Mol. Imaging 2009, 36, 1469–1474. [Google Scholar] [CrossRef] [PubMed]
- Ackermann, U.; Plougastel, L.; Wichmann, C.; Goh, Y.W.; Yeoh, S.D.; Poniger, S.S.; Tochon-Danguy, H.J.; Scott, A.M. Fully automated synthesis and coupling of [(18)F]FBEM to glutathione using the iPHASE FlexLab module. J. Labelled Comp. Radiopharm. 2014, 57, 115–120. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Tian, M.; Zhang, H. Microfluidics for synthesis of peptide-based PET tracers. Biomed. Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Richter, S.; Bouvet, V.; Wuest, M.; Bergmann, R.; Steinbach, J.; Pietzsch, J.; Neundorf, I.; Wuest, F. (18)F-Labeled phosphopeptide-cell-penetrating peptide dimers with enhanced cell uptake properties in human cancer cells. Nucl. Med. Biol. 2012, 39, 1202–1212. [Google Scholar] [CrossRef] [PubMed]
- Audrain, H. Positron emission tomography (PET) and microfluidic devices: A breakthrough on the microscale? Angew. Chem. Int. Ed. 2007, 46, 1772–1775. [Google Scholar] [CrossRef]
- Bouvet, V.R.; Wuest, F. Application of [18F]FDG in radiolabeling reactions using microfluidic technology. Lab. Chip 2013, 13, 4290–4294. [Google Scholar] [CrossRef] [PubMed]
- Wängler, C.; Niedermoser, S.; Chin, J.; Orchowski, K.; Schirrmacher, E.; Jurkschat, K.; Iovkova-Berends, L.; Kostikov, A.P.; Schirrmacher, R.; Wängler, B. One-step (18)F-labeling of peptides for positron emission tomography imaging using the SiFA methodology. Nat. Protoc. 2012, 7, 1946–1955. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Park, R.; Conti, P.S.; Li, Z. “Kit like” (18)F labeling method for synthesis of RGD peptide-based PET probes. Am. J. Nucl. Med. Mol. Imaging 2013, 3, 97–101. [Google Scholar] [PubMed]
- Wessmann, S.H.; Henriksen, G.; Wester, H.J. Cryptate mediated nucleophilic 18F-fluorination without azeotropic drying. Nuklearmedizin 2012, 51, 1–8. [Google Scholar] [PubMed]
- McBride, W.J.; Sharkey, R.M.; Goldenberg, D.M. Radiofluorination using aluminum-fluoride (Al18F). EJNMMI Res. 2013, 3, 36. [Google Scholar] [CrossRef] [PubMed]
- Laverman, P.; McBride, W.J.; Sharkey, R.M.; Goldenberg, D.M.; Boerman, O.C. Al18F labeling of peptides and proteins. J. Label. Comp. Radiopharm. 2014, 57, 219–223. [Google Scholar] [CrossRef]
- Shetty, D.; Choi, S.Y.; Jeong, J.M.; Lee, J.Y.; Hoigebazar, L.; Lee, Y.S.; Lee, D.S.; Chung, J.K.; Lee, M.C.; Chung, Y.K. Stable aluminium fluoride chelates with triazacyclononane derivatives proved by X-ray crystallography and 18F-labeling study. Chem. Commun. 2011, 47, 9732–9734. [Google Scholar] [CrossRef]
- McBride, W.J.; D’Souza, C.A.; Karacay, H.; Sharkey, R.M.; Goldenberg, D.M. New lyophilized kit for rapid radiofluorination of peptides. Bioconjugate Chem. 2012, 23, 538–547. [Google Scholar] [CrossRef]
- Wan, W.; Guo, N.; Pan, D.; Yu, C.; Weng, Y.; Luo, S.; Ding, H.; Xu, Y.; Wang, L.; Lang, L.; et al. First experience of 18F-alfatide in lung cancer patients using a new lyophilized kit for rapid radiofluorination. J. Nucl. Med. 2013, 54, 691–698. [Google Scholar] [CrossRef] [PubMed]
- Cescato, R.; Waser, B.; Fani, M.; Reubi, J.C. Evaluation of 177Lu-DOTA-sst2 antagonist versus 177Lu-DOTA-sst2 agonist binding in human cancers in vitro. J. Nucl. Med. 2011, 52, 1886–1890. [Google Scholar] [CrossRef] [PubMed]
- Nock, B.A.; Maina, T.; Krenning, E.P.; de Jong, M. “To serve and protect”: Enzyme inhibitors as radiopeptide escorts promote tumor targeting. J. Nucl. Med. 2014, 55, 121–127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergmann, R.; Scheunemann, M.; Heichert, C.; Mäding, P.; Wittrisch, H.; Kretzschmar, M.; Rodig, H.; Tourwé, D.; Iterbeke, K.; Chavatte, K.; et al. Biodistribution and catabolism of 18F-labeled neurotensin(8-13) analogs. Nucl. Med. Biol. 2002, 29, 61–72. [Google Scholar] [CrossRef] [PubMed]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Richter, S.; Wuest, F. 18F-Labeled Peptides: The Future Is Bright. Molecules 2014, 19, 20536-20556. https://doi.org/10.3390/molecules191220536
Richter S, Wuest F. 18F-Labeled Peptides: The Future Is Bright. Molecules. 2014; 19(12):20536-20556. https://doi.org/10.3390/molecules191220536
Chicago/Turabian StyleRichter, Susan, and Frank Wuest. 2014. "18F-Labeled Peptides: The Future Is Bright" Molecules 19, no. 12: 20536-20556. https://doi.org/10.3390/molecules191220536
APA StyleRichter, S., & Wuest, F. (2014). 18F-Labeled Peptides: The Future Is Bright. Molecules, 19(12), 20536-20556. https://doi.org/10.3390/molecules191220536