Cyclodepsipeptides: A Rich Source of Biologically Active Compounds for Drug Research

Abstract
:
Table of Contents
| 1. Introduction | 12369 |
| 2. Biosynthesis of Cyclodepsipeptides | 12370 |
| 3. Cyclotetradepsipeptides | 12371 |
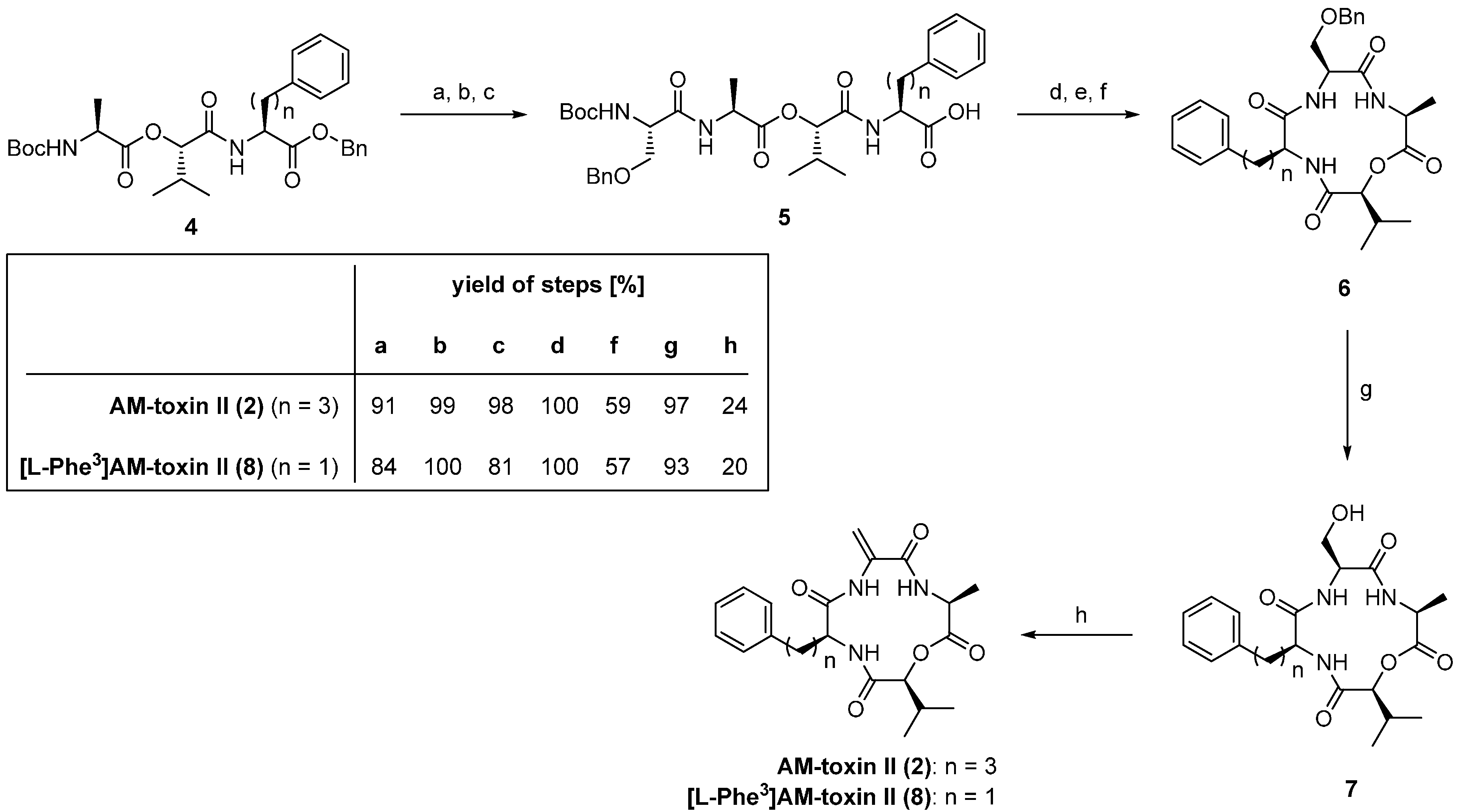
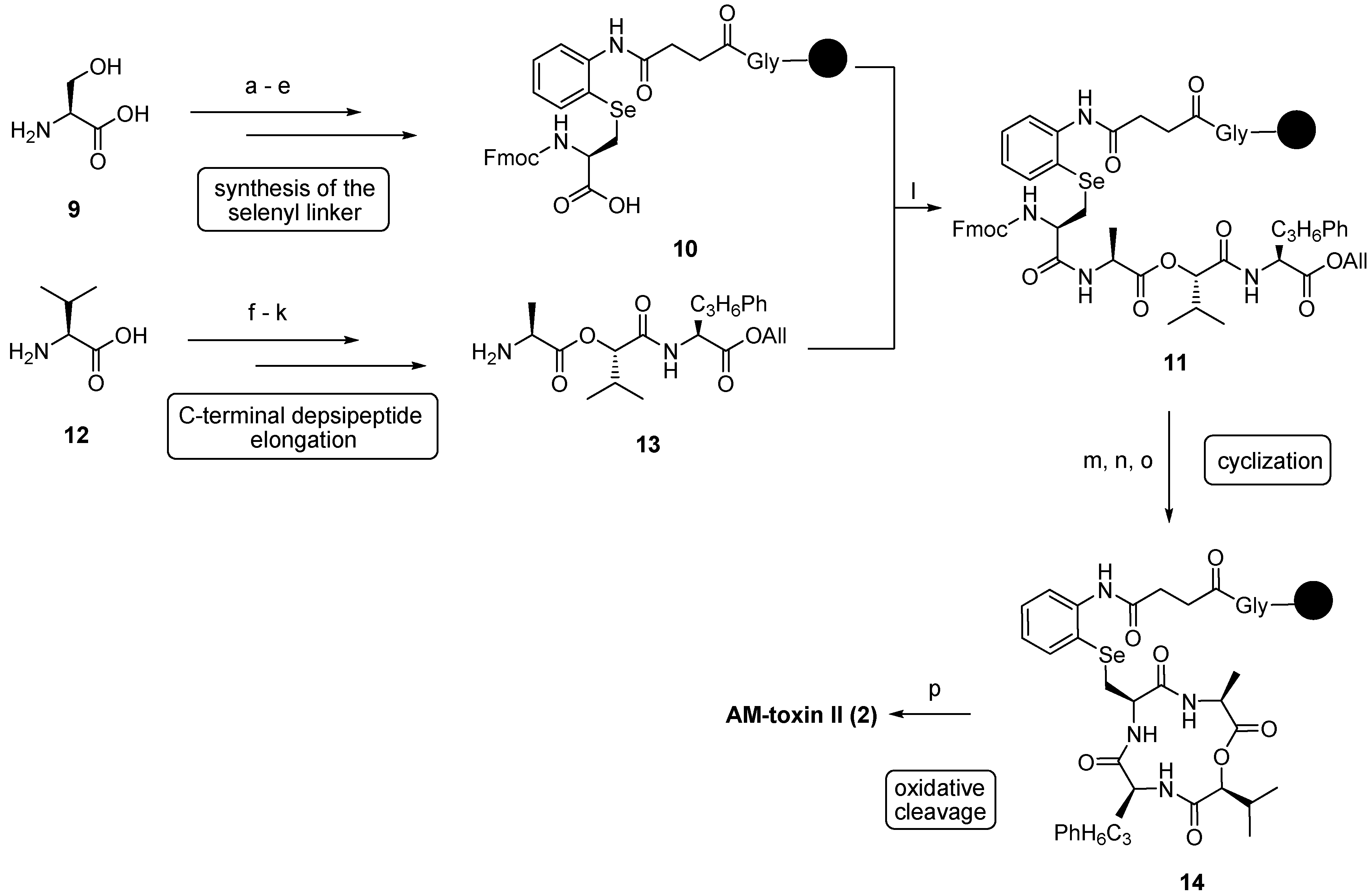
| 3.1. AM-Toxins | 12371 |
| 4. Cyclopentadepsipeptides | 12374 |
| 4.1. Sansalvamide A, N-methylsansalvamide and neo-N-methylsansalvamide | 12374 |
| 4.2. Alternaramide | 12376 |
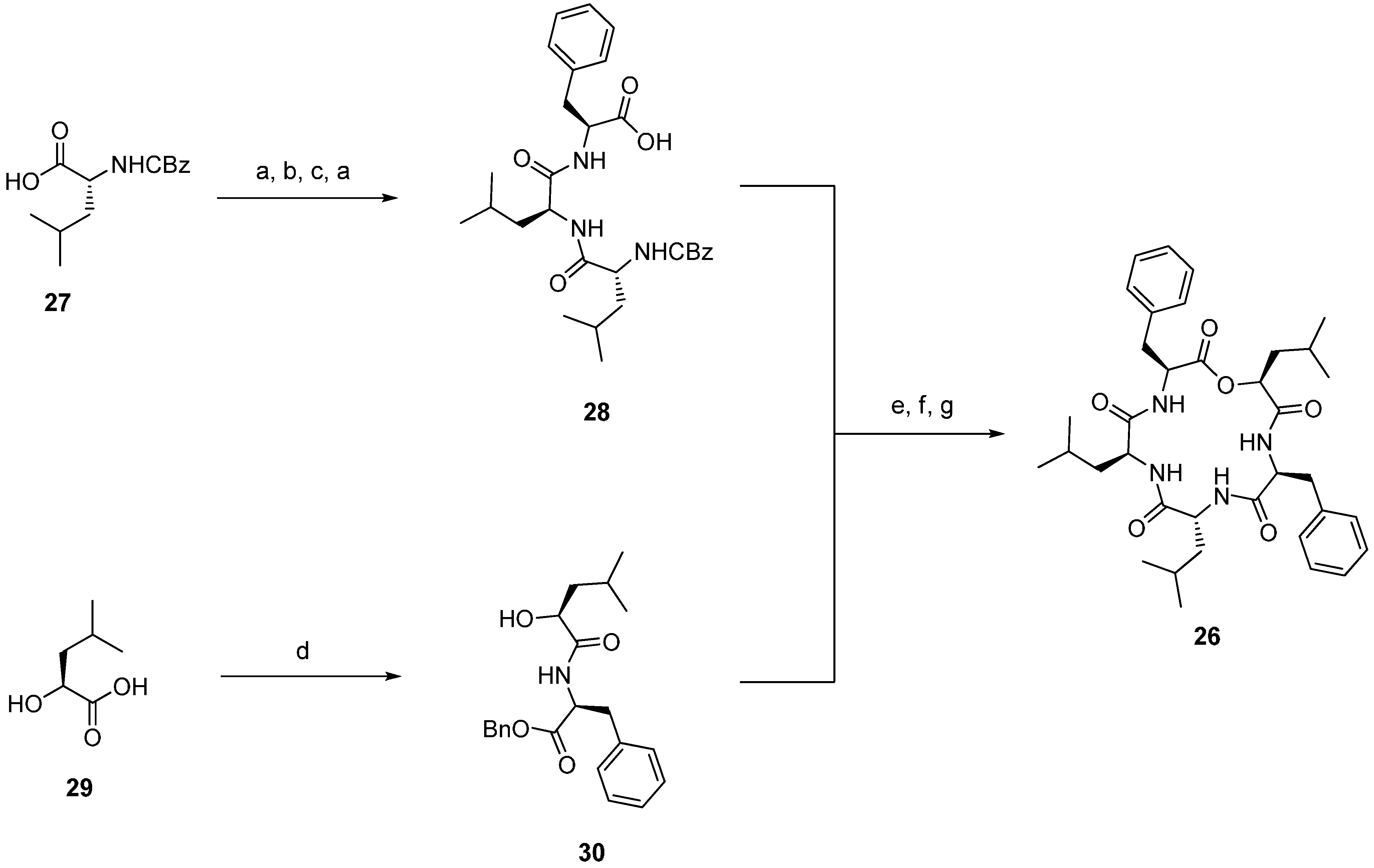
| 4.3. Zygosporamide | 12376 |
| 5. Cyclohexadepsipeptides | 12378 |
| 5.1. Enniatins and Beauvericin | 12378 |
| 5.1.1. Synthesis of Enniatins and Enniatin Derivatives | 12379 |
| 5.1.2. Biosynthesis of Enniatins and Beauvericin | 12381 |
| 5.2. Beauvenniatins | 12384 |
| 5.3. Hirsutellide A | 12385 |
| 5.4. Kutznerides | 12386 |
| 5.5. Monamycins | 12387 |
| 5.6. Himastatin | 12387 |
| 5.7. Paecilodepsipeptide A and Conoideocrellide A | 12390 |
| 5.8. Pullularins A-E | 12391 |
| 5.9. Hirsutatins A and B | 12392 |
| 6. Cycloheptadepsipeptides | 12392 |
| 6.1. HUN-7293 | 12392 |
| 7. Cyclooctadepsipeptides | 12395 |
| 7.1. Bassianolide | 12395 |
| 7.2. Verticlide | 12396 |
| 7.3. PF1022A and Emodepside | 12397 |
| 7.3.1. Syntheses of PF1022A | 12399 |
| 7.3.2. Synthesis of PF1022A-Analogues via Total Synthesis | 12401 |
| 7.3.3. PF1022A Analogues by Direct Derivatization of the Natural Product | 12404 |
| 7.3.4. Biosynthesis of PF1022A | 12406 |
| 7.3.5. Mode of Action of PF1022A and Emodepside | 12406 |
| 8. Cyclononadepsipeptides | 12408 |
| 8.1. BZR-cotoxins I-IV | 12408 |
| 8.2. Aureobasidins | 12409 |
| 9. Cyclodecadepsipeptides | 12410 |
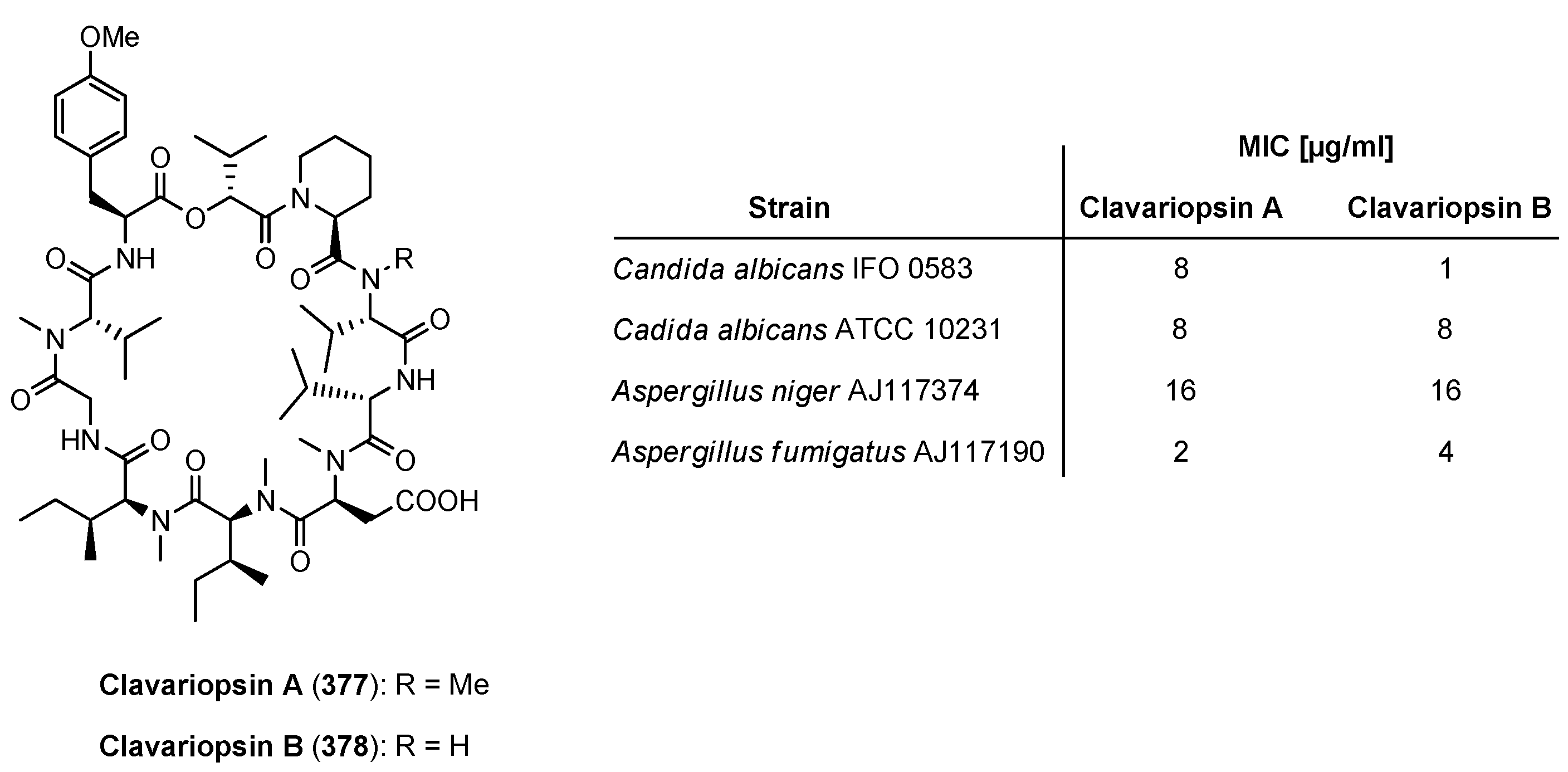
| 9.1. Clavariopsin A and B | 12410 |
| 10. Summary | 12411 |
| Conflicts of Interest | 12411 |
| References | 12412 |
1. Introduction
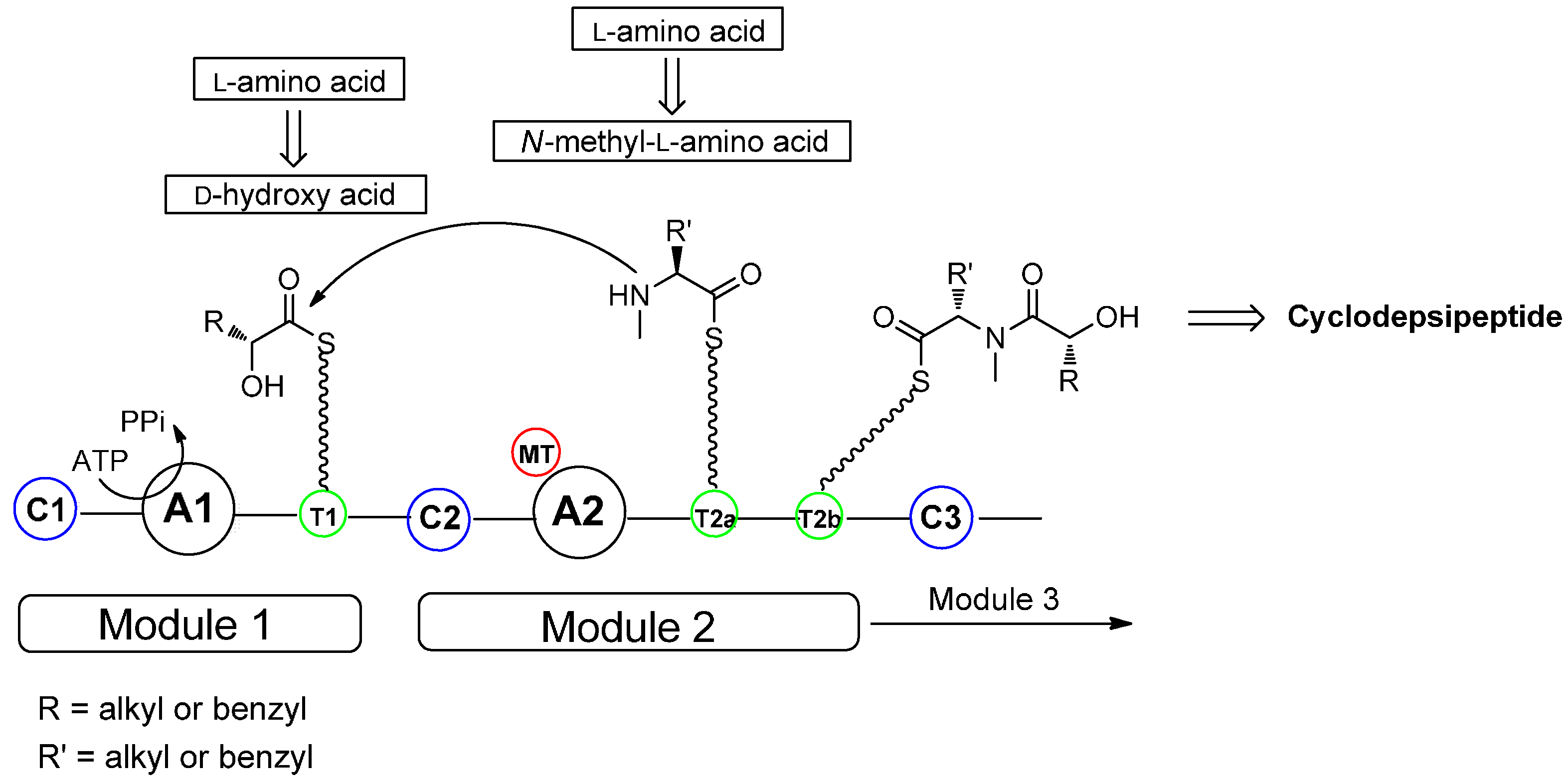
2. Biosynthesis of Cyclodepsipeptides

