Experimental and Theoretical Perspectives of the Noyori-Ikariya Asymmetric Transfer Hydrogenation of Imines

Abstract
:1. Introduction

2. The RuII Catalytic Complex
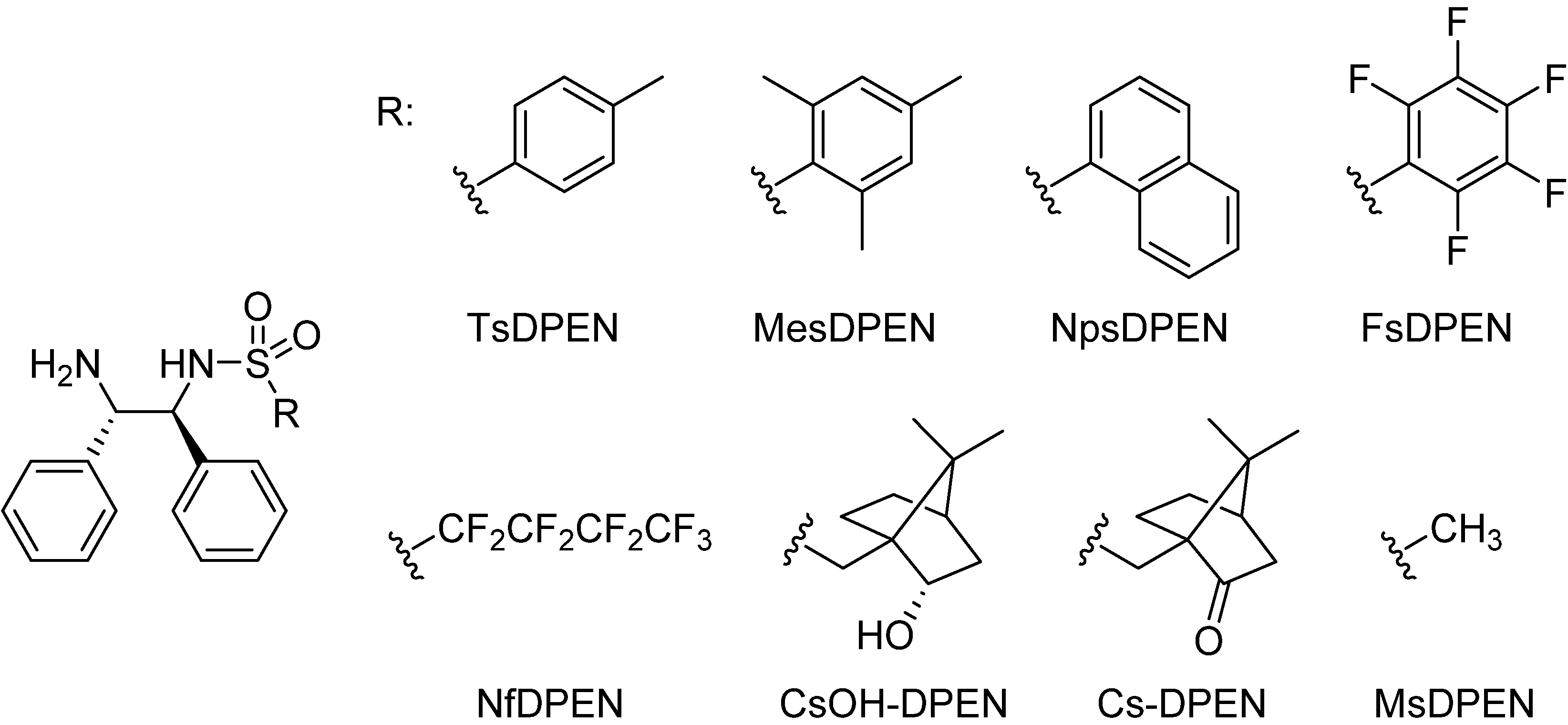
2.1. The N-R-Sulfonyl Fragment

2.2. The Sulfonamide Moiety
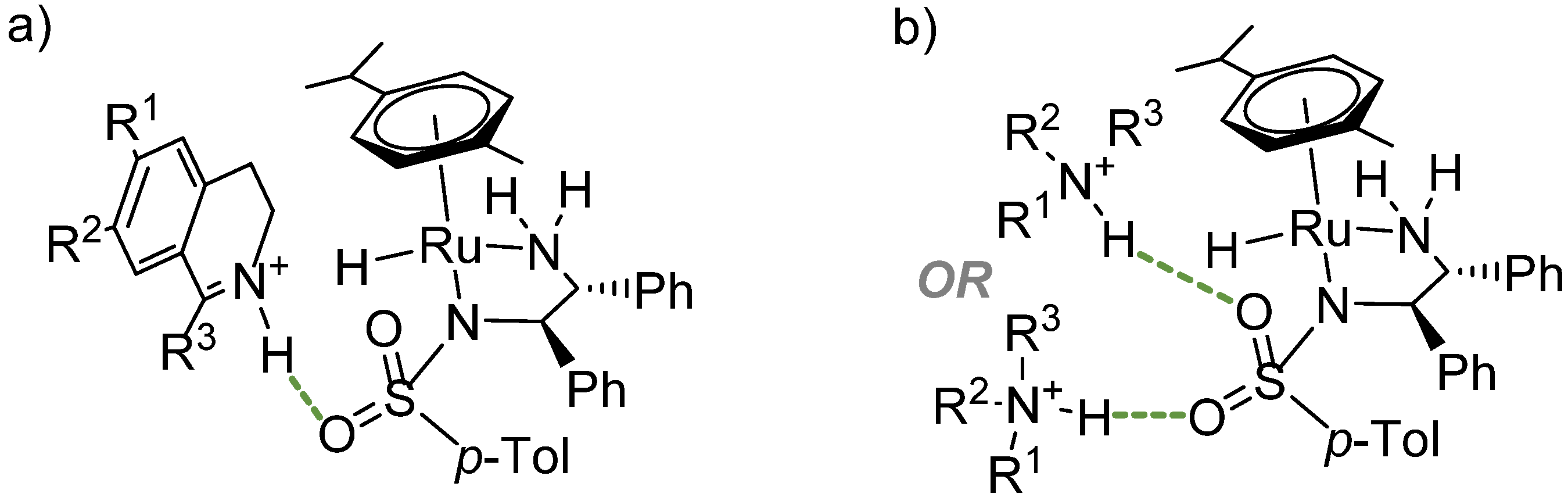
2.2.1. Interaction of the N-R-Sulfonyl Moiety with Imine Substrates

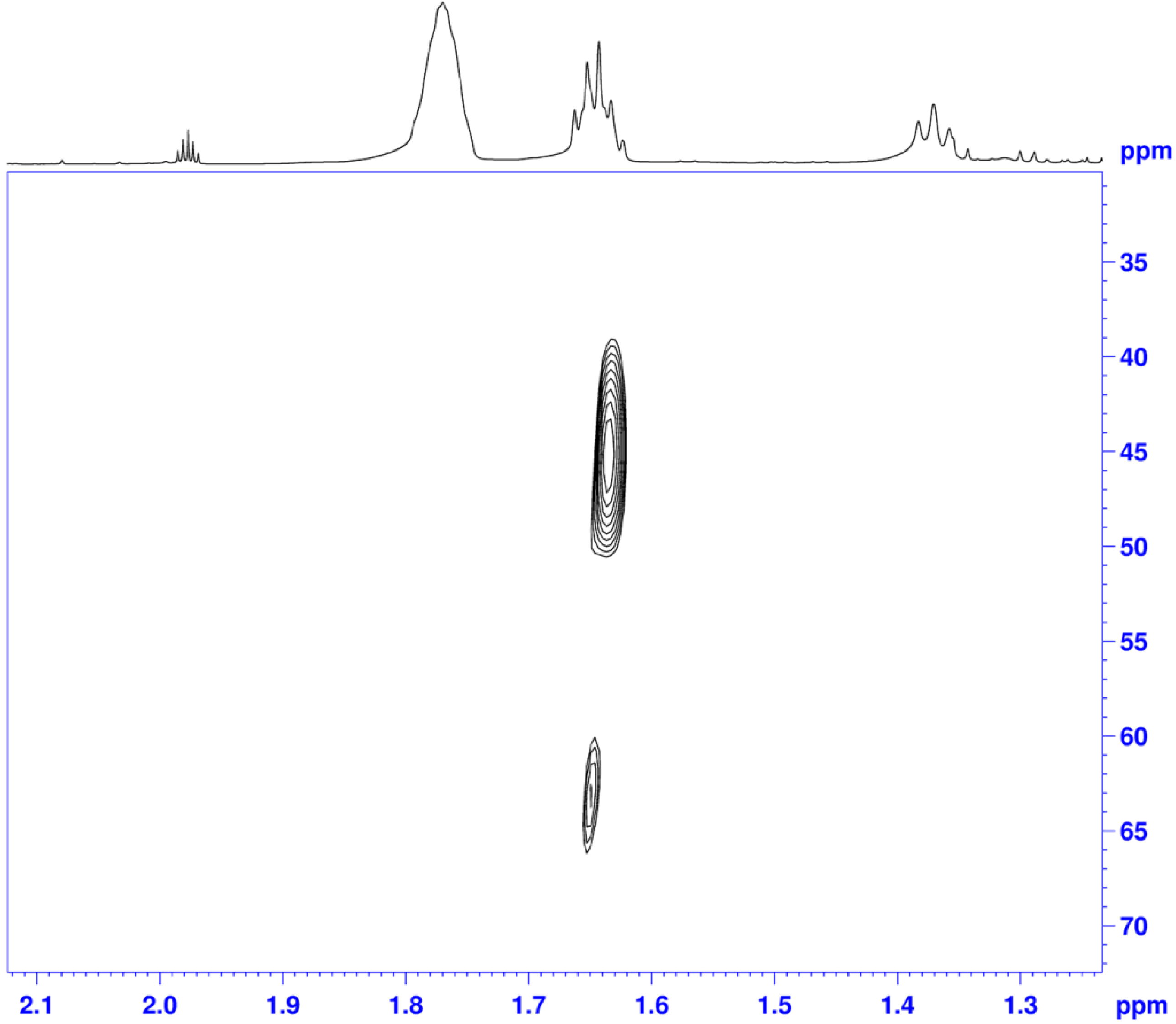
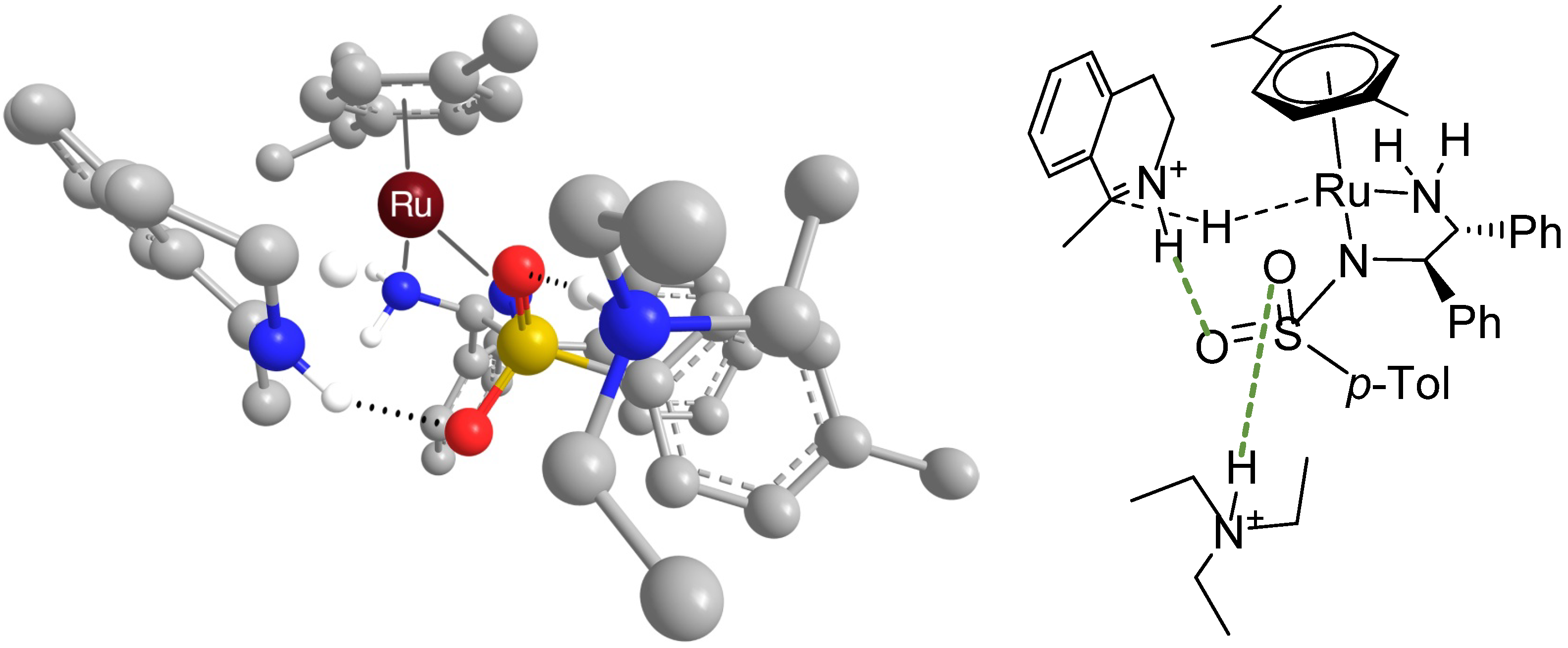
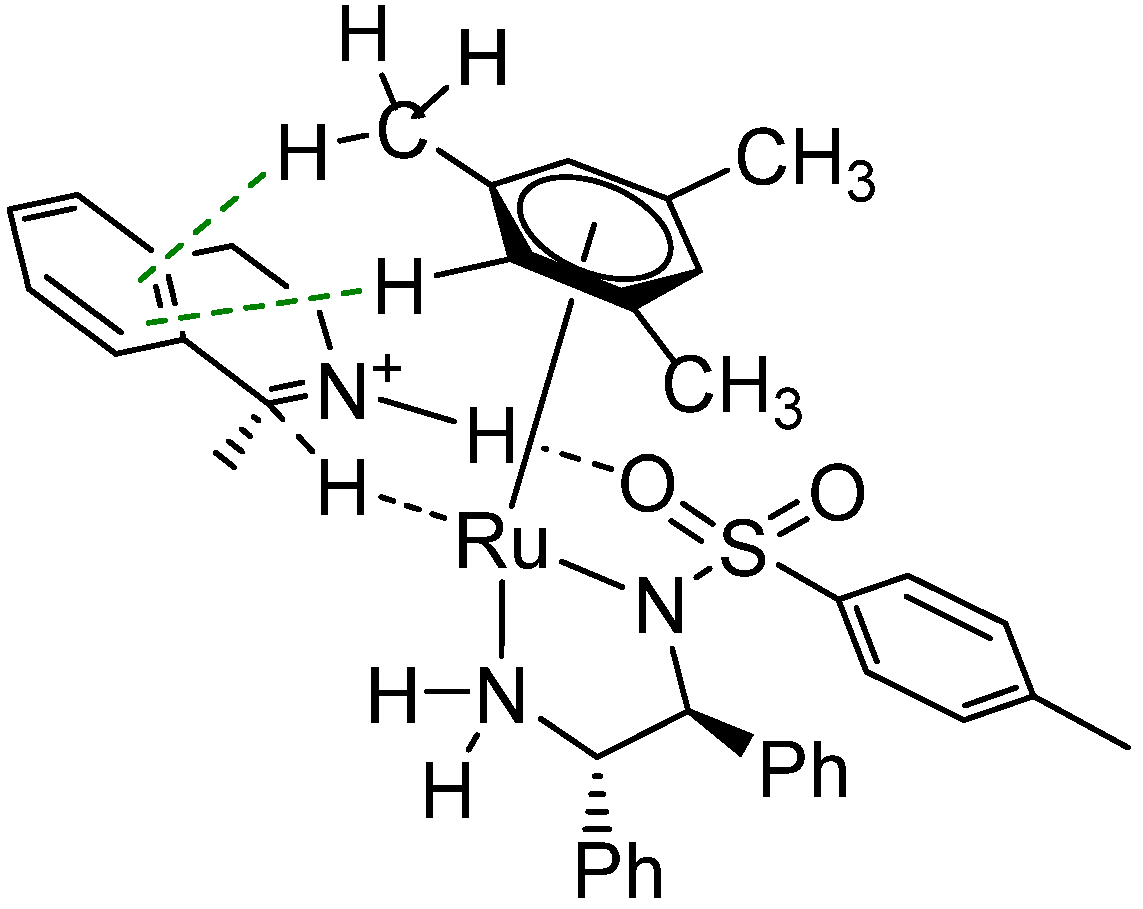
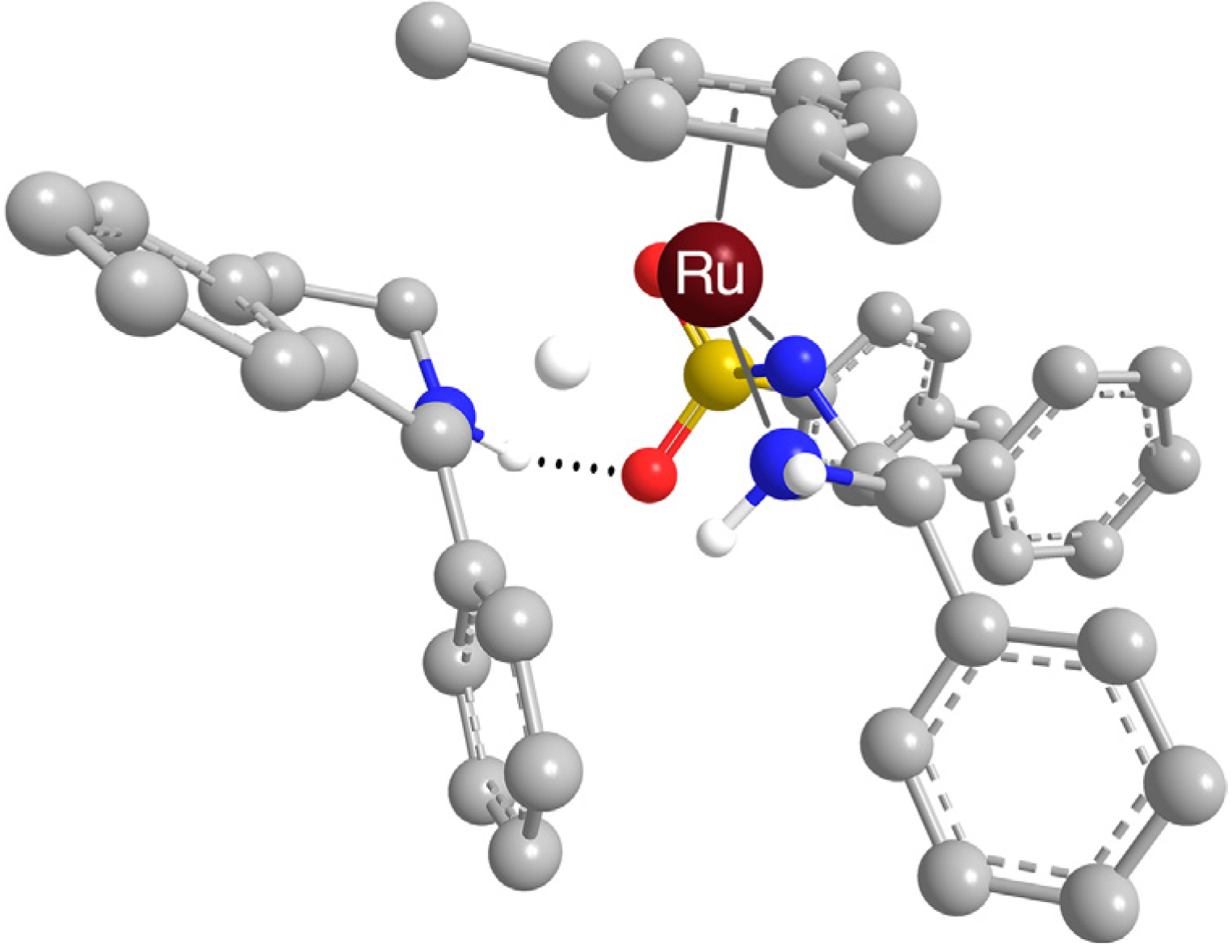
2.2.2. Interaction of the N-R-Sulfonyl Moiety with Nitrogenous Bases Present in the Hydrogen Donor Mixture


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Solvation | Amine | ΔEb [kJ·mol−1] (MP2//DFT) | ΔEb [kJ·mol−1] (DFT//DFT) | ΔGb [kJ·mol−1] (DFT//DFT) |
|---|---|---|---|---|---|
| 1 | none | n-Butylamine | –220.0 | –234.6 | –177.6 |
| 2 | none | Piperidine | –208.4 | –222.0 | –166.2 |
| 3 | none | Piperidine | –204.0 | –210.7 | –159.1 |
| 4 | none | Triethylamine | –203.1 | –210.0 | –157.3 |
| 5 | none | Tributylamine | –206.8 | –210.3 | –158.4 |
| 6 | IEFPCM | n-Butylamine | –76.2 | –81.4 | –24.1 |
| 7 | IEFPCM | Piperidine | –79.6 | –81.5 | –25.9 |
| 8 | IEFPCM | Piperidine | –78.2 | –77.2 | –26.7 |
| 9 | IEFPCM | Triethylamine | –89.4 | –87.3 | –31.0 |
| 10 | IEFPCM | Tributylamine | –95.5 | –93.5 | –46.5 |

| Entry | Amine | Initial reaction rate [mmol∙min−1∙mmolcat−1] | ee [%] [b] |
|---|---|---|---|
| 1 | n-Butylamine | 1.73 | 85 |
| 2 | Piperidine | 2.75 | 85 |
| 3 | Triethylamine | 2.86 | 83 |
| 4 | Tributylamine | 2.55 | 84 |

2.3. The η6-Arene Ligand

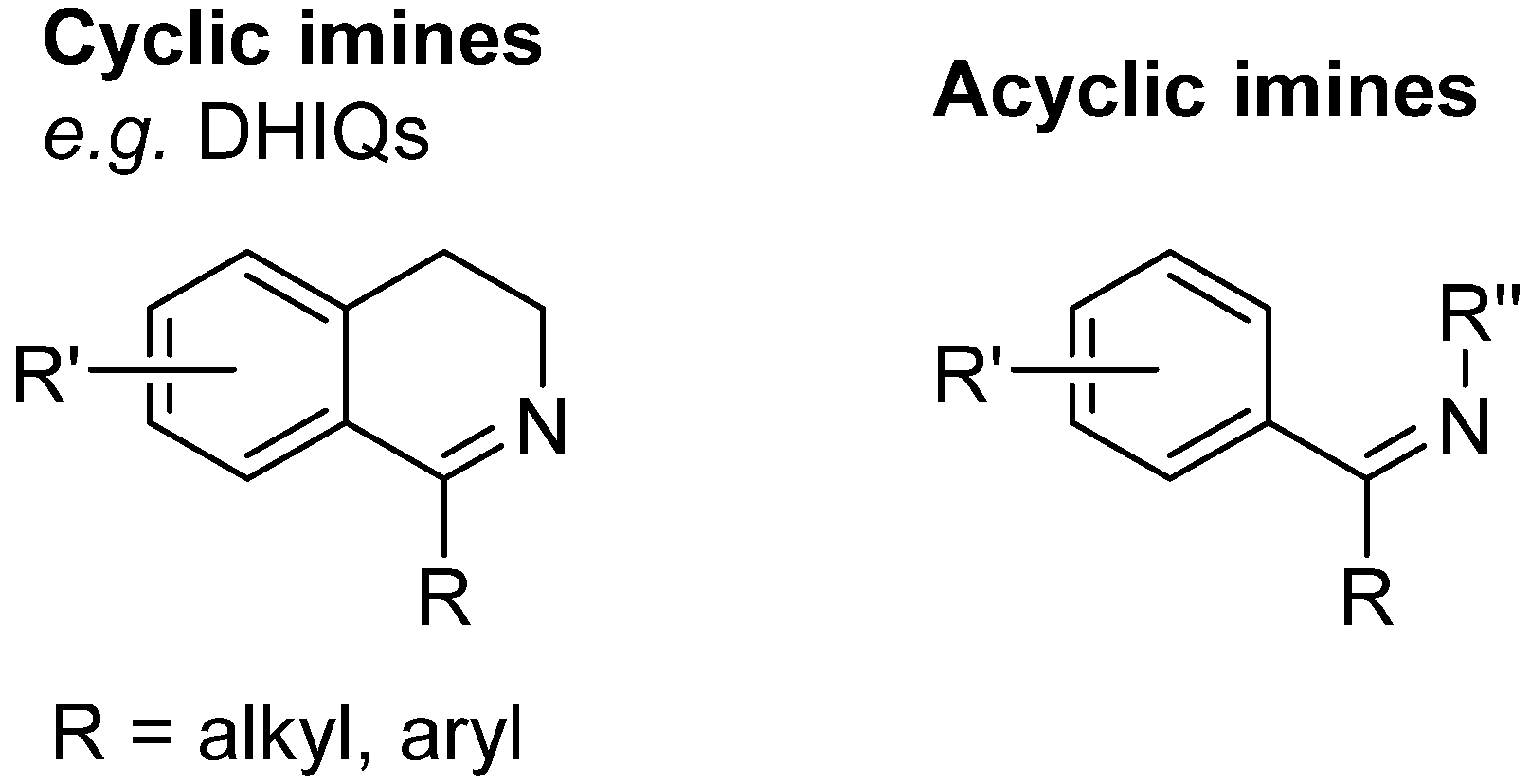
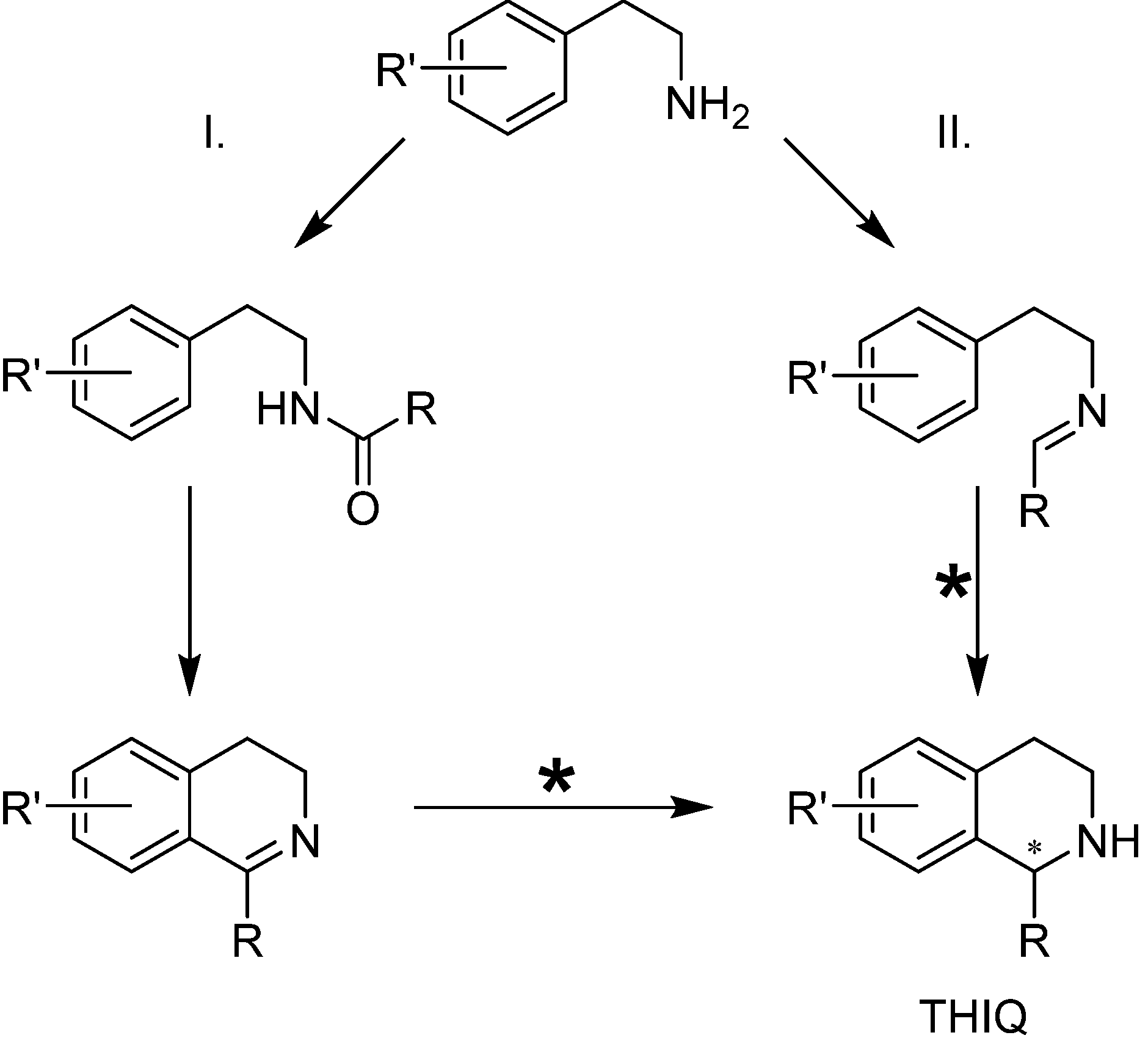
3. The Imine Substrate

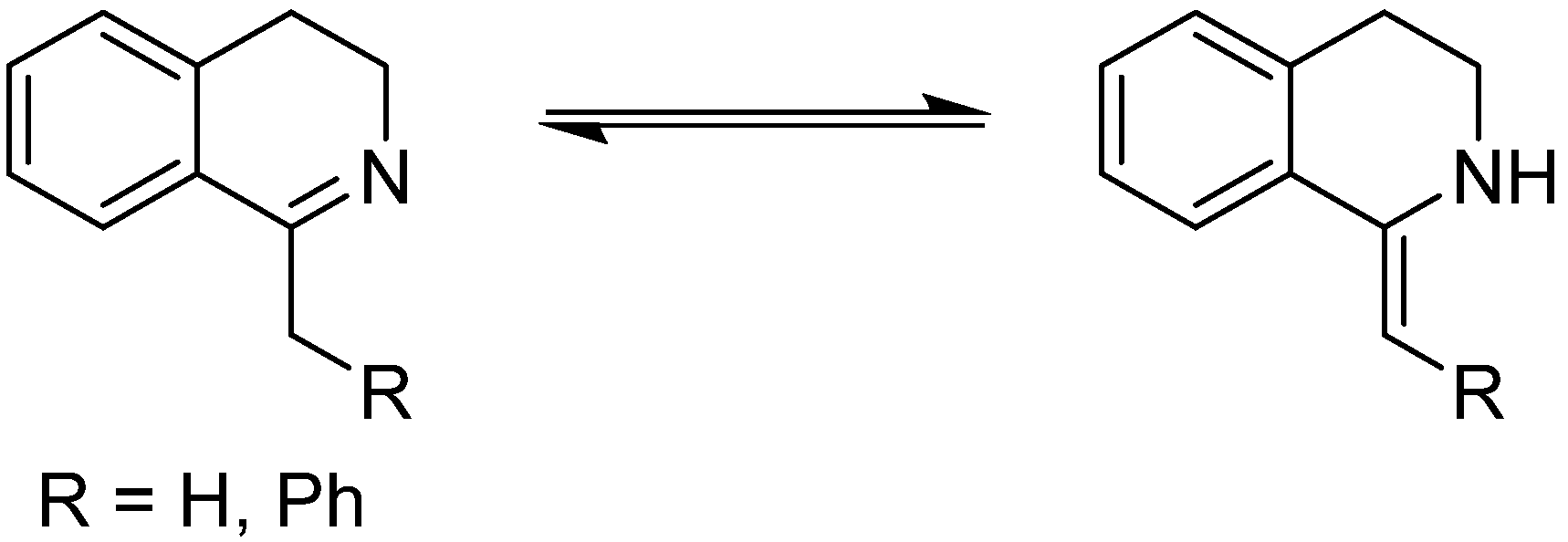
3.1. 1-Aryl-3,4-Dihydroisoquinolines


| 1-Me | 6-MeO-1-Me | 7-MeO-1-Me | 6,7-diMeO-1-Me | 1-Ph | |
|---|---|---|---|---|---|
| N–H [Å] | 1.5196 | 1.0675 | 1.0763 | 1.0702 | 1.5677 |
| H–O [Å] | 1.0522 | 1.5868 | 1.5556 | 1.5764 | 1.0416 |
| C=N [Å] [a] | 1.2785 | 1.2799 | 1.2784 | 1.2788 | 1.2795 |
| C=N [Å] [b] | 1.2825 | 1.2943 | 1.2900 | 1.2929 | 1.2828 |
| C=N+ [Å] [c] | 1.2977 | 1.3026 | 1.2978 | 1.3020 | 1.3020 |
| Activation [%] [d] | 21.3 | 63.4 | 59.8 | 60.8 | 14.7 |

3.3. Acyclic Imines

4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Ivanov, I.; Nikolova, S.; Aladjov, D.; Stefanova, I.; Zagorchev, P. Synthesis and contractile activity of substituted 1,2,3,4-tetrahydroisoquinolines. Molecules 2011, 16, 7019–7042. [Google Scholar] [CrossRef]
- Whaley, W.M.; Govindachari, T.R. The Pictet-Spengler synthesis of tetrahydroisoquinolines and related compounds. In Organic Reactions; Adams, R., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1951; Volume 6, pp. 151–190. [Google Scholar]
- Whaley, W.M.; Govindachari, T.R. Preparation of 3,4-dihydroisoquinolines and related compounds by the Bischler-Napieralski reaction. In Organic Reactions; Adams, R., Ed.; John Wiley & Sons, Inc.: New York, NY, USA, 1951; Volume 6, pp. 74–150. [Google Scholar]
- Lorenz, H.; Seidel-Morgenstern, A. Processes to separate enantiomers. Angew. Chemie. Int. Ed. 2014, 53, 1218–1250. [Google Scholar] [CrossRef]
- Gawley, R.E.; Aubé, J. Principles of Asymmetric Synthesis, 2nd ed; Elsevier: Oxford, UK, 2012. [Google Scholar]
- Ojima, I. Catalytic Asymmetric Synthesis, 3rd ed; Ojima, I., Ed.; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2010. [Google Scholar]
- Seayad, J.; Seayad, A.M.; List, B. Catalytic asymmetric Pictet-Spengler reaction. J. Am. Chem. Soc. 2006, 128, 1086–1087. [Google Scholar] [CrossRef]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Václavík, J.; Kačer, P.; Kuzma, M.; Červený, L. Opportunities offered by chiral η6-arene/N-arylsulfonyl-diamine-RuII catalysts in the asymmetric transfer hydrogenation of ketones and imines. Molecules 2011, 16, 5460–5495. [Google Scholar] [CrossRef]