3. Cyclotetradepsipeptides
3.1. AM-Toxins



4. Cyclopentadepsipeptides
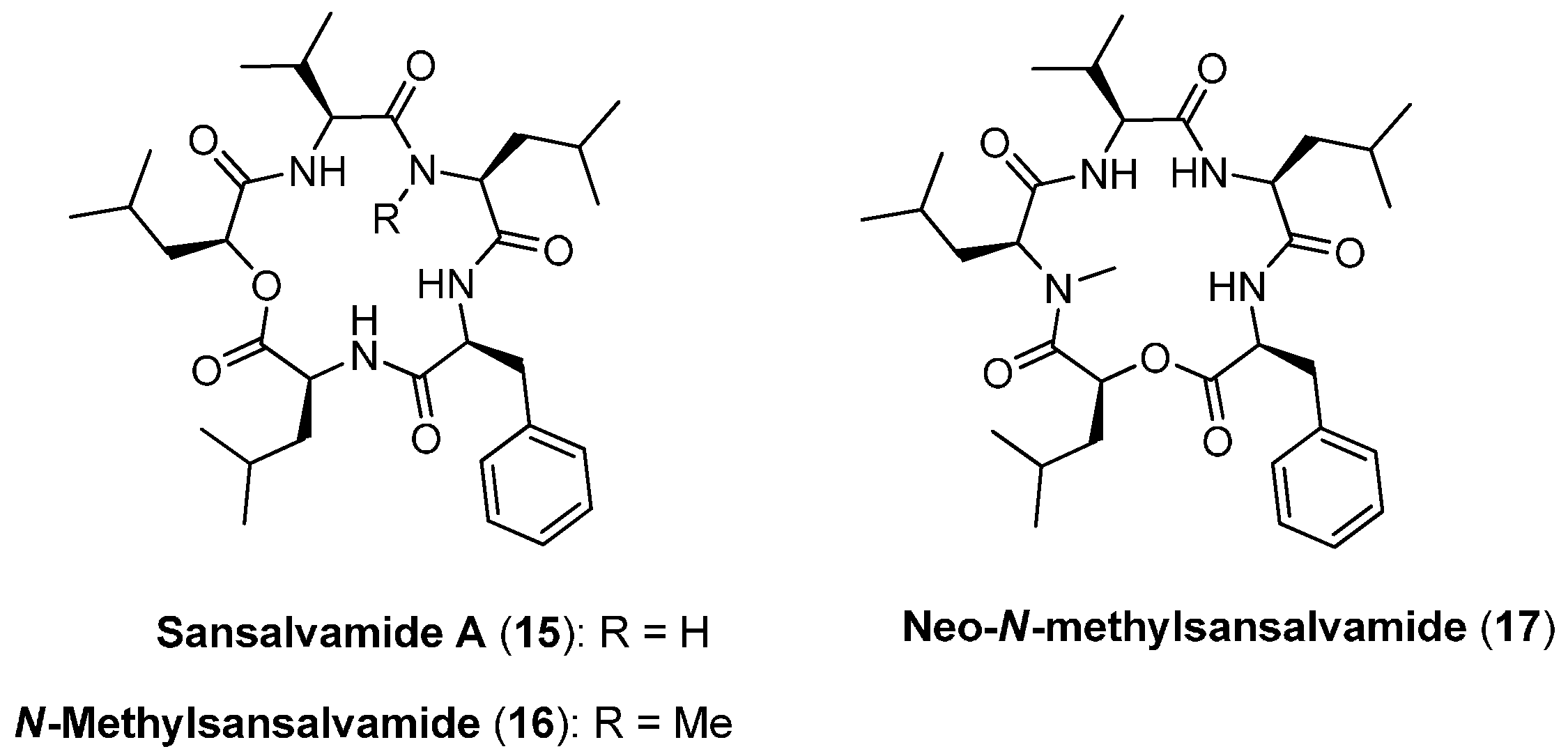
4.1. Sansalvamide A, N-methylsansalvamide and neo-N-methylsansalvamide


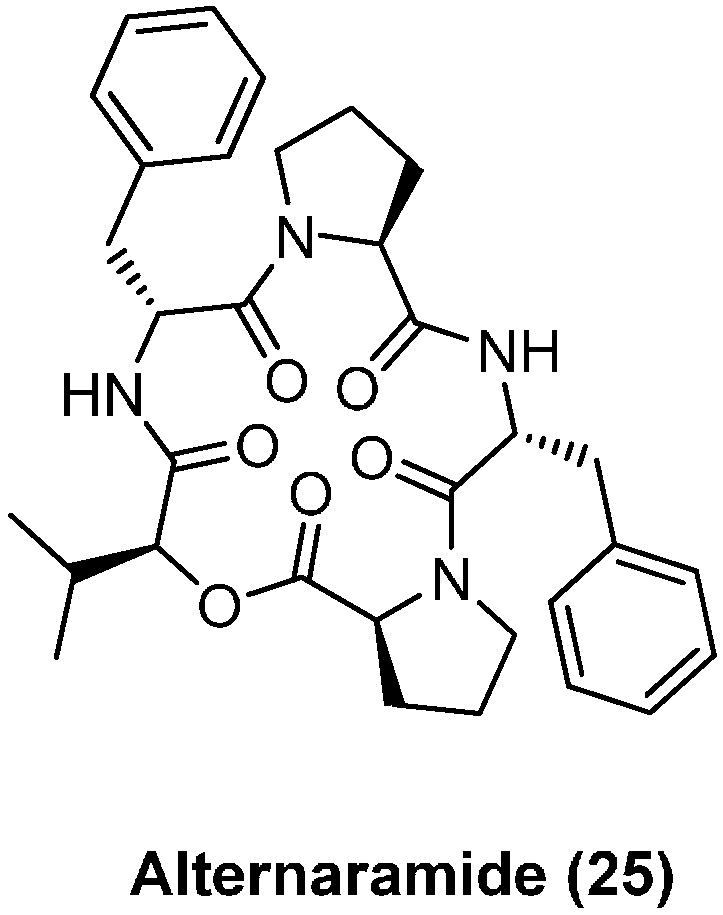
4.2. Alternaramide

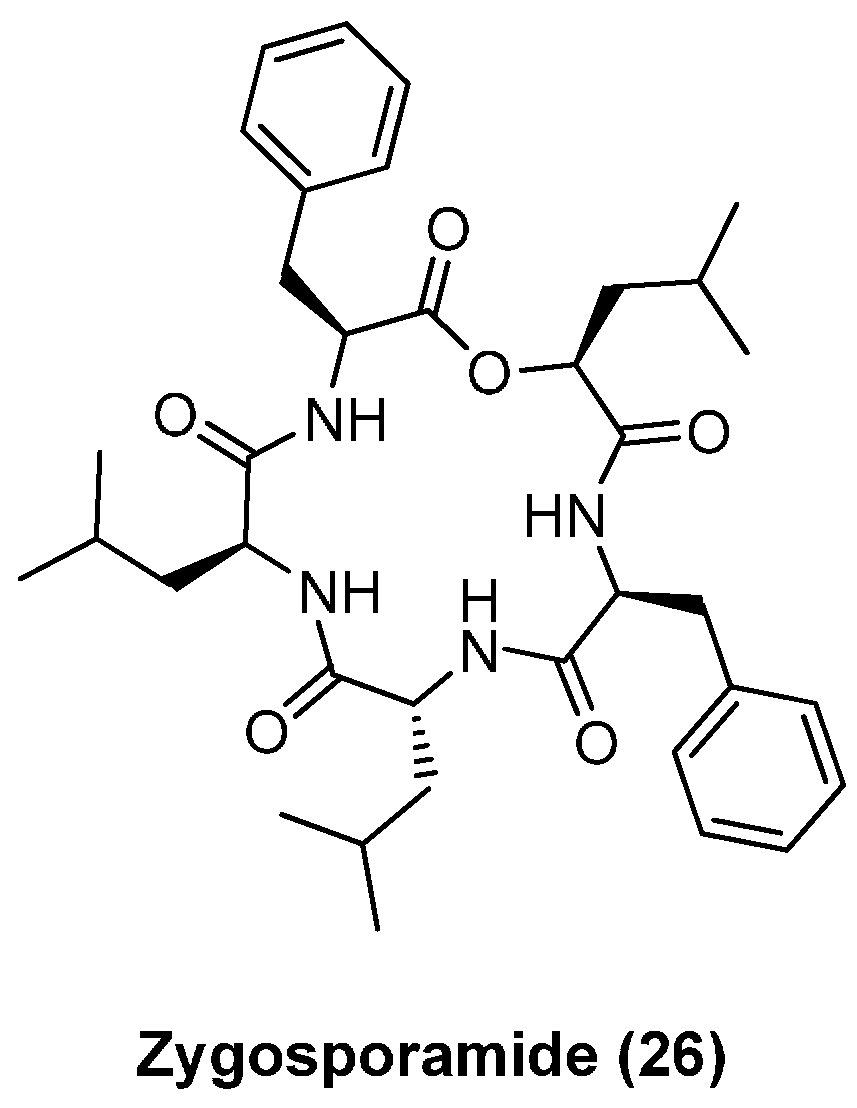
4.3. Zygosporamide



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
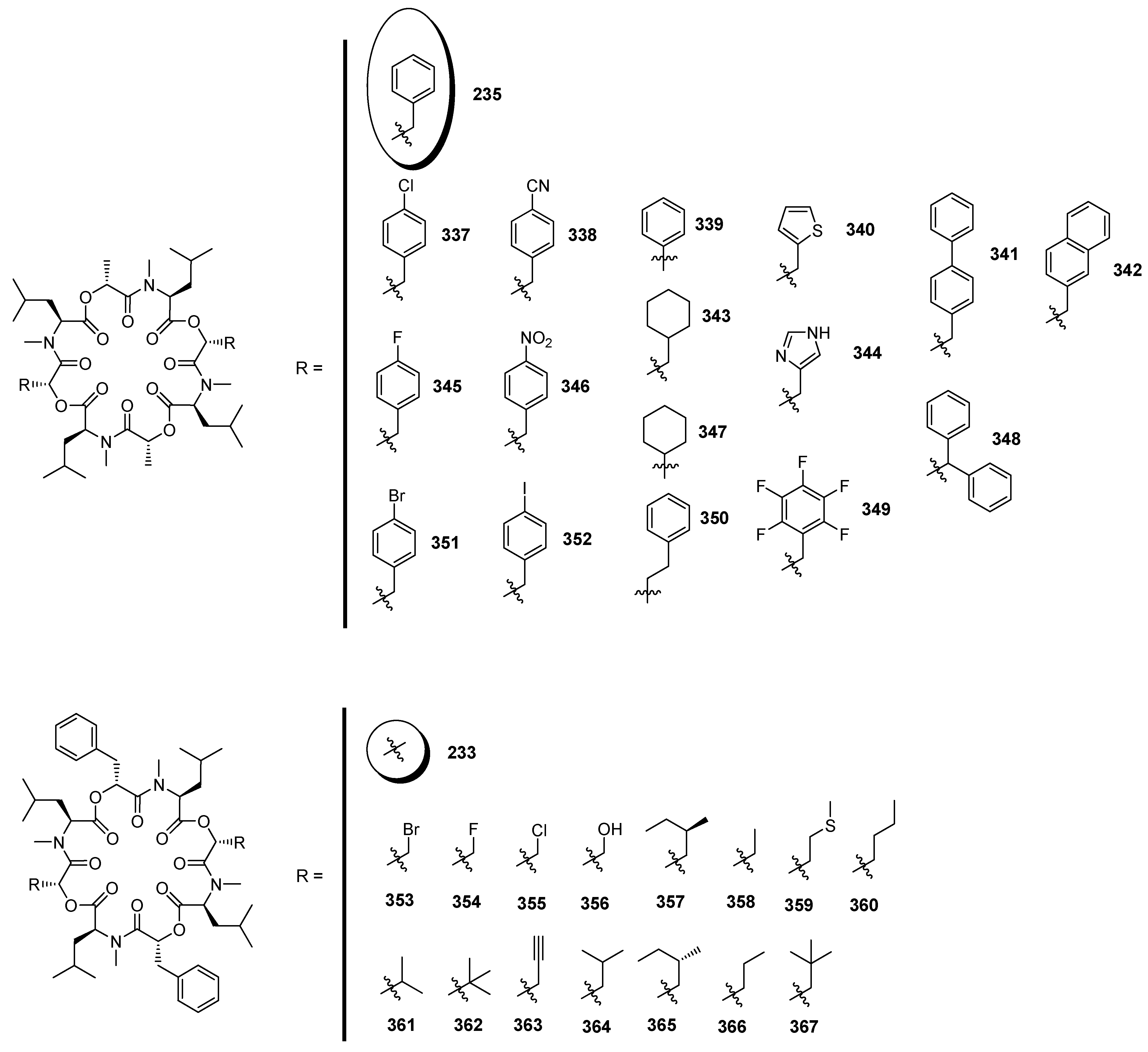{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | IC50 (µM) | ||||
|---|---|---|---|---|---|
| SF-268 | SF-295 | A549 | MDA-MB-231 | HCT-116 | |
| Zygosporamide (26) | 0.0065 | 15 | 7.4 | 8.5 | 11.5 |
| 31 | ~35 | 19.7 ± 8.8 | ~31 | 7.5 ± 6.3 | >50 |
| 32 | 10.4 ± 1.5 | 8.7 ± 0.9 | >50 | 5.0 ± 1.8 | 2.1 ± 0.4 |
| 33 | >50 | >50 | >50 | >50 | >50 |
| 34 | 8.8 ± 1.5 | ~6 | >50 | 2.8 ± 2.1 | 2.7 ± 0.6 |
| 35 | ~31 | ~32 | >50 | >50 | 7.8 ± 6.0 |
| 36 | 2.05 ± 2.0 | 14.4 | 21.0 ± 5.9 | >50 | 1.9 ± 1.3 |