- Václavík, J.; Šot, P.; Vilhanová, B.; Pecháček, J.; Kuzma, M.; Kačer, P. Practical aspects and mechanism of asymmetric hydrogenation with chiral half-sandwich complexes. Molecules 2013, 18, 6804–6828. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; King, F.; Xiao, J. Insight into and practical application of pH-controlled asymmetric transfer hydrogenation of aromatic ketones in water. Angew. Chem. Int. Ed. 2005, 44, 3407–3411. [Google Scholar] [CrossRef]
- Soni, R.; Cheung, F.K.; Clarkson, G.C.; Martins, J.E.D.; Graham, M.A.; Wills, M. The importance of the N-H bond in Ru/TsDPEN complexes for asymmetric transfer hydrogenation of ketones and imines. Org. Biomol. Chem. 2011, 9, 3290–3294. [Google Scholar] [CrossRef]
- Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of imines. J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar] [CrossRef]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid−triethylamine mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Wu, X.; Li, X.; Hems, W.; King, F.; Xiao, J. Accelerated asymmetric transfer hydrogenation of aromatic ketones in water. Org. Biomol. Chem. 2004, 2, 1818–1821. [Google Scholar] [CrossRef]
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral ruthenium(II) complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Haack, K.-J.; Hashiguchi, S.; Fujii, A.; Ikariya, T.; Noyori, R. The catalyst precursor, catalyst, and intermediate in the Ru(II)-promoted asymmetric hydrogen transfer between alcohols and ketones. Angew. Chem. Int. Ed. Engl. 1997, 36, 285–288. [Google Scholar] [CrossRef]
- Wu, Z.; Perez, M.; Scalone, M.; Ayad, T.; Ratovelomanana-Vidal, V. Ruthenium-catalyzed asymmetric transfer hydrogenation of 1-aryl-substituted dihydroisoquinolines: Access to valuable chiral 1-aryl-tetrahydroisoquinoline scaffolds. Angew. Chem. Int. Ed. Engl. 2013, 52, 4925–4928. [Google Scholar] [CrossRef]