5. Cyclohexadepsipeptides
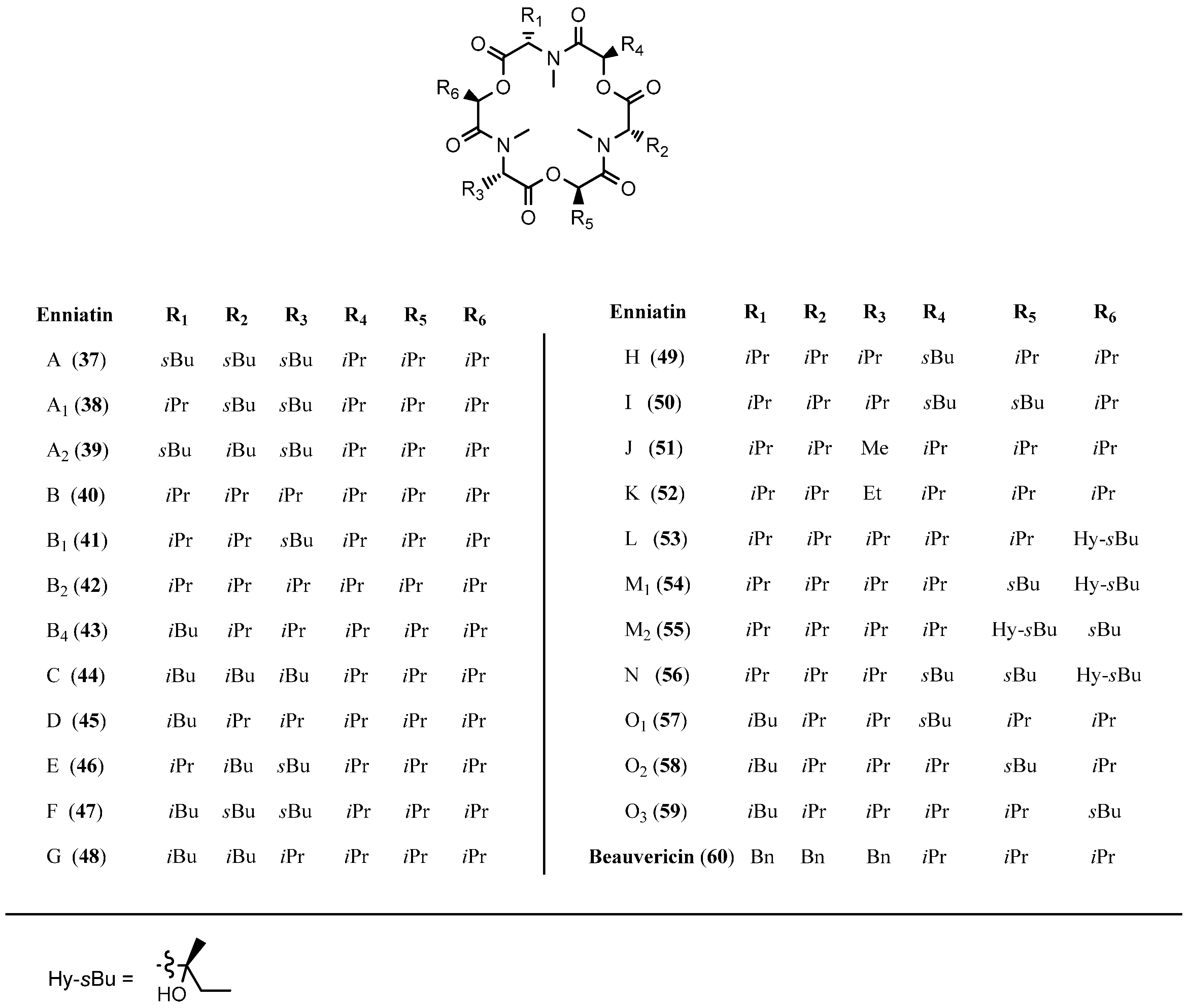
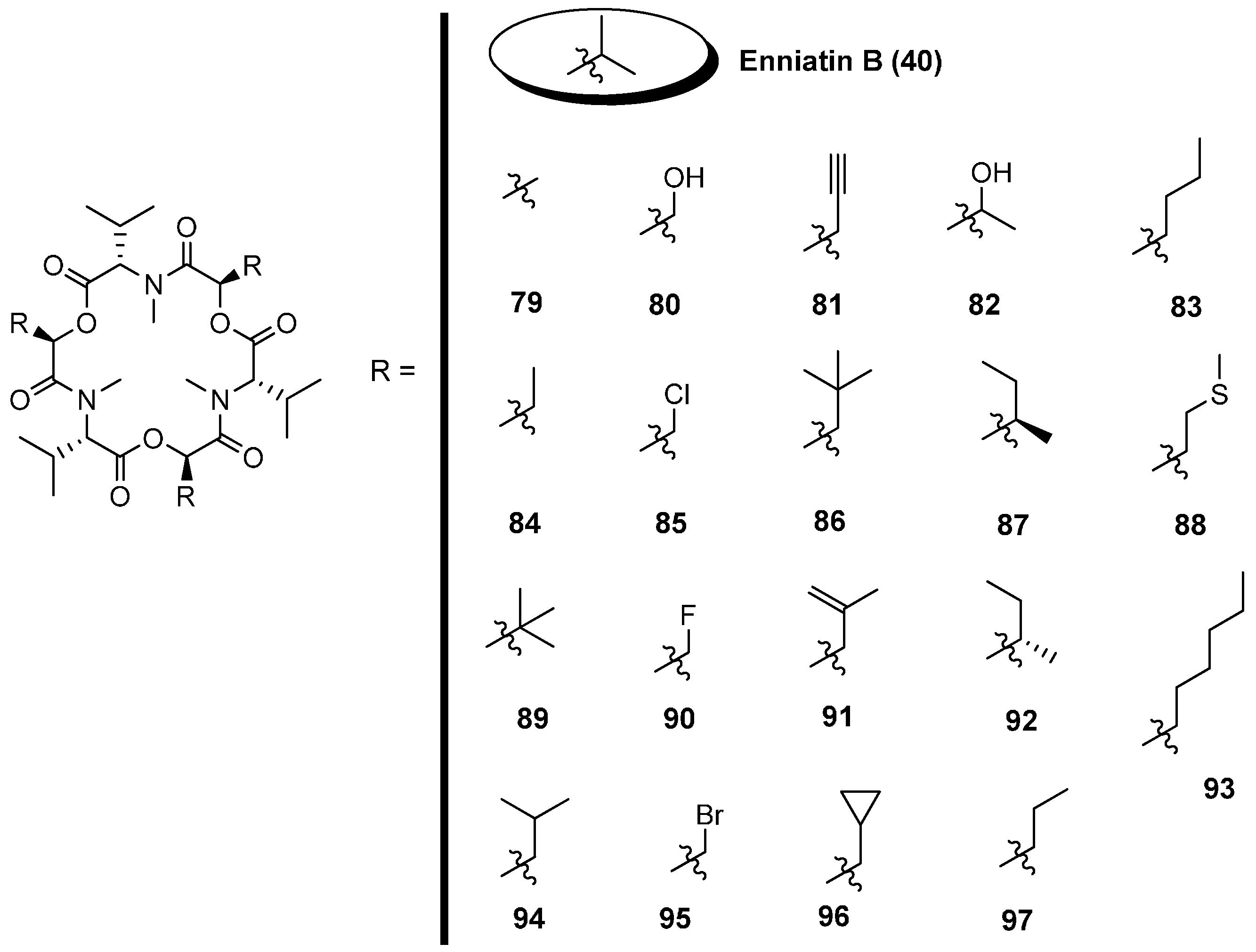
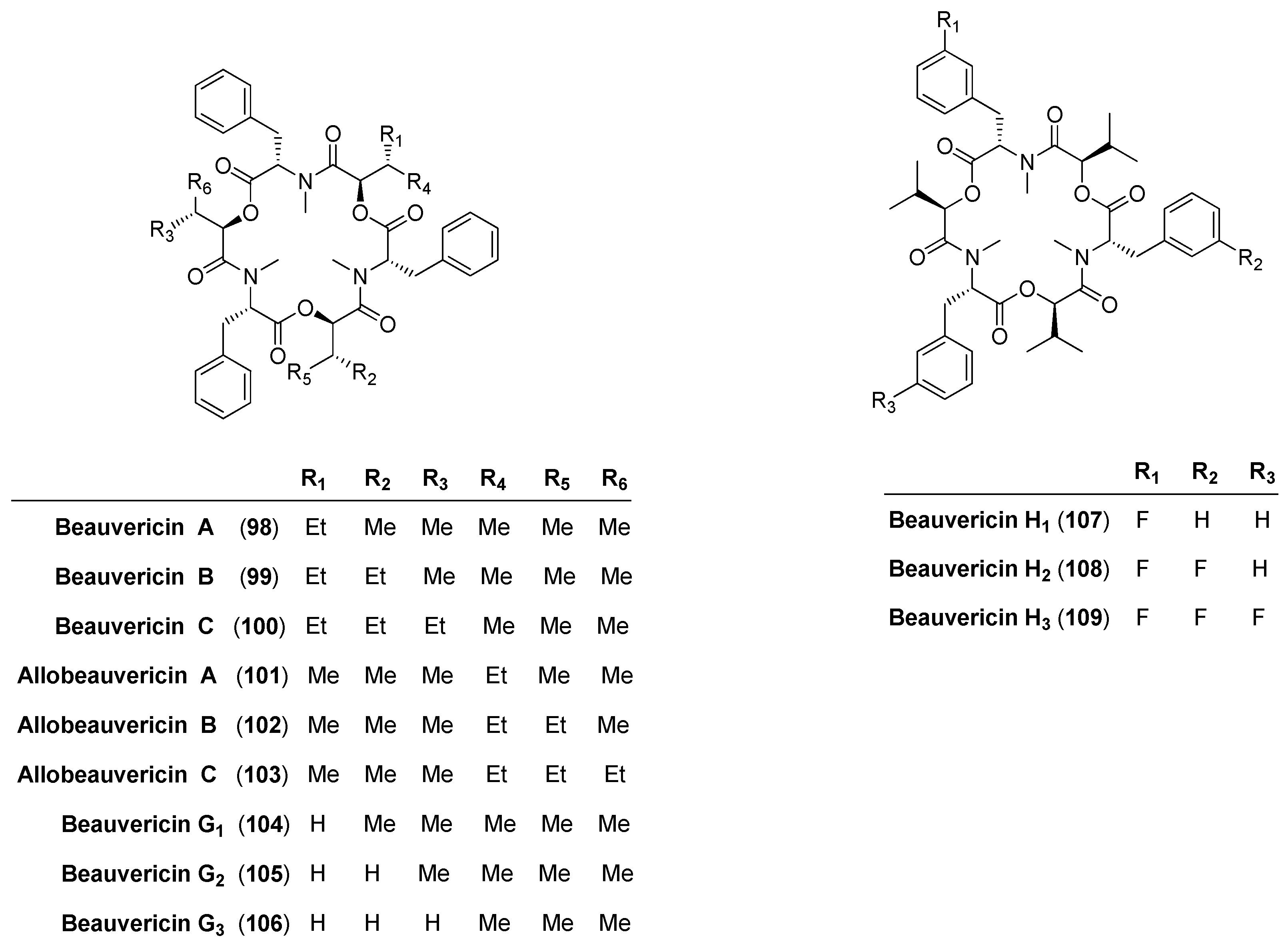
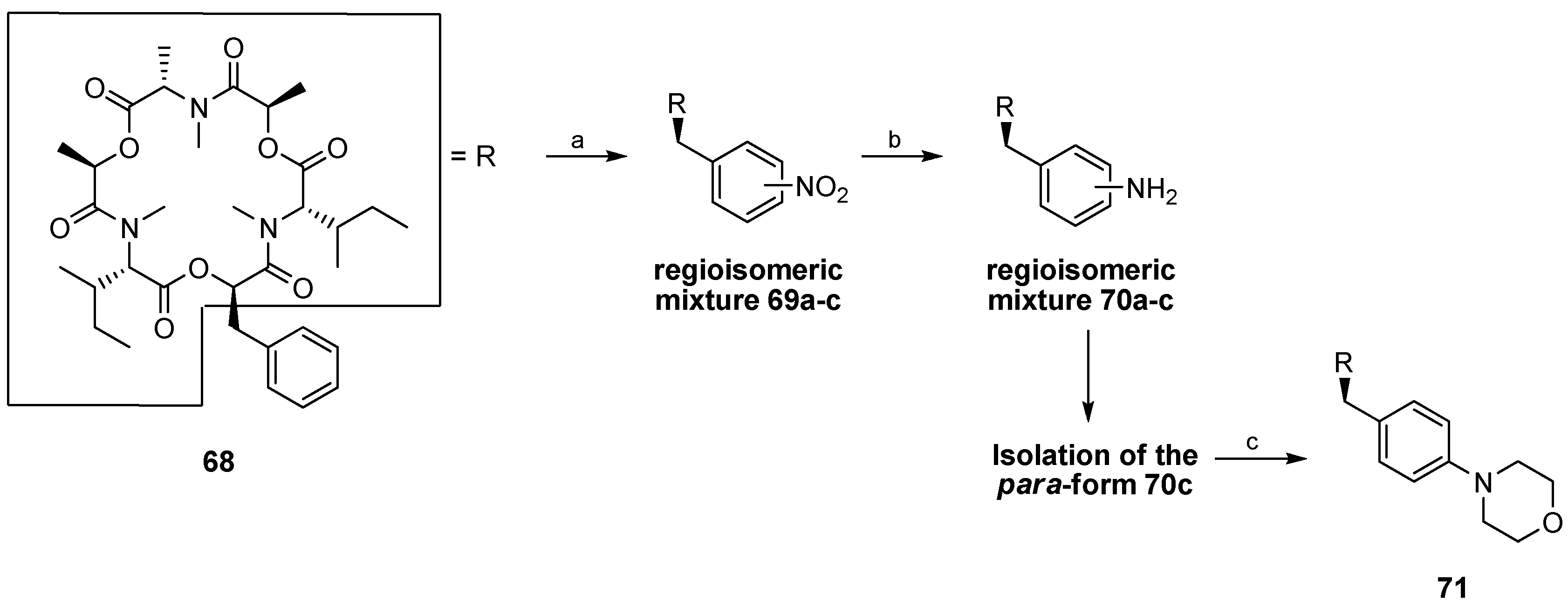
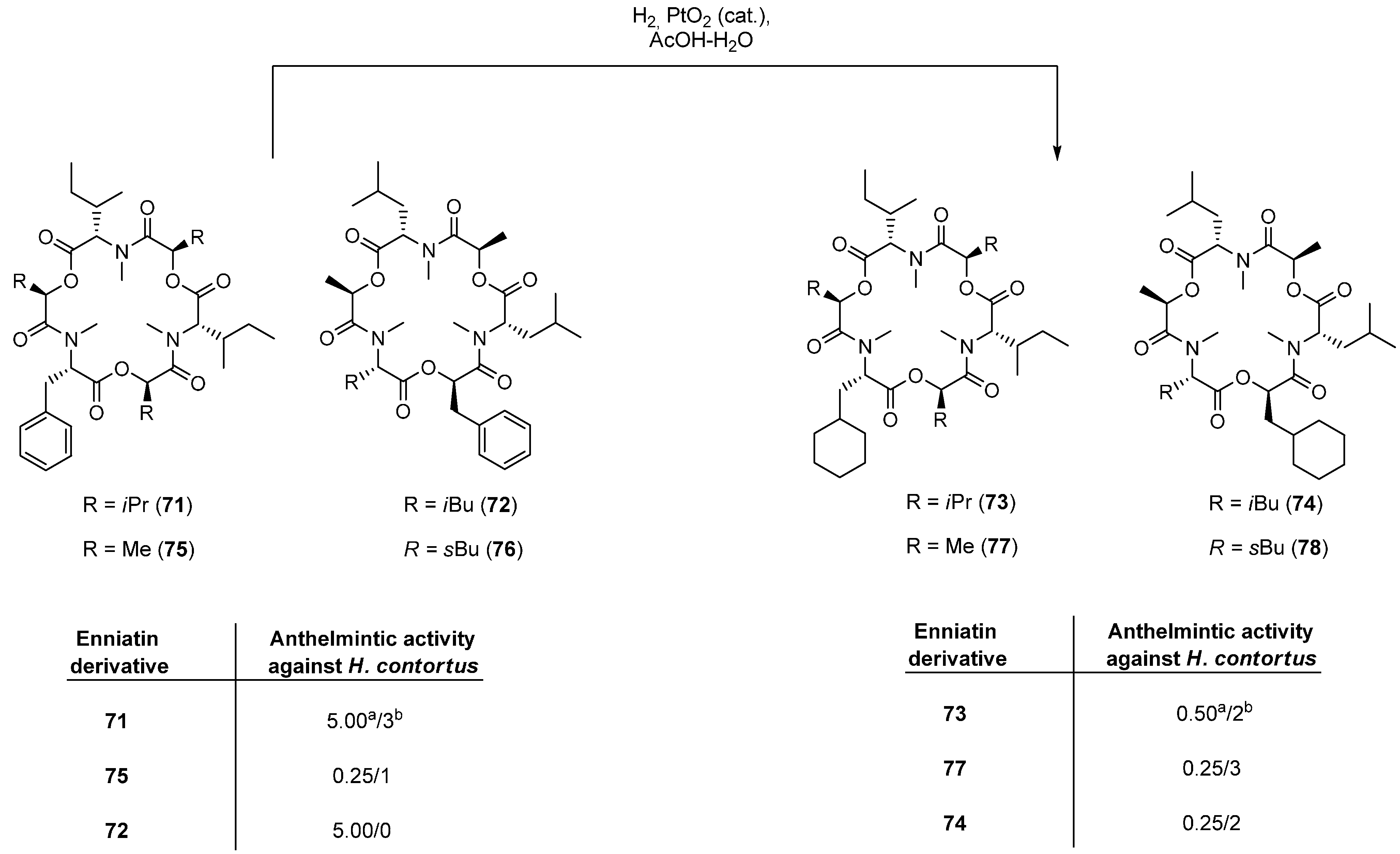
5.1. Enniatins and Beauvericin