- Ohkuma, T.; Utsumi, N.; Watanabe, M.; Tsutsumi, K.; Arai, N.; Murata, K. Asymmetric hydrogenation of α-hydroxy ketones catalyzed by MsDPEN-Cp*Ir(III) complex. Org. Lett. 2007, 9, 2565–2567. [Google Scholar] [CrossRef]
- Chen, F.; Ding, Z.; Qin, J.; Wang, T.; He, Y.; Fan, Q. Highly effective asymmetric hydrogenation of cyclic N-alkyl imines with chiral cationic Ru-MsDPEN catalysts. Org. Lett. 2011, 13, 4348–4351. [Google Scholar] [CrossRef]
- Li, X.; Blacker, J.; Houson, I.; Wu, X.; Xiao, J. An efficient Ir(III) catalyst for the asymmetric transfer hydrogenation of ketones in neat water. Synlett 2006, 2006, 1155–1160. [Google Scholar] [CrossRef]
- Yin, L.; Zheng, Y.; Jia, X.; Li, X.; Chan, A.S.C. Efficient and promising asymmetric preparation of enantiopure tolvaptan via transfer hydrogenation with robust catalysts. Tetrahedron: Asymmetry 2010, 21, 2390–2393. [Google Scholar] [CrossRef]
- Lu, C.; Luo, Z.; Huang, L.; Li, X. The Ru-catalyzed enantioselective preparation of chiral halohydrins and their application in the synthesis of (R)-clorprenaline and (S)-sotalol. Tetrahedron: Asymmetry 2011, 22, 722–727. [Google Scholar] [CrossRef]
- Luo, Z.; Qin, F.; Yan, S.; Li, X. An efficient and promising method to prepare Ladostigil (TV3326) via asymmetric transfer hydrogenation catalyzed by Ru–Cs-DPEN in an HCOONa–H2O–surfactant system. Tetrahedron: Asymmetry 2012, 23, 333–338. [Google Scholar] [CrossRef]
- Přech, J.; Václavík, J.; Šot, P.; Pecháček, J.; Vilhanová, B.; Januščák, J.; Syslová, K.; Pažout, R.; Maixner, J.; Zápal, J.; et al. Asymmetric transfer hydrogenation of 1-phenyl dihydroisoquinolines using Ru(II) diamine catalysts. Catal. Commun. 2013, 36, 67–70. [Google Scholar] [CrossRef]
- Mohar, B.; Valleix, A.; Desmurs, J.-R.; Felemez, M.; Wagner, A.; Mioskowski, C. Highly enantioselective synthesis via dynamic kinetic resolution under transfer hydrogenation using Ru(η6-arene)-N.-perfluorosulfonyl-1,2-diamine catalysts: A first insight into the relationship of the ligand’s pKa and the catalyst activity. Chem. Commun. 2001, 2572–2573. [Google Scholar]
- Šterk, D.; Stephan, M.S.; Mohar, B. New chiral N-(N,N-dialkylamino)sulfamoyl-1,2-diamine ligands for highly enantioselective transfer hydrogenation of ketones. Tetrahedron: Asymmetry 2002, 13, 2605–2608. [Google Scholar] [CrossRef]
- Šterk, D.; Stephan, M.; Mohar, B. Highly enantioselective transfer hydrogenation of fluoroalkyl ketones. Org. Lett. 2006, 8, 5935–5938. [Google Scholar] [CrossRef]
- Strotman, N.A.; Baxter, C.A.; Brands, K.M.J.; Cleator, E.; Krska, S.W.; Reamer, R.A.; Wallace, D.J.; Wright, T.J. Reaction development and mechanistic study of a ruthenium catalyzed intramolecular asymmetric reductive amination en route to the dual Orexin inhibitor Suvorexant (MK-4305). J. Am. Chem. Soc. 2011, 133, 8362–8371. [Google Scholar] [CrossRef]
- Šterk, D.; Stephan, M.S.; Mohar, B. Transfer hydrogenation of activated ketones using novel chiral Ru(II)-N-arenesulfonyl-1,2-diphenylethylenediamine complexes. Tetrahedron Lett. 2004, 45, 535–537. [Google Scholar] [CrossRef]
- Martins, J.E.D.; Wills, M. Ir(III) complexes of diamine ligands for asymmetric ketone hydrogenation. Tetrahedron 2009, 65, 5782–5786. [Google Scholar] [CrossRef]
- Åberg, J.B.; Samec, J.S.M.; Bäckvall, J.-E. Mechanistic investigation on the hydrogenation of imines by [p-(Me2CH)C6H4Me]RuH(NH2CHPhCHPhNSO2C6H4-p-CH3). Experimental support for an ionic pathway. Chem. Commun. 2006, 2771–2773. [Google Scholar]
- Martins, J.E.D.; Clarkson, G.J.; Wills, M. Ru(II) complexes of N-alkylated TsDPEN ligands in asymmetric transfer hydrogenation of ketones and imines. Org. Lett. 2009, 11, 847–850. [Google Scholar] [CrossRef]
- Václavík, J.; Kuzma, M.; Přech, J.; Kačer, P. Asymmetric transfer hydrogenation of imines and ketones using chiral RuIICl(η6-p-cymene)[(S,S)-N-TsDPEN] as a catalyst: A computational study. Organometallics 2011, 30, 4822–4829. [Google Scholar] [CrossRef]