5.1.1. Synthesis of Enniatins and Enniatin Derivatives

| Enniatin Derivative | Anthelminctic Activity against H. Contortus |
|---|---|
| 68 | 0.25 a/0 b |
| 69c (para) | 0.10/1 |
| 70a (ortho) | 0.05/3 |
| 70b (meta) | 0.05/3 |
| 70c (para) | 0.10/1 |
| 71 | 0.10/3 |


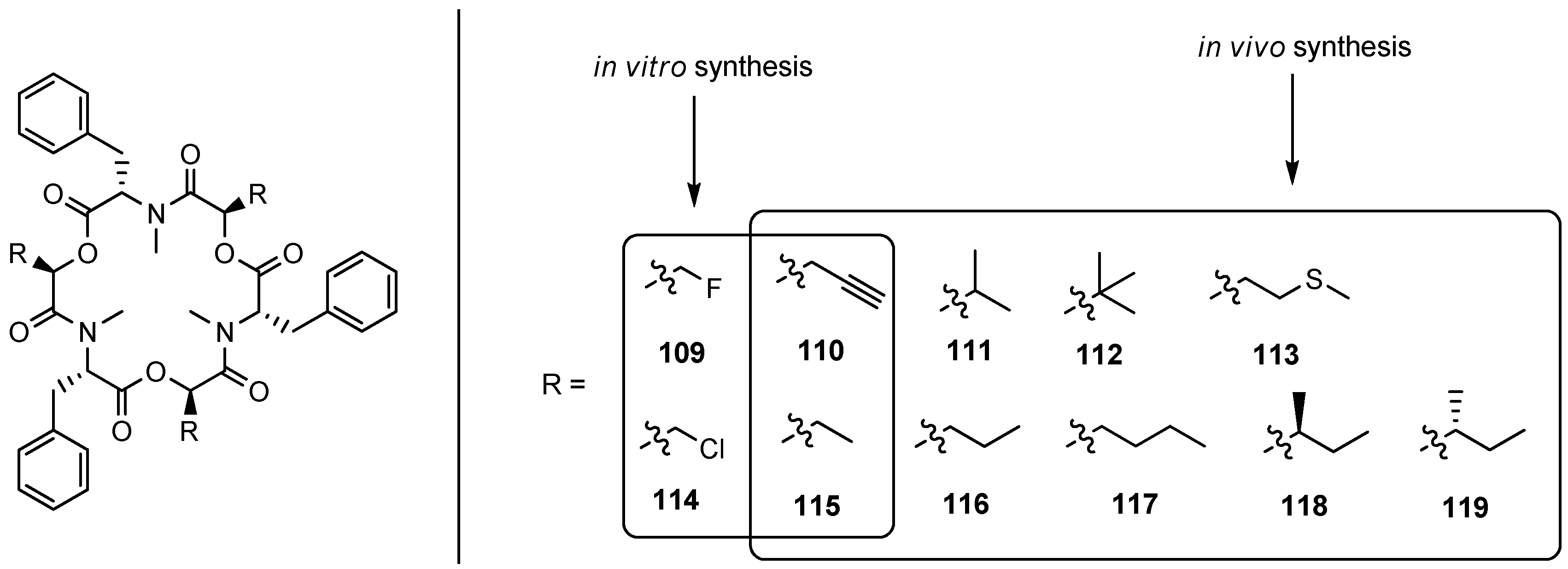
5.1.2. Biosynthesis of Enniatins and Beauvericin



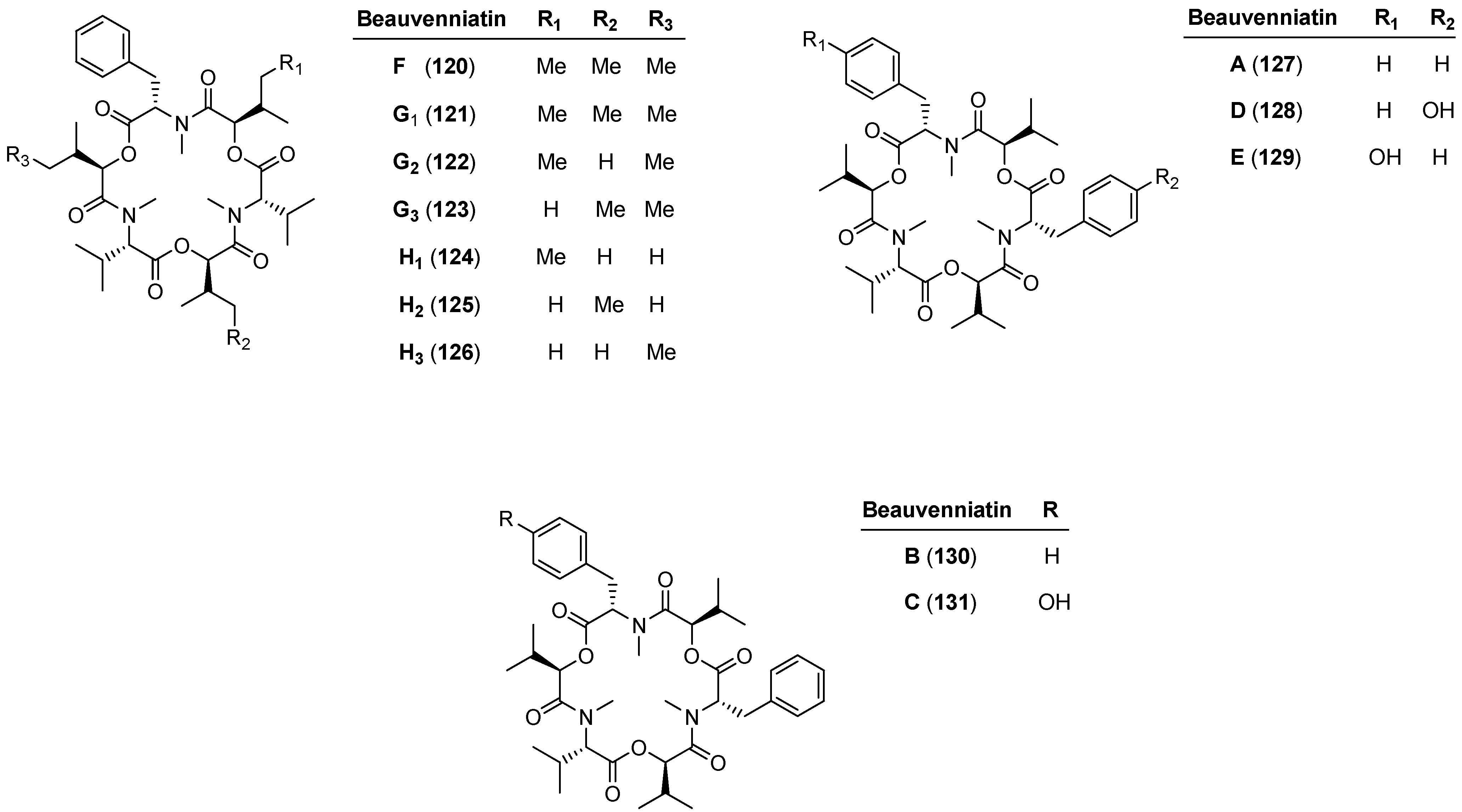
5.2. Beauvenniatins

| Beauvenniatin | Cytotoxicity (IC50) | Antimalaria (IC50) | Antituberculosis (IC50) | ||
|---|---|---|---|---|---|
| NCI-H187 g | KB h | Vero i | |||
| 127 | 1.0 a | 1.3 a | 2.2 a | 3.0 c | 3.13 e |
| 130 | 0.92 a | 1.6 a | 1.3 a | 3.0 c | 3.13 e |
| 131 | 6.6 a | 14 a | 7.0 a | 3.4 c | ˃50 e |
| 128 | ˃50 a | ˃50 a | ˃50 a | ˃10 c | ˃50 e |
| 129 | 7.6 a | 9.0 a | 8.3 a | 2.9 c | 25 e |
| 120 | 2.29 ± 1.26 b | 1.05 ± 0.05 b | 5.5 b | 3.8 ± 0.1 d | 1.07 f |
| 121–123 | 1.23 ± 0.49 b | 1.06 ± 0.06 b | 4.1 b | 3.9 ± 0.4 d | 2.18 f |
| 124–126 | 1.45 ± 0.38 b | 1.15 ± 0.11 b | 1.9 b | 3.6 ± 0.9 d | 4.45 f |
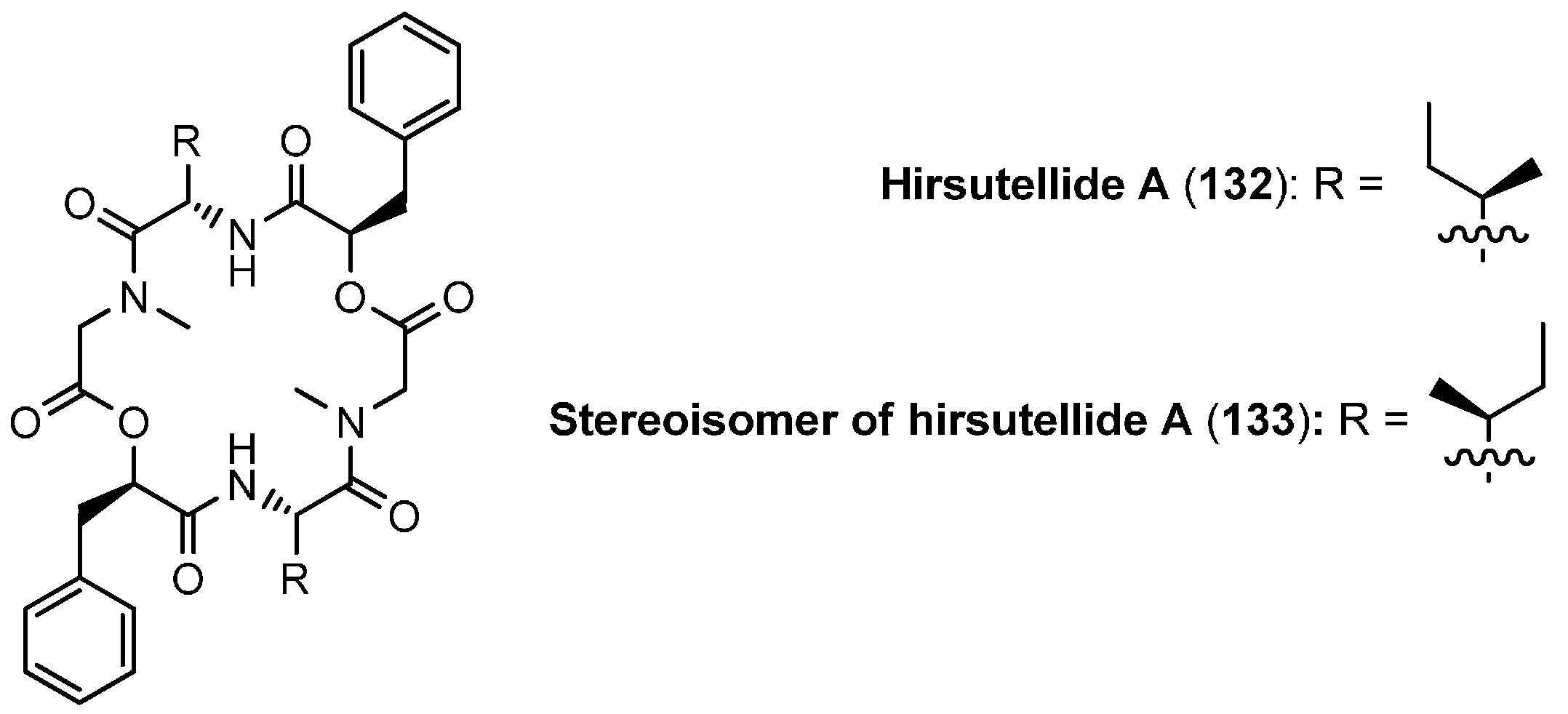
5.3. Hirsutellide A

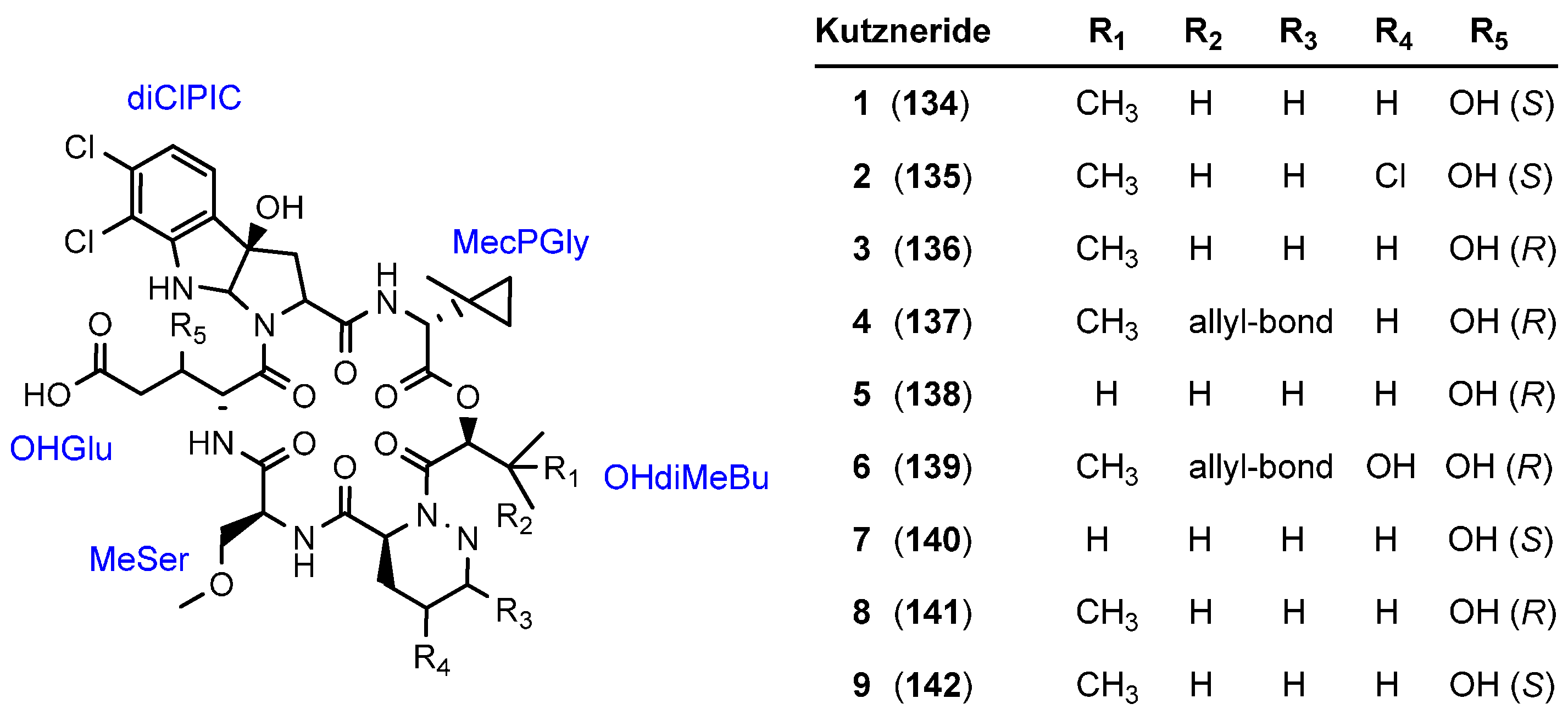
5.4. Kutznerides

| Kutzneride | MIC values (µM) of Pathogens | ||
|---|---|---|---|
| Drechslera sorokiniana (agricultural fungus) | Erwina carotovora (agricultural bacterium) | Staphylococcus aureus (human bacterium) | |
| 1 (134) | 230 | 60 | 12 |
| 2 (135) | 110 | 6 | 6 |
| 3 (136) | 260 | 12 | 9 |
| 4 (137) | ˃590 a | 120 | 140 |
| 5 (138) | ˃240 a | 120 | 120 |
| 6 (139) | ˃400 a | ˃230 a | ˃230 a |
| 7 (140) | ˃420 a | 180 | 120 |
| 8 (141) | 230 | 6 | 6 |
| 9 (142) | ˃230 | 230 | 60 |
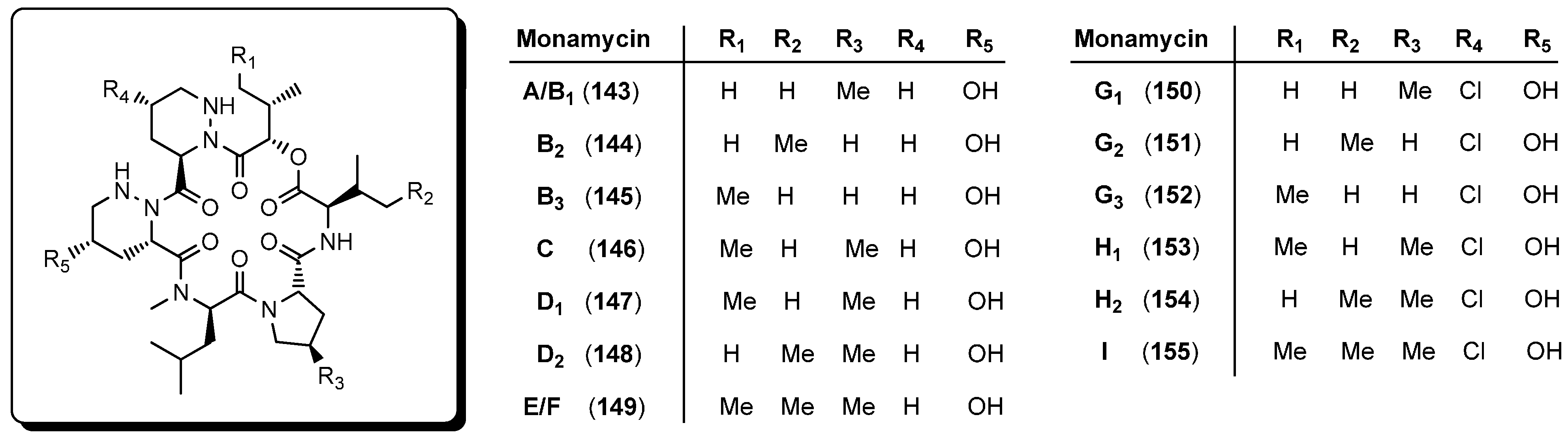
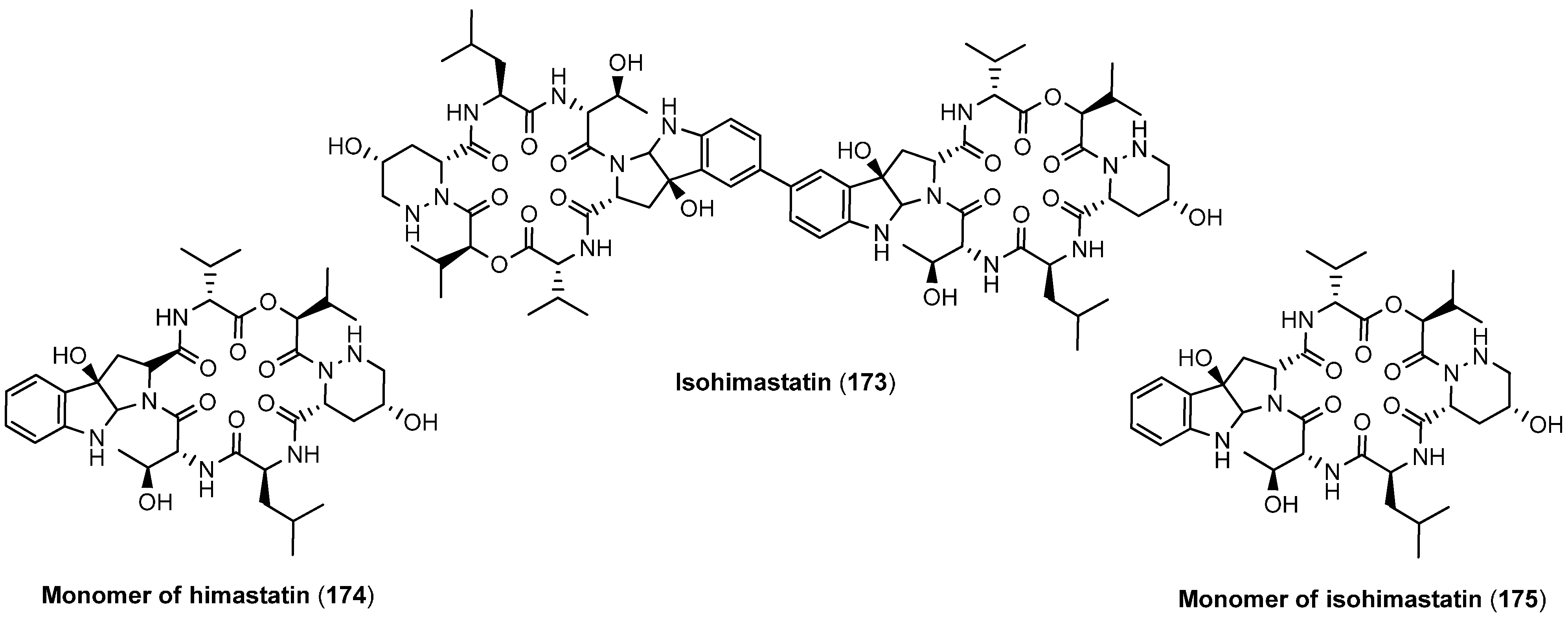
5.5. Monamycins

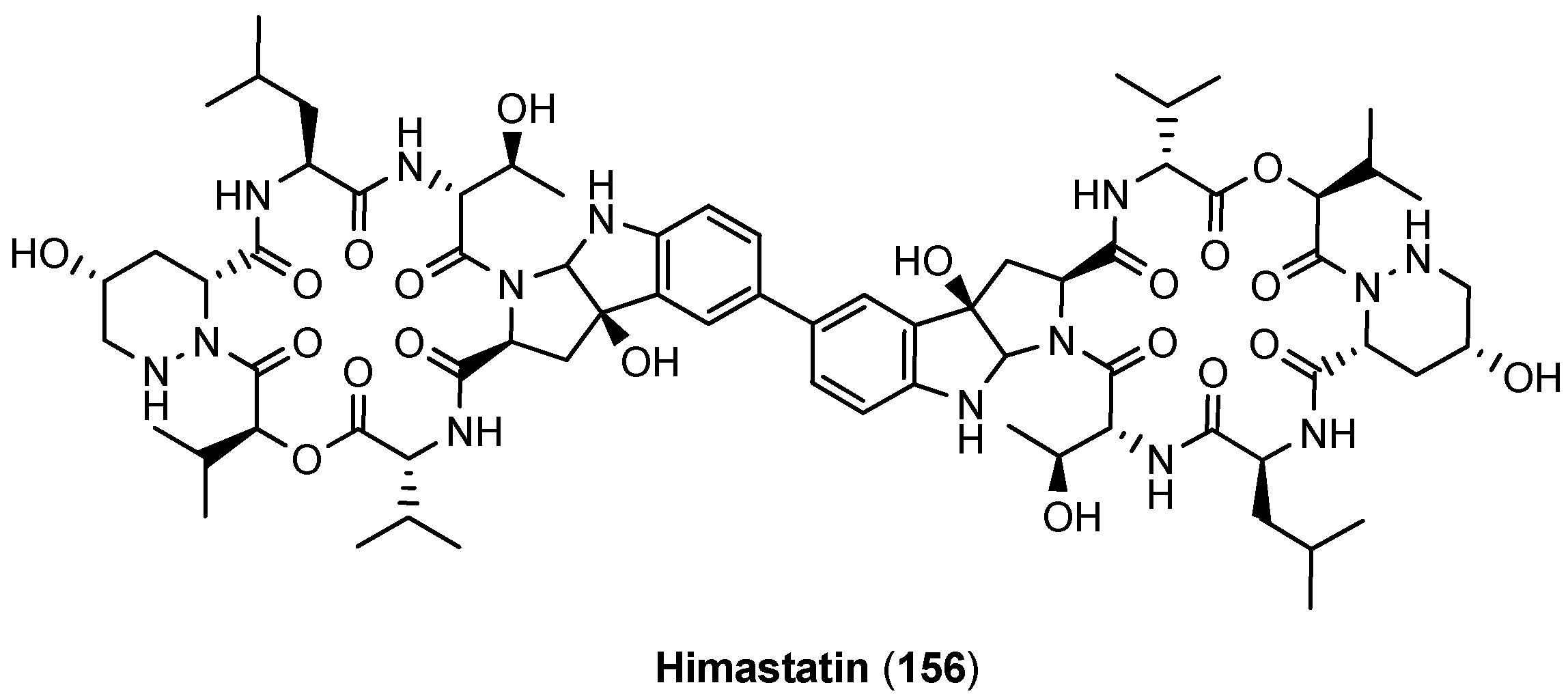
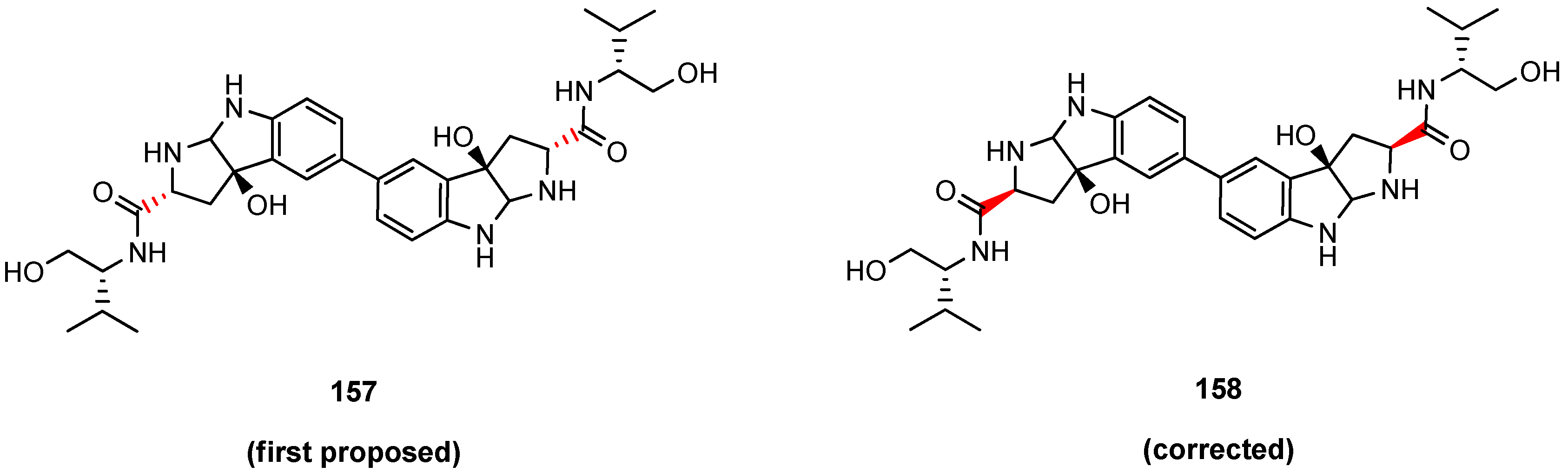
5.6. Himastatin

| Organism | MIC (µg/mL) | Organism | MIC (µg/mL) |
|---|---|---|---|
| E. faecalis A20688 | 0.5 | E. coli A20697 | ˃500 |
| E. faecalis A25707 | 0.5 | E. coli A9751 | 2 |
| E. faecalis A25707 | 0.25 | Klebsiella pneumoniae A9664 | ˃500 |
| S.aureus A9537 | 0.5 | K. pneumonia A20468 | ˃500 |
| S. aures A20698 | 1 | Proteus vulgaris A21559 | ˃500 |
| S. aureus A24407 | 1 | Pseudonomas aeruginosa A9843 | ˃500 |
| Bacillus subtilis A9506-A | 2 | P. aeruginosa A20235) | ˃500 |
| Escherichia coli A15119 | ˃500 | P. aeruginosa A20235 | ˃500 |