- Muñoz Robles, V.; Vidossich, P.; Lledós, A.; Ward, T.R.; Maréchal, J.-D. Computational insights on an artificial imine reductase based on the biotin−streptavidin technology. ACS Catal. 2014, 4, 833–842. [Google Scholar] [CrossRef]
- Kuzma, M.; Václavík, J.; Novák, P.; Přech, J.; Januščák, J.; Červený, J.; Pecháček, J.; Šot, P.; Vilhanová, B.; Matoušek, V.; et al. New insight into the role of a base in the mechanism of imine transfer hydrogenation on a Ru(II) half-sandwich complex. Dalton Trans. 2013, 42, 5174–5182. [Google Scholar] [CrossRef]
- Blackmond, D.G.; Ropic, M.; Stefinovic, M. Kinetic studies of the asymmetric transfer hydrogenation of imines with formic acid catalyzed by Rh-diamine catalysts. Org. Proc. Res. Dev. 2006, 10, 457–463. [Google Scholar] [CrossRef]
- See Supplementary materials for further details
- Dokalik, A.; Kalchhauser, H.; Mikenda, W.; Schweng, G. NMR spectra of nitrogen-containing compounds. Correlations between experimental and GIAO calculated data. Magn. Reson. Chem. 1999, 37, 895–902. [Google Scholar] [CrossRef]
- Rao, N.S.; Rao, G.B.; Murthy, B.N.; Das, M.M.; Prabhakar, T.; Lalitha, M. Natural abundance nitrogen-15 nuclear magnetic resonance spectral studies on selected donors. Spectrochim. Acta. A. Mol. Biomol. Spectrosc. 2002, 58, 2737–2757. [Google Scholar] [CrossRef]
- Marek, R.; Lyčka, A. 15N-NMR spectroscopy in structural analysis. Curr. Org. Chem. 2002, 6, 35–66. [Google Scholar] [CrossRef]
- Pecháček, J.; Šot, P.; Václavík, J.; Kuzma, M.; Kačer, P.; Department of Organic Technology, Institute of Chemical Technology, Czech Republic and Laboratory of Molecular Structure Characterization, Institute of Microbiology, v.v.i., Academy of Sciences of the Czech Republic, Vídeňská 1083, CZ-142 20, Prague, Czech Republic. Unpublished results. 2014.