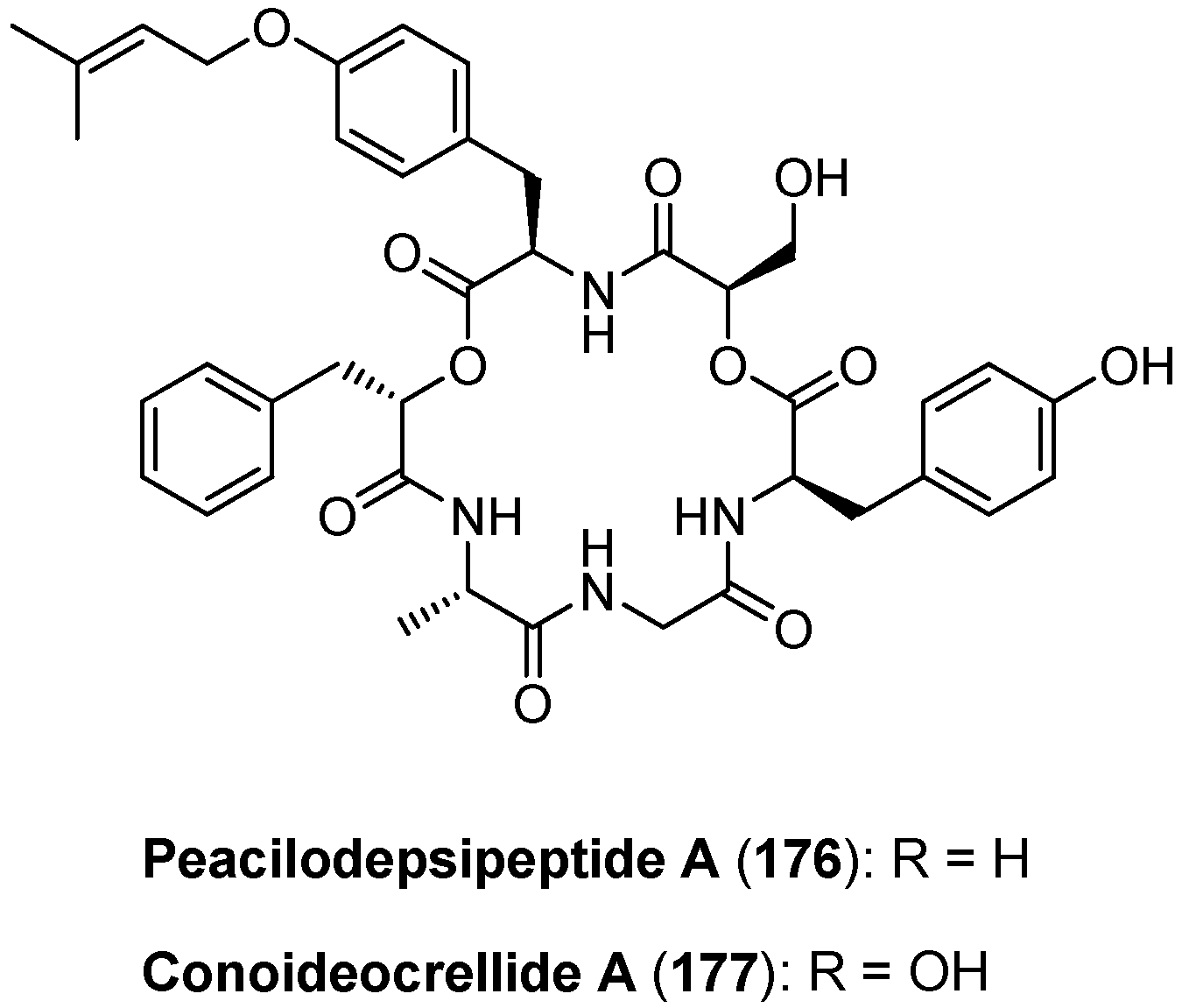
5.7. Paecilodepsipeptide A and Conoideocrellide A


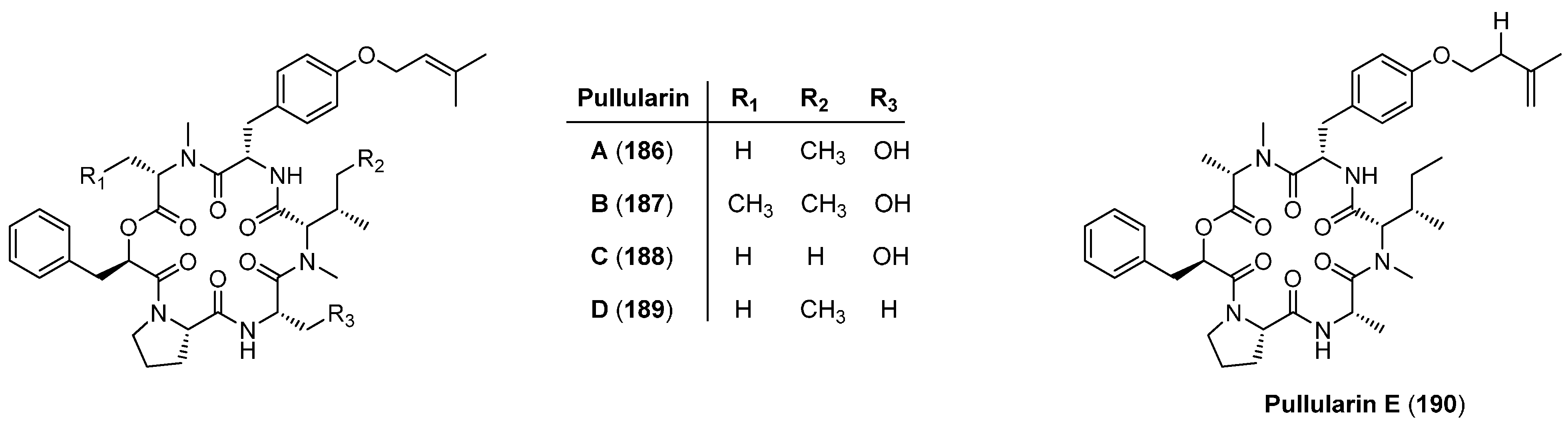
5.8. Pullularins A-E

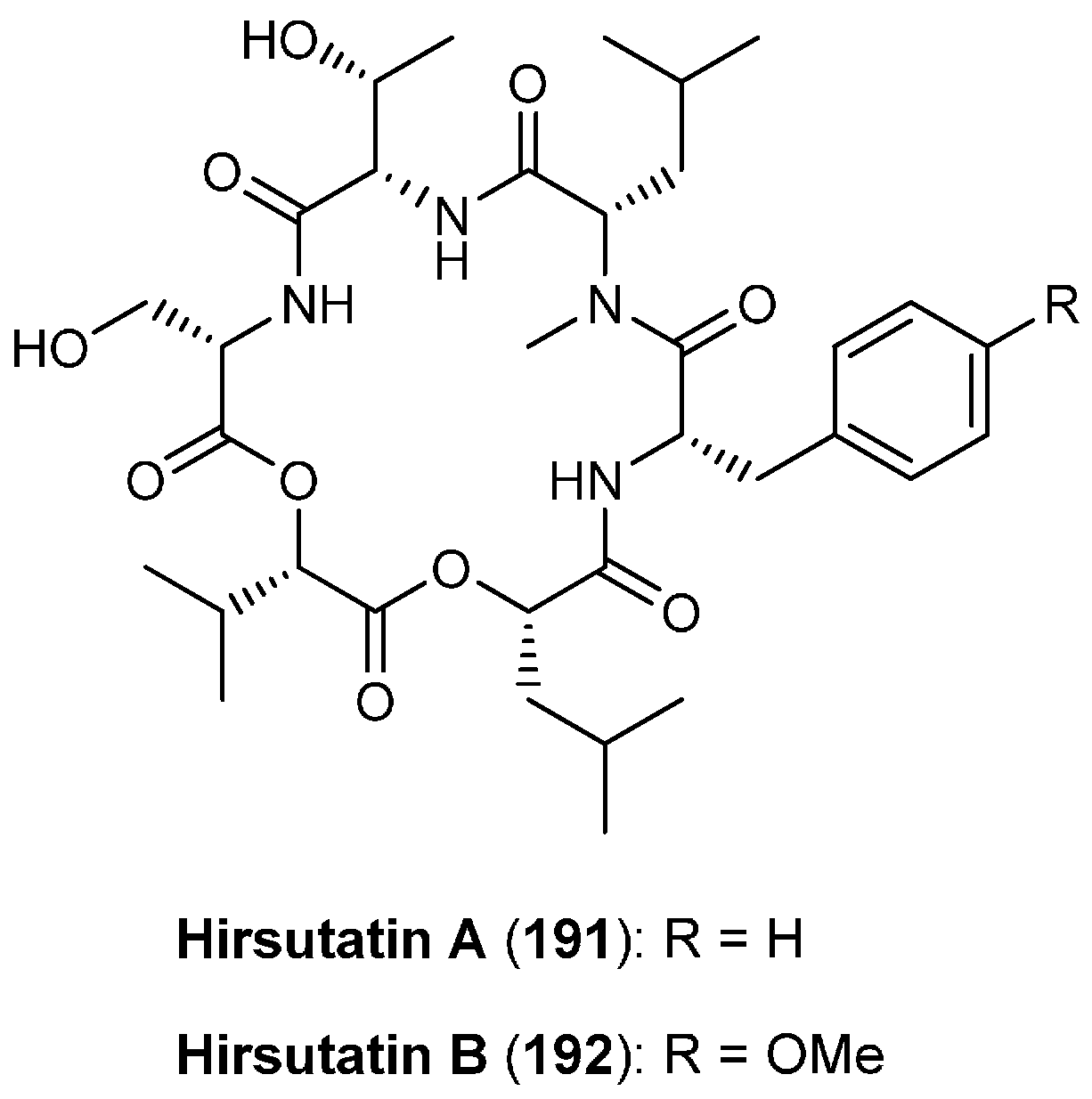
5.9. Hirsutatins A and B

6. Cycloheptadepsipeptides
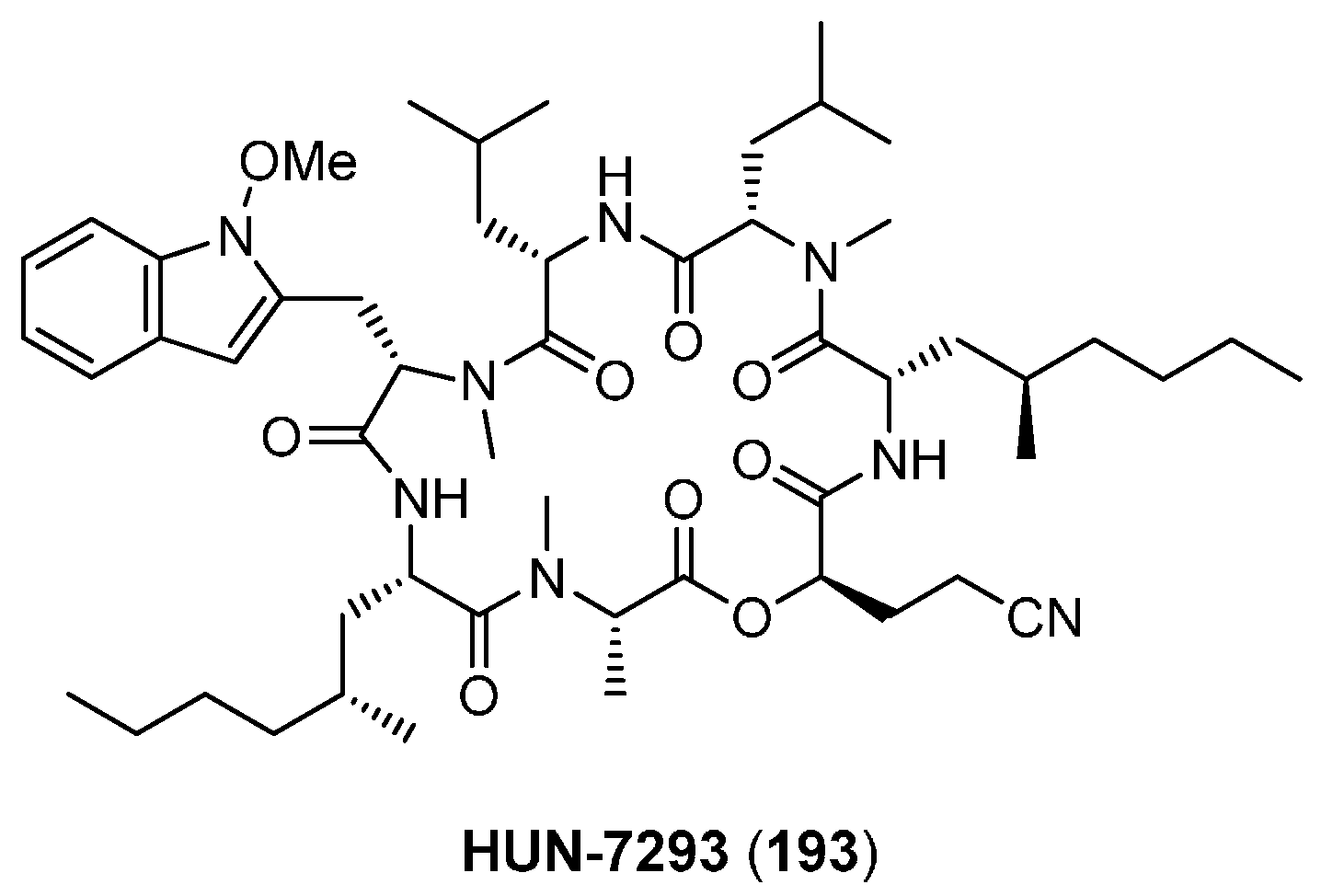
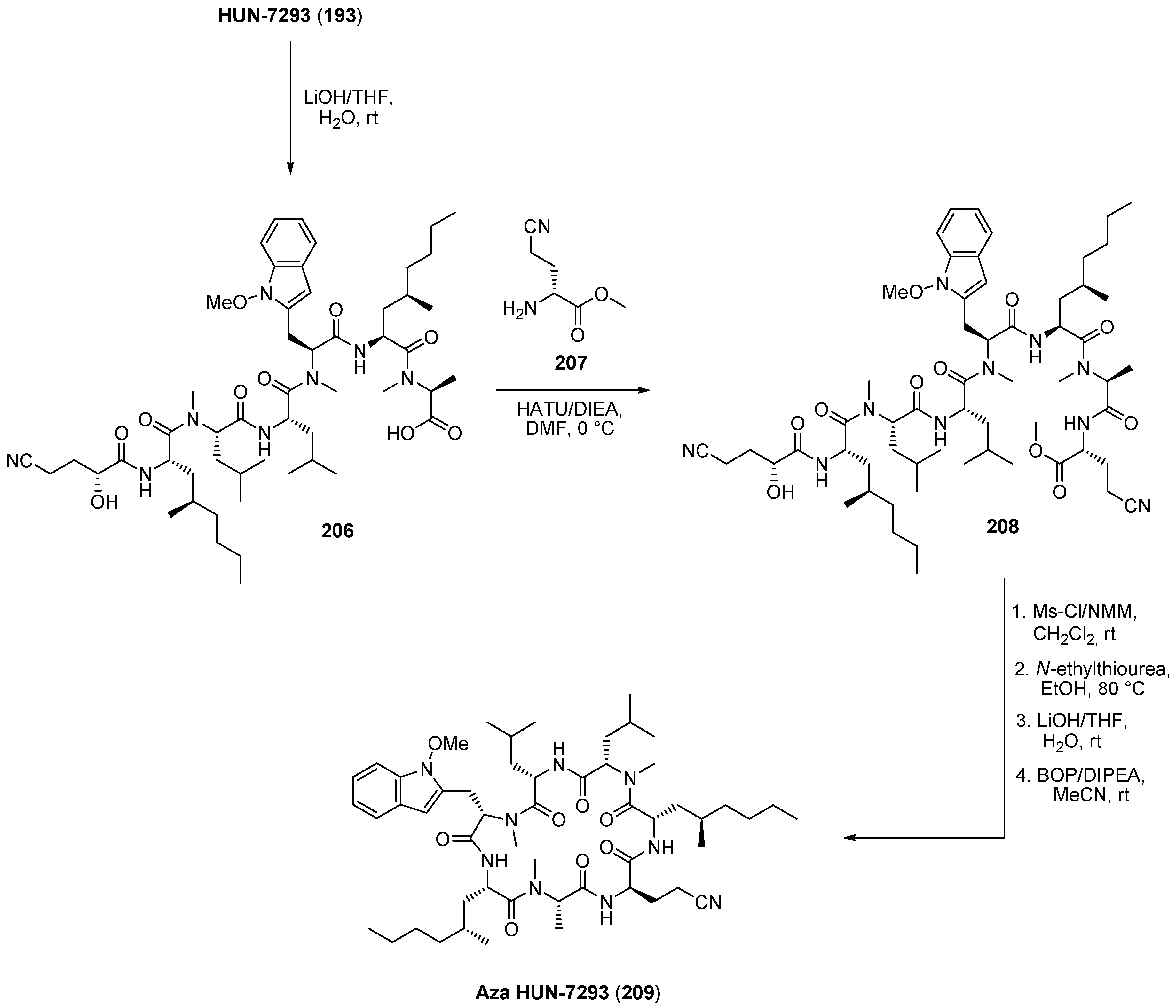
6.1. HUN-7293




7. Cyclooctadepsipeptides
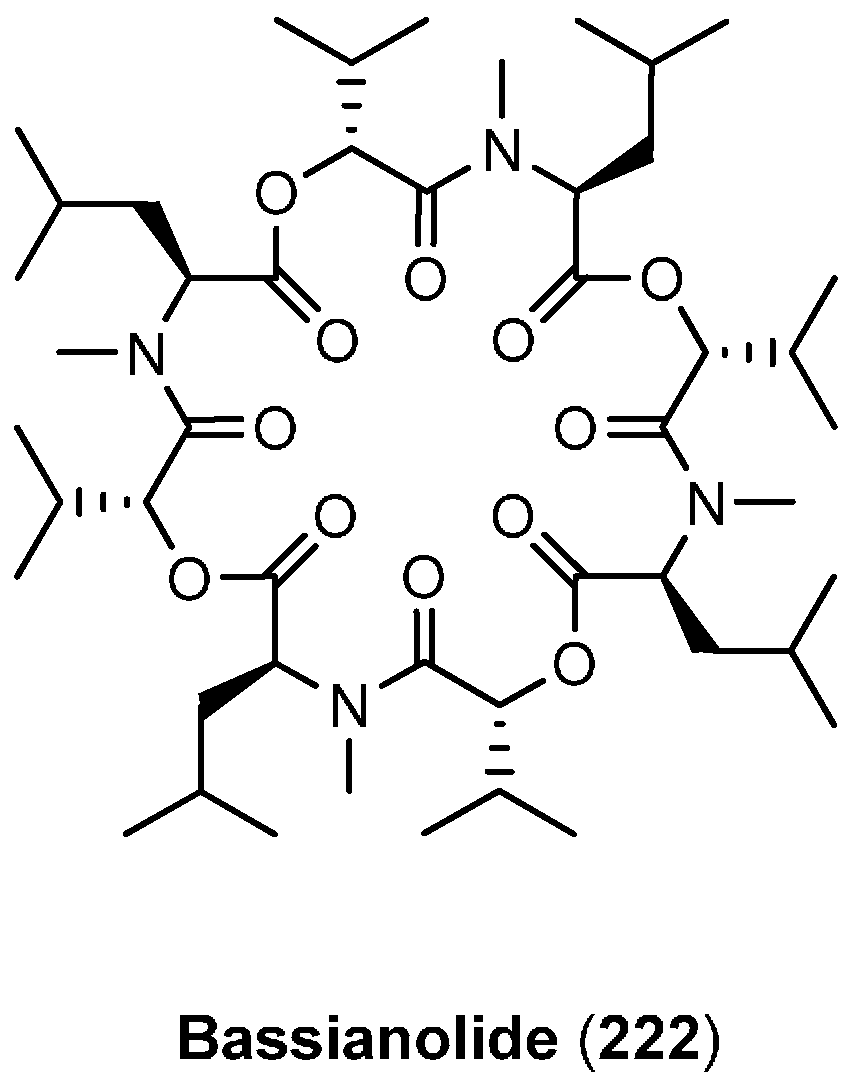
7.1. Bassianolide

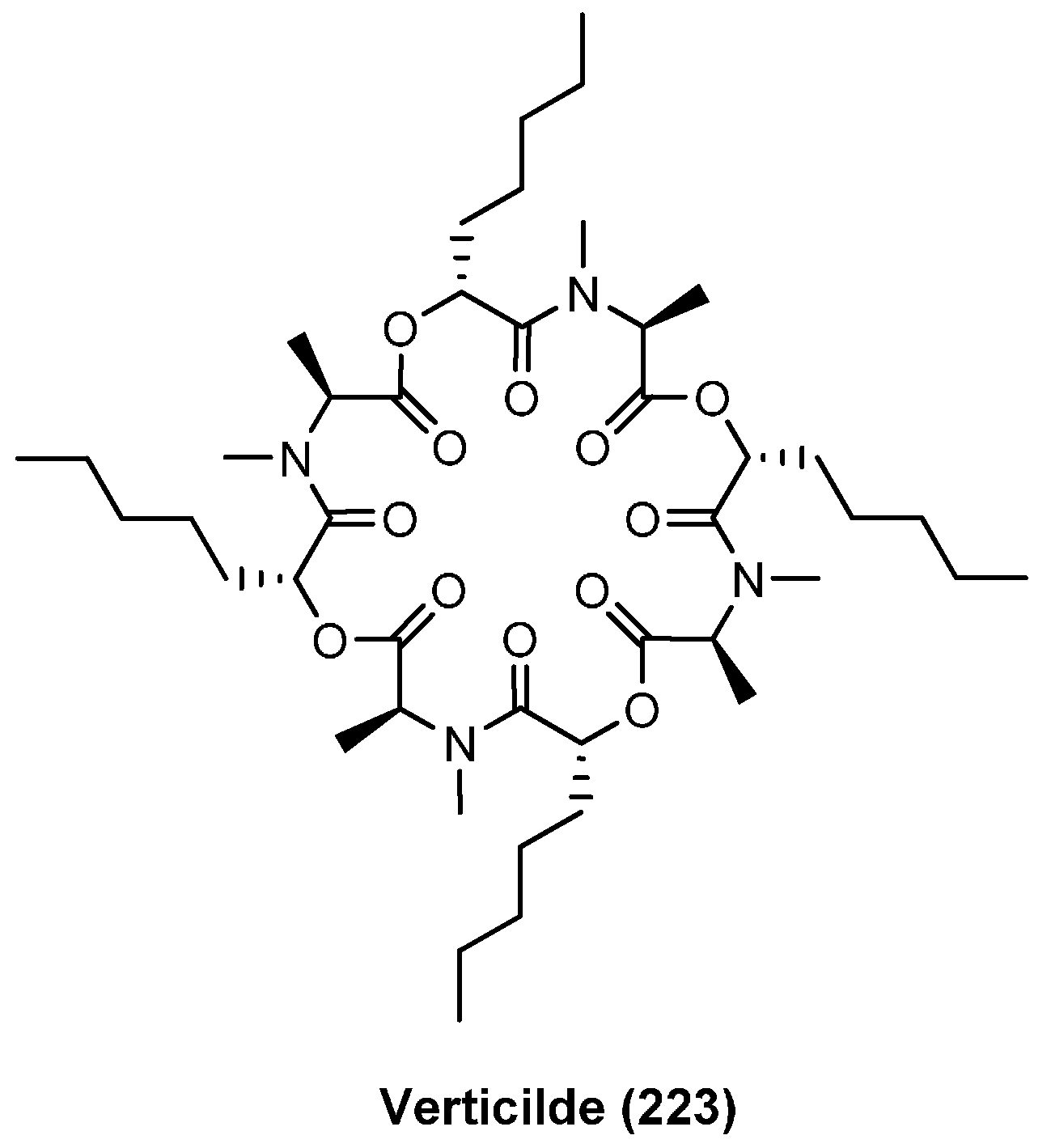
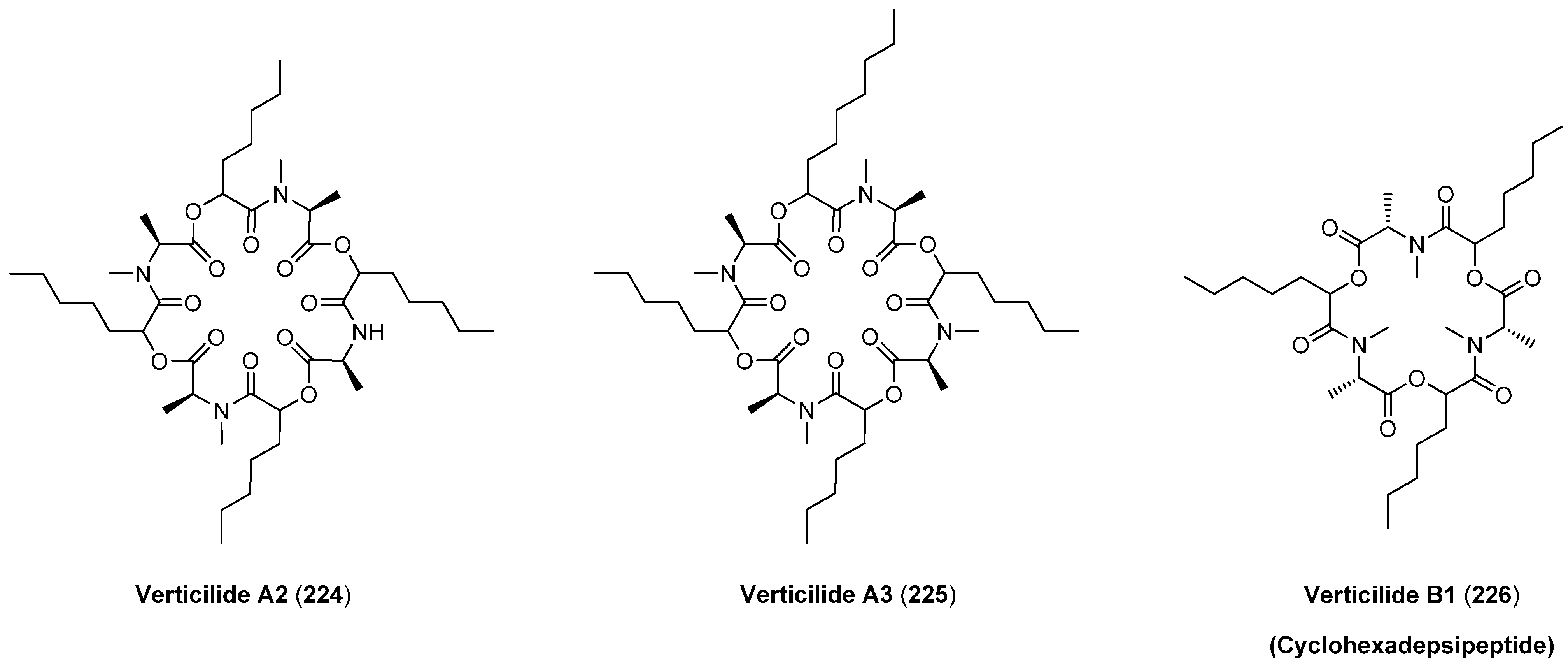
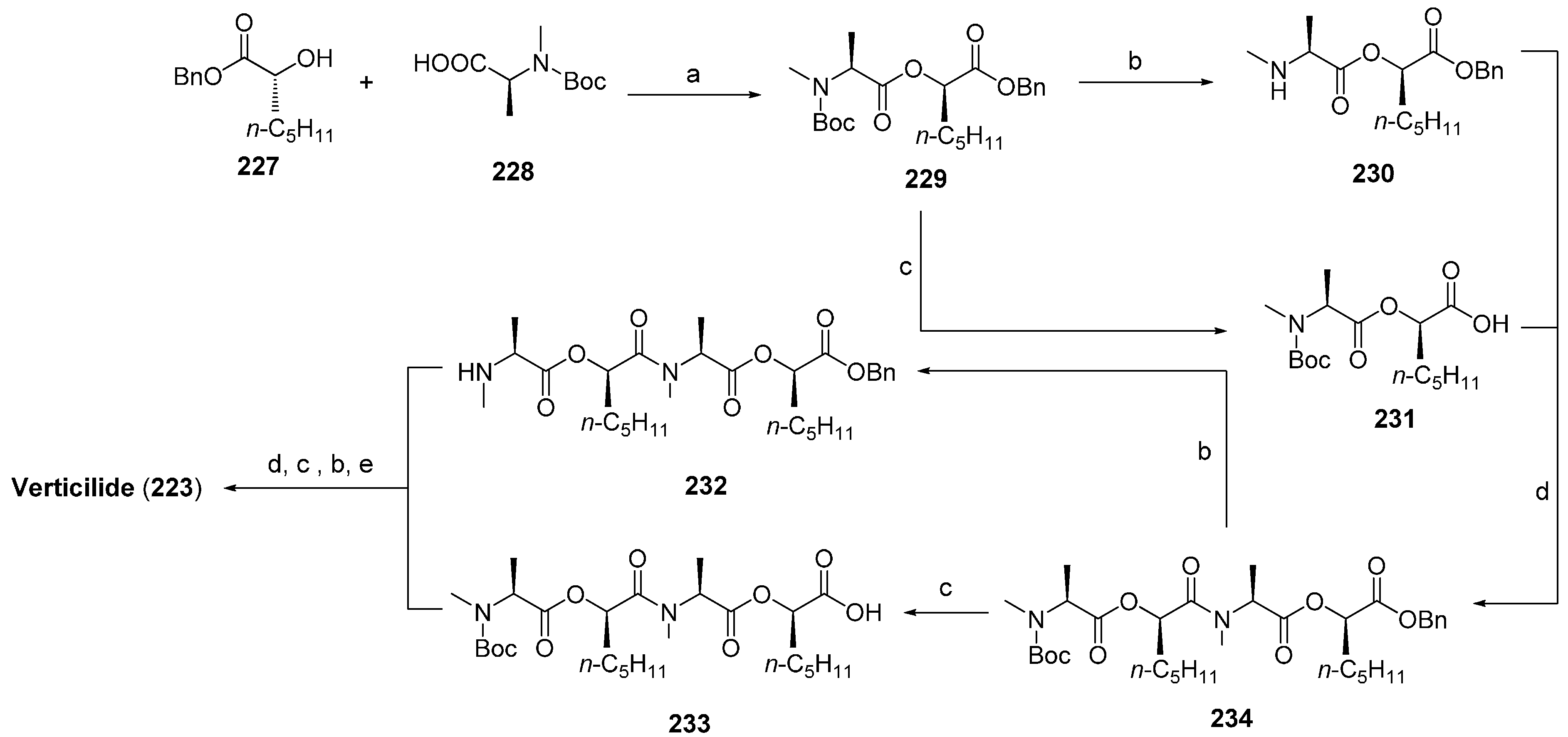
7.2. Verticlide

| Verticilde | IC50 (µM) | |
|---|---|---|
| ACAT1 | ACAT2 | |
| A1 (223) | 2.5 | 0.23 |
| A2 (224) | 4.8 | 0.55 |
| A3 (225) | 3.5 | 0.36 |
| B1 (226) | 11 | 1.3 |