- It should be noted that the bond between n-butylamine and the SO2 group was bifurcated (see the Supporting information). It was also attempted to obtain a geometry based on two hydrogen bonds between protonated piperidine and the catalytic complex, but two distinct structures with only one hydrogen bond were obtained instead (hence entries 2, 3, 7 and 8 belong to piperidine)
- Pecháček, J.; Václavík, J.; Přech, J.; Šot, P.; Januščák, J.; Vilhanová, B.; Vavřík, J.; Kuzma, M.; Kačer, P. Asymmetric transfer hydrogenation of imines catalyzed by a Noyori-type Ru(II) complex – a parametric study. Tetrahedron: Asymmetry 2013, 24, 233–239. [Google Scholar] [CrossRef]
- Přech, J.; Matoušek, V.; Václavík, J.; Pecháček, J.; Syslová, K.; Šot, P.; Januščák, J.; Vilhanová, B.; Kuzma, M.; Kačer, P. Determination of enantiomeric composition of substituted tetrahydroisoquinolines based on derivatization with menthyl chloroformate. Am. J. Anal. Chem. 2013, 4, 125–133. [Google Scholar] [CrossRef]
- Yamakawa, M.; Yamada, I.; Noyori, R. CH/π Attraction: The origin of enantioselectivity in transfer hydrogenation of aromatic carbonyl compounds catalyzed by chiral η6arene-ruthenium(II) complexes. Angew. Chem. Int. Ed. 2001, 40, 2818–2821. [Google Scholar] [CrossRef]
- Arai, N.; Satoh, H.; Utsumi, N.; Murata, K. Asymmetric hydrogenation of alkynyl ketones with the η6-arene/TsDPEN-ruthenium (II) catalyst. Org. Lett. 2013, 15, 3030–3033. [Google Scholar] [CrossRef]
- Fang, Z.; Wills, M. Asymmetric transfer hydrogenation of functionalized acetylenic ketones. J. Org. Chem. 2013, 78, 8594–8605. [Google Scholar] [CrossRef]
- Fang, Z.; Wills, M. Asymmetric reduction of diynones and the total synthesis of (S.)-Panaxjapyne A. Org. Lett. 2014, 16, 374–377. [Google Scholar]
- Dub, P.A.; Ikariya, T. Quantum chemical calculations with the inclusion of nonspecific and specific solvation: Asymmetric transfer hydrogenation with bifunctional ruthenium catalysts. J. Am. Chem. Soc. 2013, 135, 2604–2619. [Google Scholar] [CrossRef]
- Takehara, J.; Hashiguchi, S.; Fujii, A.; Inoue, S.-I.; Ikariya, T.; Noyori, R. Amino alcohol effects on the ruthenium(II)-catalysed asymmetric transfer hydrogenation of ketones in propan-2-ol. Chem. Commun. 1996, 233–234. [Google Scholar]
- Available from Strem Chemicals, Inc. (Newburyport, MA, USA) or Sigma-Aldrich Co. LLC. (St. Louis, MO, USA)
- Bennett, M.A.; Smith, A.K. Arene ruthenium (II) complexes formed by dehydrogenation of cyclohexadienes with ruthenium(II) trichloride. J. Chem. Soc. Dalt. Trans. 1974, 233–241. [Google Scholar] [CrossRef]
- Cheung, F.K.; Hayes, A.M.; Morris, D.J.; Wills, M. The use of a [4 + 2] cycloaddition reaction for the preparation of a series of “tethered” Ru(II)-diamine and aminoalcohol complexes. Org. Biomol. Chem. 2007, 5, 1093–1103. [Google Scholar] [CrossRef]
- Touge, T.; Hakamata, T.; Hideki, N.; Kobayashi, T.; Sayo, N.; Saito, T.; Kayaki, Y.; Ikariya, T. Oxo-tethered ruthenium(II) complex as a bifunctional catalyst for asymmetric transfer hydrogenation and H2 hydrogenation. J. Am. Chem. Soc. 2011, 133, 14960–14963. [Google Scholar]
- Bennett, M.A.; Matheson, T.W.; Robertson, G.B.; Smith, A.K.; Tucker, P.A. Highly fluxional arene cyclooctatetraene complexes of zerovalent iron, ruthenium, and osmium. Single-crystal X-ray ctudy of (Cyclooctatetraene)(hexamethylbenzene)ruthenium(0), Ru(η6-HMB)(1–4-η-COT). Inorg. Chem. 1980, 19, 1014–1021. [Google Scholar]
- Hull, J.W.; Gladfelter, W.L. η4-Bonding in (arene) ruthenium complexes of octamethylnaphthalene. Organometallics 1984, 3, 605–613. [Google Scholar] [CrossRef]
- Soni, R.; Jolley, K.E.; Clarkson, G.J.; Wills, M. Direct formation of tethered Ru(II) catalysts using arene exchange. Org. Lett. 2013, 15, 5110–5113. [Google Scholar] [CrossRef]
- Šot, P.; Vilhanová, B.; Pecháček, J.; Václavík, J.; Januščák, J.; Zápal, J.; Kuzma, M.; Kačer, P. Role of the aromatic ligand in the asymmetric transfer hydrogenation of C=N bond on chiral Ru Noyori’s catalysts. J. Organomet. Chem. 2014. submitted. [Google Scholar]
- Cheng, P.; Huang, N.; Jiang, Z.-Y.; Zhang, Q.; Zheng, Y.-T.; Chen, J.-J.; Zhang, X.-M.; Ma, Y.-B. 1-Aryl-tetrahydroisoquinoline analogs as active anti-HIV agents in vitro. Bioorg. Med. Chem. Lett. 2008, 18, 2475–2478. [Google Scholar] [CrossRef]
- Christopher, J.A.; Atkinson, F.L.; Bax, B.D.; Brown, M.J.B.; Champigny, A.C.; Chuang, T.T.; Jones, E.J.; Mosley, J.E.; Musgrave, J.R. 1-Aryl-3,4-dihydroisoquinoline inhibitors of JNK3. Bioorg. Med. Chem. Lett. 2009, 19, 2230–2234. [Google Scholar] [CrossRef]
- Gitto, R.; Caruso, R.; Orlando, V.; Quartarone, S.; Barreca, M.L.; Ferreri, G.; Russo, E.; de Sarro, G.; Chimirri, A. Synthesis and anticonvulsant properties of tetrahydroisoquinoline derivatives. IlFarmaco 2004, 59, 7–12. [Google Scholar] [CrossRef]
- Abrams, P.; Andersson, K.E. Muscarinic receptor antagonists for overactive bladder. BJU Int. 2007, 100, 987–1006. [Google Scholar] [CrossRef]
- Vedejs, E.; Trapencieris, P.; Suna, E. Substituted isoquinolines by Noyori transfer hydrogenation: Enantioselective synthesis of chiral diamines containing an aniline subunit. J. Org. Chem. 1999, 64, 6724–6729. [Google Scholar] [CrossRef]
- Wu, J.; Wang, F.; Ma, Y.; Cui, X.; Cun, L.; Zhu, J.; Deng, J.; Yu, B. Asymmetric transfer hydrogenation of imines and iminiums catalyzed by a water-soluble catalyst in water. Chem. Commun. 2006, 1766–1768. [Google Scholar]
- Chang, M.; Li, W.; Zhang, X. A highly efficient and enantioselective access to tetrahydroisoquinoline alkaloids: Asymmetric hydrogenation with an iridium catalyst. Angew. Chemie. Int. Ed. 2011, 50, 10679–10681. [Google Scholar] [CrossRef]
- Núñez-Rico, J.L.; Vidal-Ferran, A. Ir(P−OP)]-Catalyzed aymmetric hydrogenation of diversely substituted C=N-containing heterocycles. Org. Lett. 2013, 15, 2066–2069. [Google Scholar] [CrossRef]
- Šot, P.; Kuzma, M.; Kačer, P.; Department of Organic Technology, Institute of Chemical Technology, Czech Republic and Laboratory of Molecular Structure Characterization, Institute of Microbiology, v.v.i., Academy of Sciences of the Czech Republic, Vídeňská 1083, CZ-142 20, Prague, Czech Republic. Unpublished results. 2014.
- Rappoport, D.; Furche, F. Property-optimized Gaussian basis sets for molecular response calculations. J. Chem. Phys. 2010, 133, 134105. [Google Scholar] [CrossRef]
- Samano, V.; Ray, J.A.; Thompson, J.B.; Mook, R.A.; Jung, D.K.; Koble, C.S.; Martin, M.T.; Bigham, E.C.; Regitz, C.S.; Feldman, P.L.; et al. Synthesis of ultra-short-acting neuromuscular blocker GW 0430: A remarkably stereo- and regioselective Tetrahydroisoquinolinium. Org. Lett. 1999, 1, 1993–1996. [Google Scholar] [CrossRef]
- Vilhanová, B.; Matoušek, V.; Václavík, J.; Syslová, K.; Přech, J.; Pecháček, J.; Šot, P.; Januščák, J.; Toman, J.; Zápal, J.; et al. Two optimized synthetic pathways toward a chiral precursor of Mivacurium chloride and other skeletal muscle relaxants. Tetrahedron: Asymmetry 2013, 24, 50–55. [Google Scholar] [CrossRef]
- Kuzma, M.; Laboratory of Molecular Structure Characterization, Institute of Microbiology, v.v.i., Academy of Sciences of the Czech Republic, Vídeňská 1083, CZ-142 20, Prague, Czech Republic. Unpublished results. 2014.
- Budnikova, M.V; Rubinov, D.B.; Mikhal’chuk, A.L. Annelation of 3,4-dihydroisoquinolines with 3-acylthiotetronic acids: Synthesis and properties of 8-aza-16-thiagona-12,17-diones and 3,4-dihydroisoquinolinium 3-acetylthiotetronate. Chem. Heterocycl. Compd. 2002, 38, 929–939. [Google Scholar] [CrossRef]
- Martin, N.H.; Champion, S.L.; Belt, P.B. Regiospecific oxidation of substituted 1-benzyl-3,4-dihydroisoquinolines using singlet oxygen. Tetrahedron Lett. 1980, 21, 2613–2616. [Google Scholar] [CrossRef]
- Martin, N.H.; Jefford, C.W. Evidence for a charge-transfer mechanism in the photo-oxygenation of an enamine. Tetrahedron Lett. 1981, 22, 3949–3952. [Google Scholar] [CrossRef]
- Martin, N.H.; Jefford, C.W. Synthesis and photo-oxygenation of some substituted 1-benzyl-3,4-dihydroisoquinolines. Mechanism of enamine photo-oxygenation. Helv. Chim. Acta 1982, 65, 762–774. [Google Scholar] [CrossRef]
- Pitacco, G.; Valentin, E. Oxidation and reduction of enamines. In The Chemistry of Enamines; Rappoport, Z., Ed.; John Wiley & Sons, Inc.: Chichester, UK, 1994; pp. 923–992. [Google Scholar]
- Weisbach, J.A.; Kirkpatrick, J.L.; Macko, E.; Douglas, B. Synthesis and parmacology of some α-oxy- and α-hydroxy-1-benzyltetrahydroisoquinolines. J. Med. Chem. 1968, 11, 752–760. [Google Scholar]
- Lebœuf, M.; Ranaivo, A.; Cavé, A.; Moskowitz, H. La velucryptine, nouvel alcaloïde isoquinoléique isolé de Cryptocarya velutinosta. J. Nat. Prod. 1989, 52, 516–521. [Google Scholar] [CrossRef]
- Cho, S.-D.; Kweon, D.-H.; Kang, Y.-J.; Lee, S.-G.; Lee, W.S.; Yoon, Y.-J. Synthesis of 6,7-dimethoxy-1-halobenzyl-1,2,3,4-tetrahydroisoquinolines. J. Heterocycl. Chem. 1999, 36, 1151–1156. [Google Scholar] [CrossRef]
- Shklyaev, Y.V.; Yeltsov, M.A.; Rozhkova, Y.S.; Tolstikov, A.G.; Dembitsky, V.M. A new approach to synthesis of 3,3-dialkyl-3,4-dihydroisoquinoline derivatives. Heteroat. Chem. 2004, 15, 486–493. [Google Scholar] [CrossRef]
- Amaravathi, M.; Kumari, L.K.; Pardhasaradhi, M. Oxidation of 1-benzyl-3,4-dihydroisoquinolines using active manganese-dioxide. Indian J. Chem. Sect. B 1983, 22B, 1246–1247. [Google Scholar]
- Hubert, M.; Herrmann, R. Oxidation of imines by selenium dioxide. Z. Naturforsch. B 1986, 41B, 1260–1264. [Google Scholar]
- Andreu, I.; Cabedo, N.; Atassi, G.; Pierré, A.; Caignard, D.H.; Renard, P.; Cortes, D.; Bermejo, A. An efficient method for the preparation of antitumoral α-keto-imines benzyldihydroisoquinolines by selective benzylic oxidation with C/Pd in acetonitrile. Tetrahedron Lett. 2002, 43, 757–759. [Google Scholar] [CrossRef]
- Bermejo, A.; Andreu, I.; Suvire, F.; Léonce, S.; Caignard, D.H.; Renard, P.; Pierré, A.; Enriz, R.D.; Cortes, D.; Cabedo, N. Syntheses and antitumor targeting G1 phase of the cell cycle of benzoyldihydroisoquinolines and related 1-substituted isoquinolines. J. Med. Chem. 2002, 45, 5058–5068. [Google Scholar] [CrossRef]
- McMahon, R.M.; Thornber, C.W.; Ruchirawat, S. Rearrangement of 1-(α-hydroxybenzyl)-1,2,3,4-tetrahydroisoquinolines to 1-phenyl-2,3,4,5-tetrahydro-1H-3-benzazepines. J. Chem. Soc. Perkin Trans. 1 1982, 2163–2167. [Google Scholar]
- Canonica, L.; Galliani, G.; Rindone, B.; Tollari, S.; Andreoni, V.; Galli, E. The microbial oxygenation of the benzylisoquinoline alkaloid laudanosine. Experientia 1983, 39, 1273–1275. [Google Scholar] [CrossRef]
- Šot, P.; Kuzma, M.; Václavík, J.; Pecháček, J.; Přech, J.; Januščák, J.; Kačer, P. Asymmetric transfer hydrogenation of acetophenone N-benzylimine using [RuIICl(S,S)-N-(TsDPEN)η6-(p-cymene)]: A DFT study. Organometallics 2012, 31, 6496–6499. [Google Scholar] [CrossRef]
- Chen, F.; Wang, T.; He, Y.; Ding, Z.; Li, Z.; Xu, L.; Fan, Q.-H. Asymmetric hydrogenation of N-alkyl ketimines with phosphine-free, chiral, cationic Ru-MsDPEN catalysts. Chem. Eur. J. 2011, 17, 1109–1113. [Google Scholar]
- Chen, F.; Ding, Z.; He, Y.; Qin, J.; Wang, T.; Fan, Q.-H. Asymmetric hydrogenation of N-alkyl and N-aryl ketimines using chiral cationic Ru(diamine) complexes as catalysts: The counteranion and solvent effects, and substrate scope. Tetrahedron 2012, 68, 5248–5257. [Google Scholar] [CrossRef]
© 2014 by the authors. licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Václavík, J.; Šot, P.; Pecháček, J.; Vilhanová, B.; Matuška, O.; Kuzma, M.; Kačer, P. Experimental and Theoretical Perspectives of the Noyori-Ikariya Asymmetric Transfer Hydrogenation of Imines. Molecules 2014, 19, 6987-7007. https://doi.org/10.3390/molecules19066987
Václavík J, Šot P, Pecháček J, Vilhanová B, Matuška O, Kuzma M, Kačer P. Experimental and Theoretical Perspectives of the Noyori-Ikariya Asymmetric Transfer Hydrogenation of Imines. Molecules. 2014; 19(6):6987-7007. https://doi.org/10.3390/molecules19066987
Chicago/Turabian StyleVáclavík, Jiří, Petr Šot, Jan Pecháček, Beáta Vilhanová, Ondřej Matuška, Marek Kuzma, and Petr Kačer. 2014. "Experimental and Theoretical Perspectives of the Noyori-Ikariya Asymmetric Transfer Hydrogenation of Imines" Molecules 19, no. 6: 6987-7007. https://doi.org/10.3390/molecules19066987
APA StyleVáclavík, J., Šot, P., Pecháček, J., Vilhanová, B., Matuška, O., Kuzma, M., & Kačer, P. (2014). Experimental and Theoretical Perspectives of the Noyori-Ikariya Asymmetric Transfer Hydrogenation of Imines. Molecules, 19(6), 6987-7007. https://doi.org/10.3390/molecules19066987