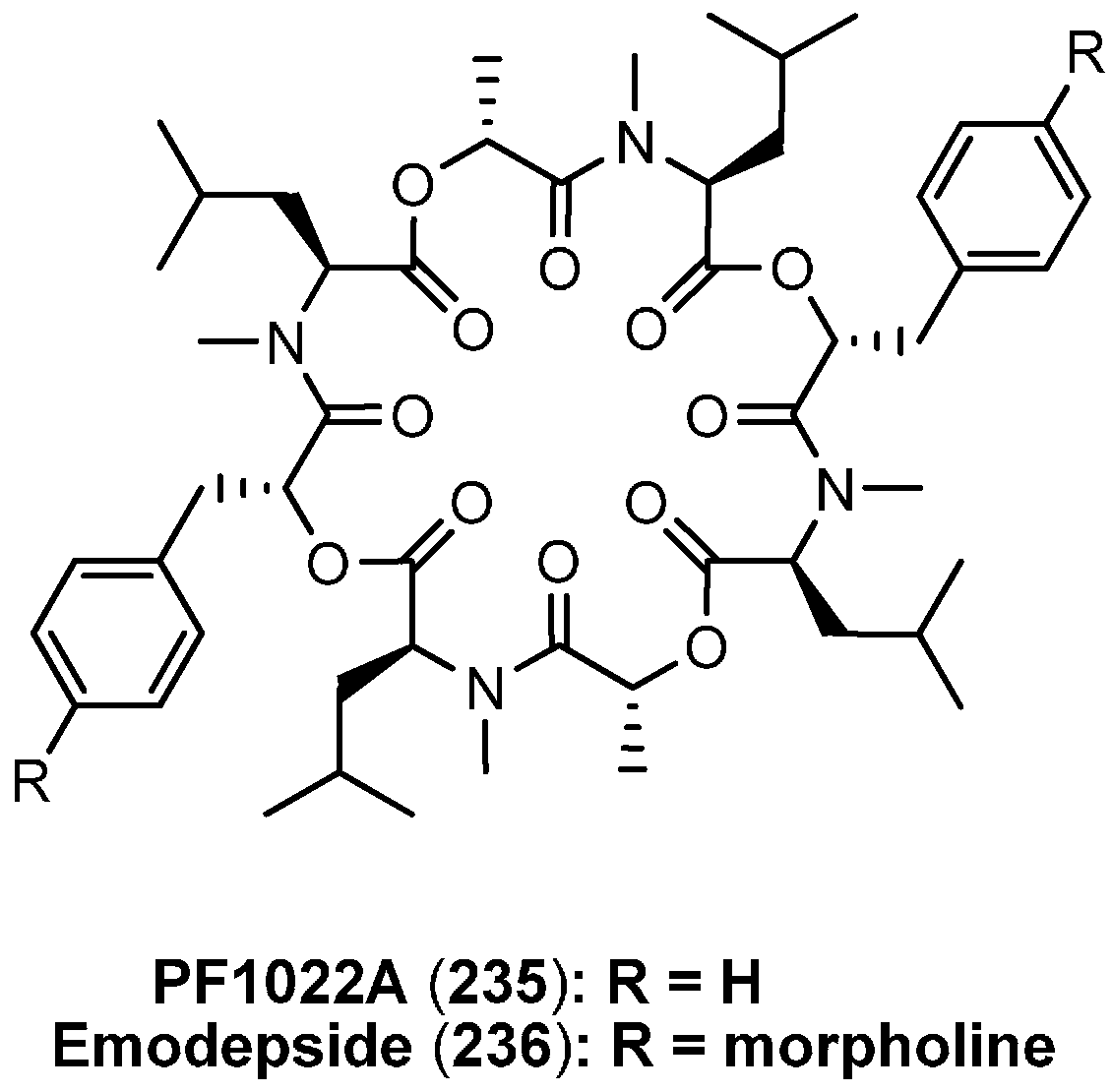
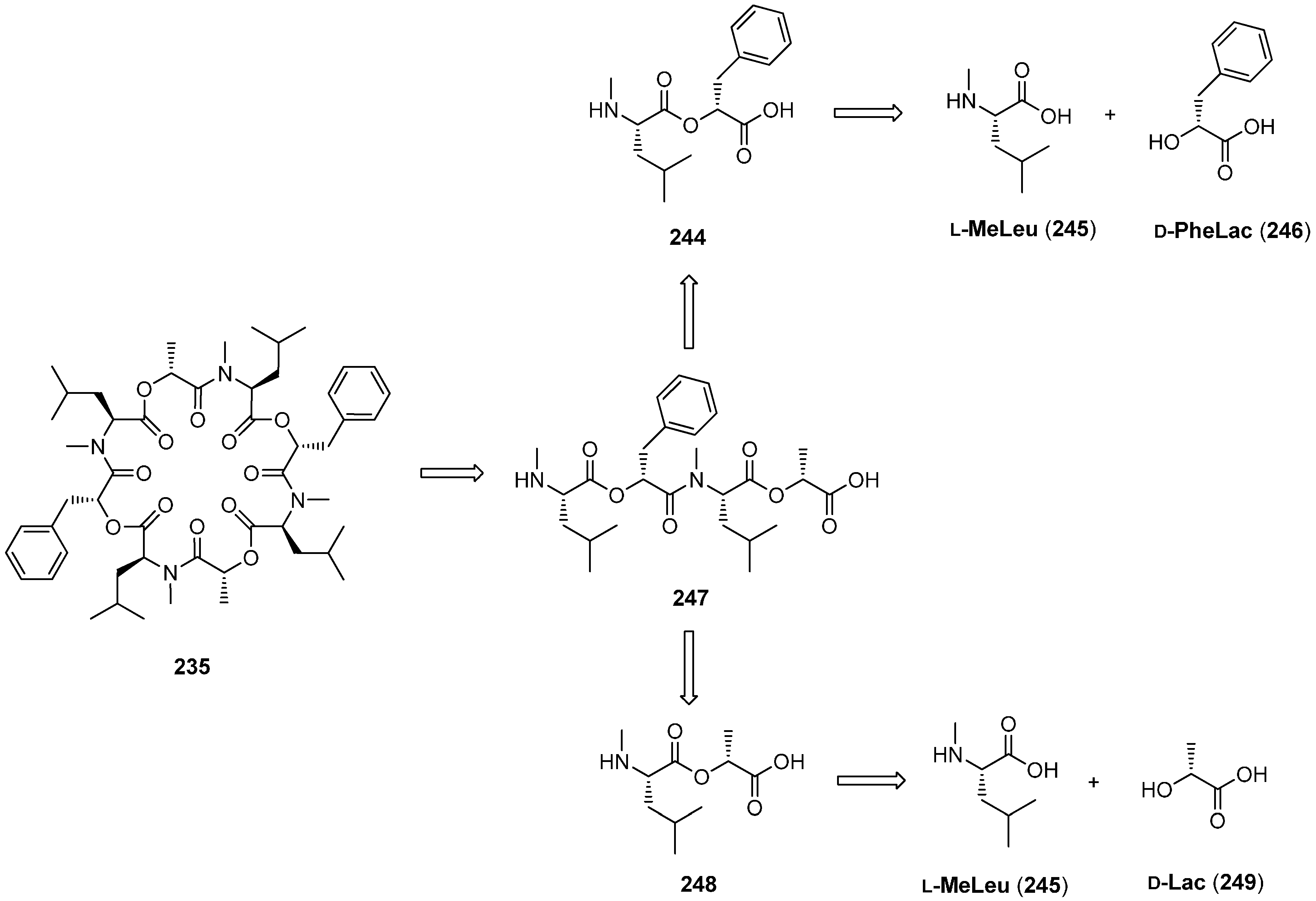
7.3. PF1022A and Emodepside


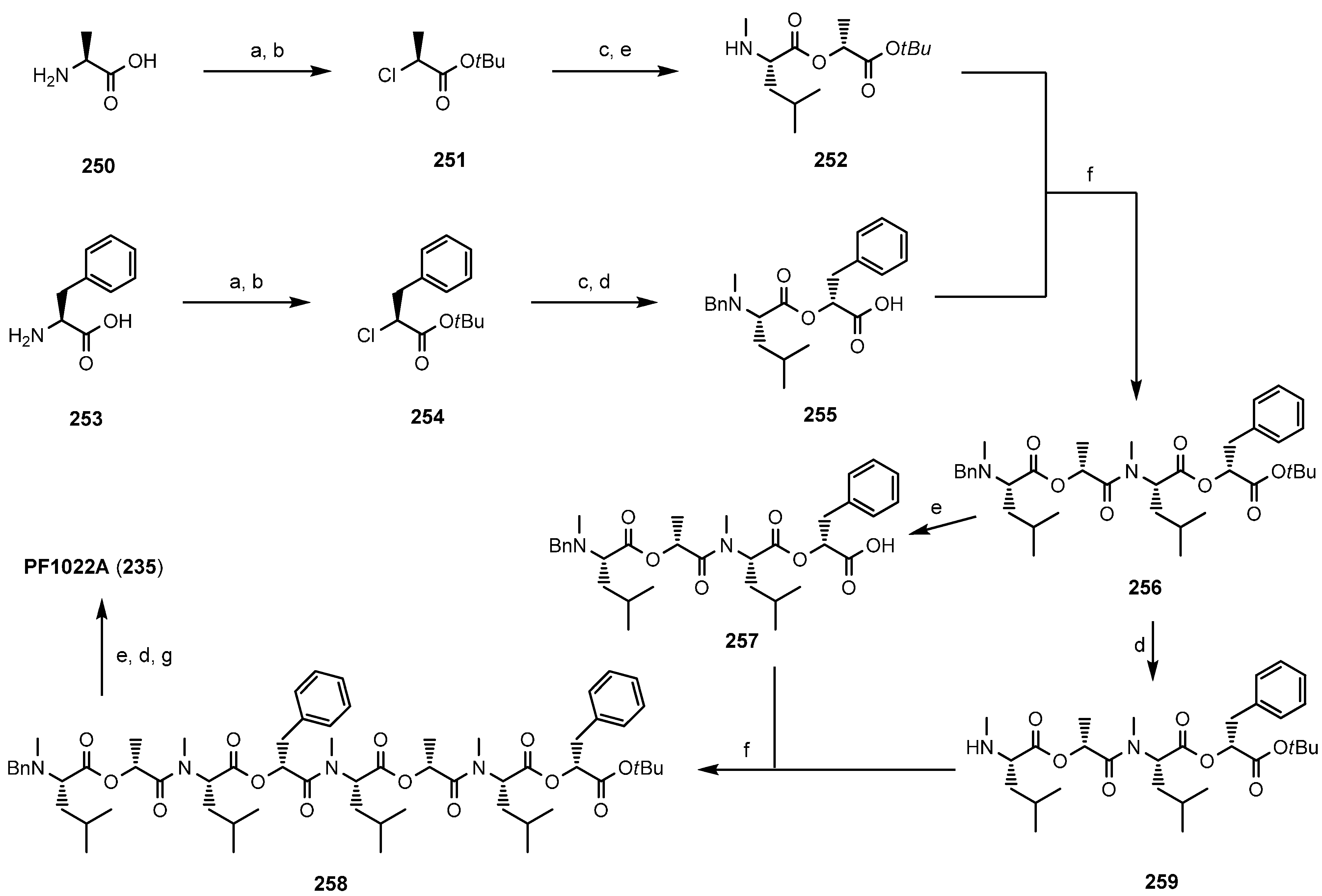
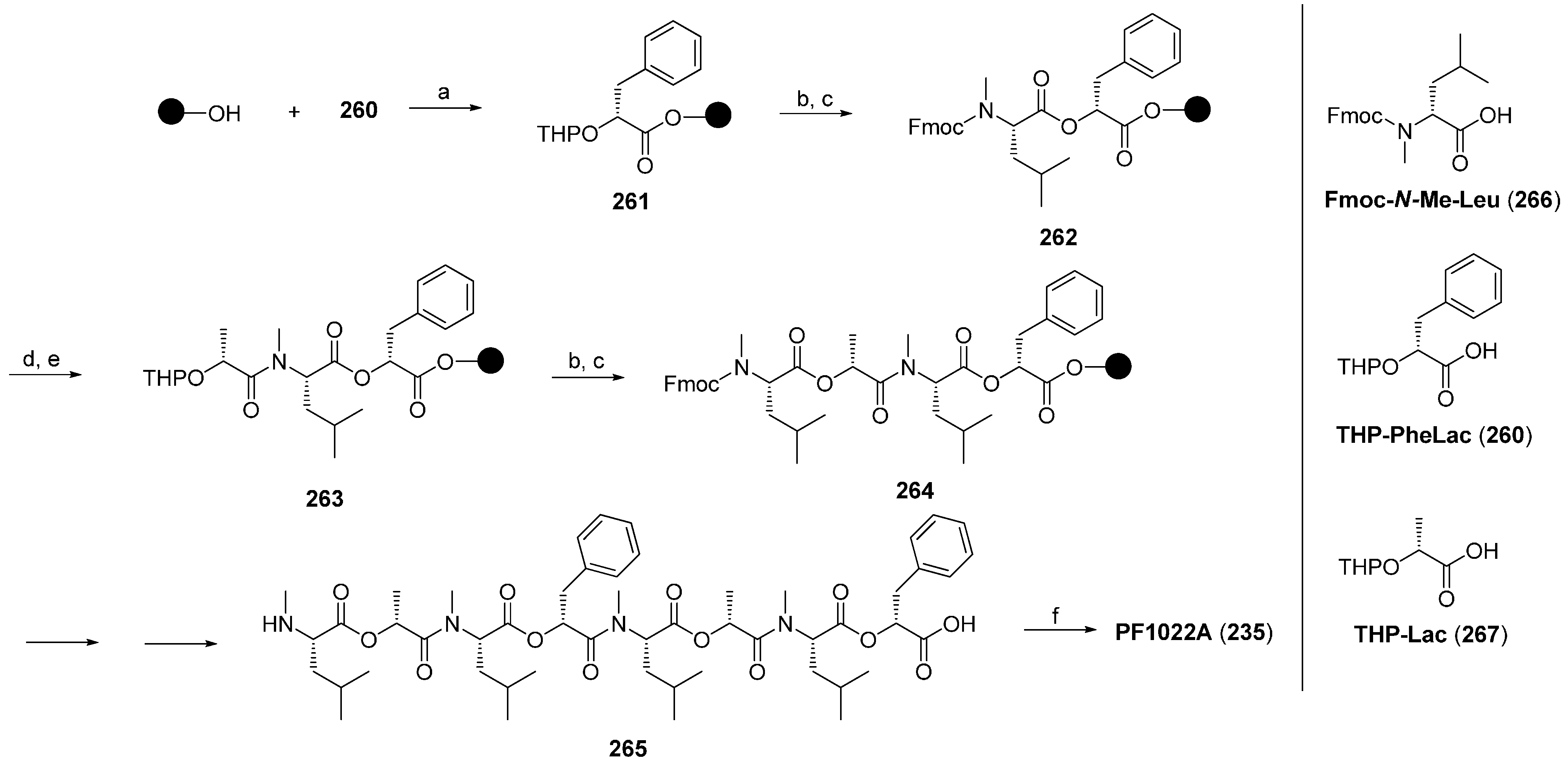
7.3.1. Syntheses of PF1022A



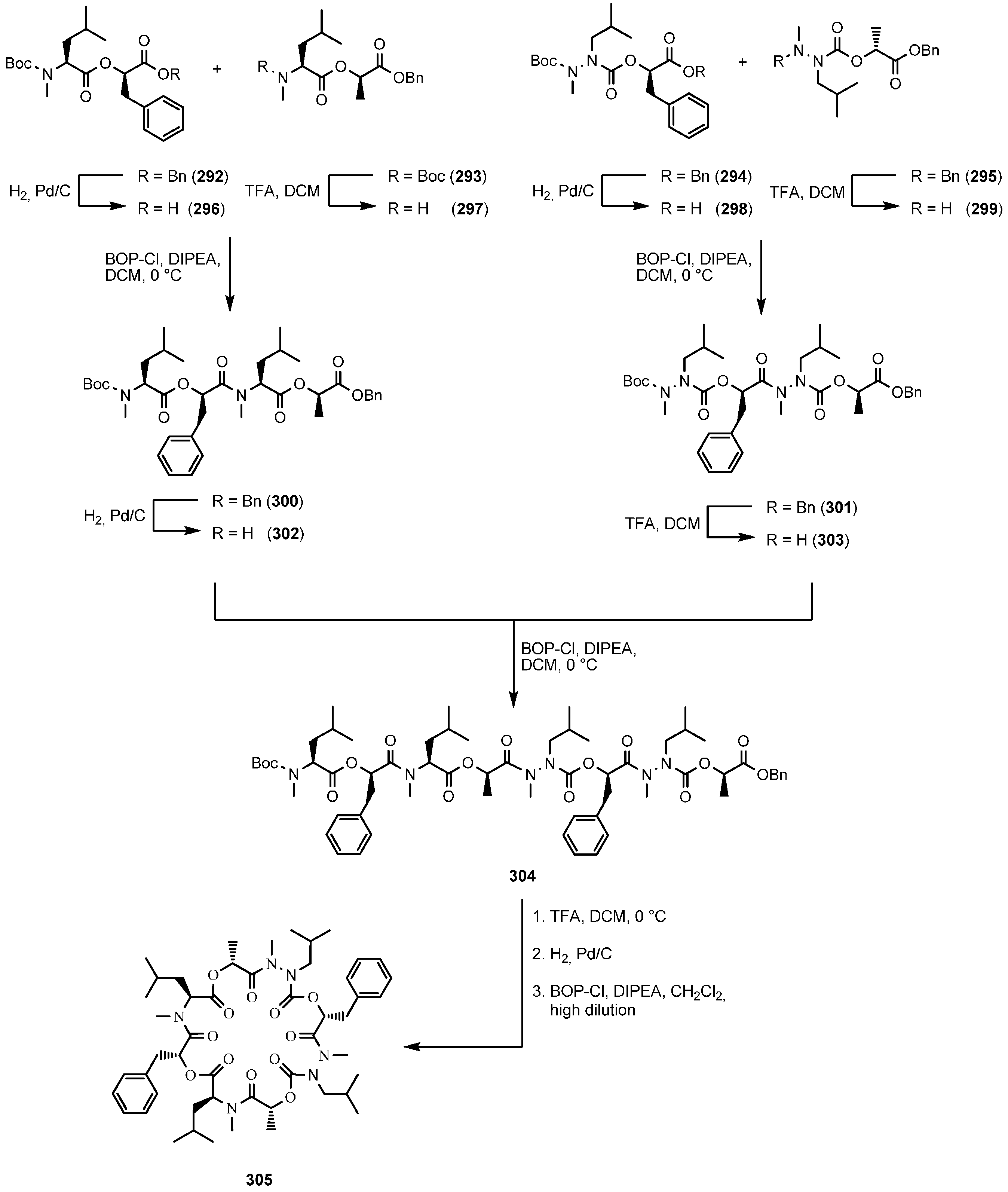
7.3.2. Synthesis of PF1022A-Analogues via Total Synthesis




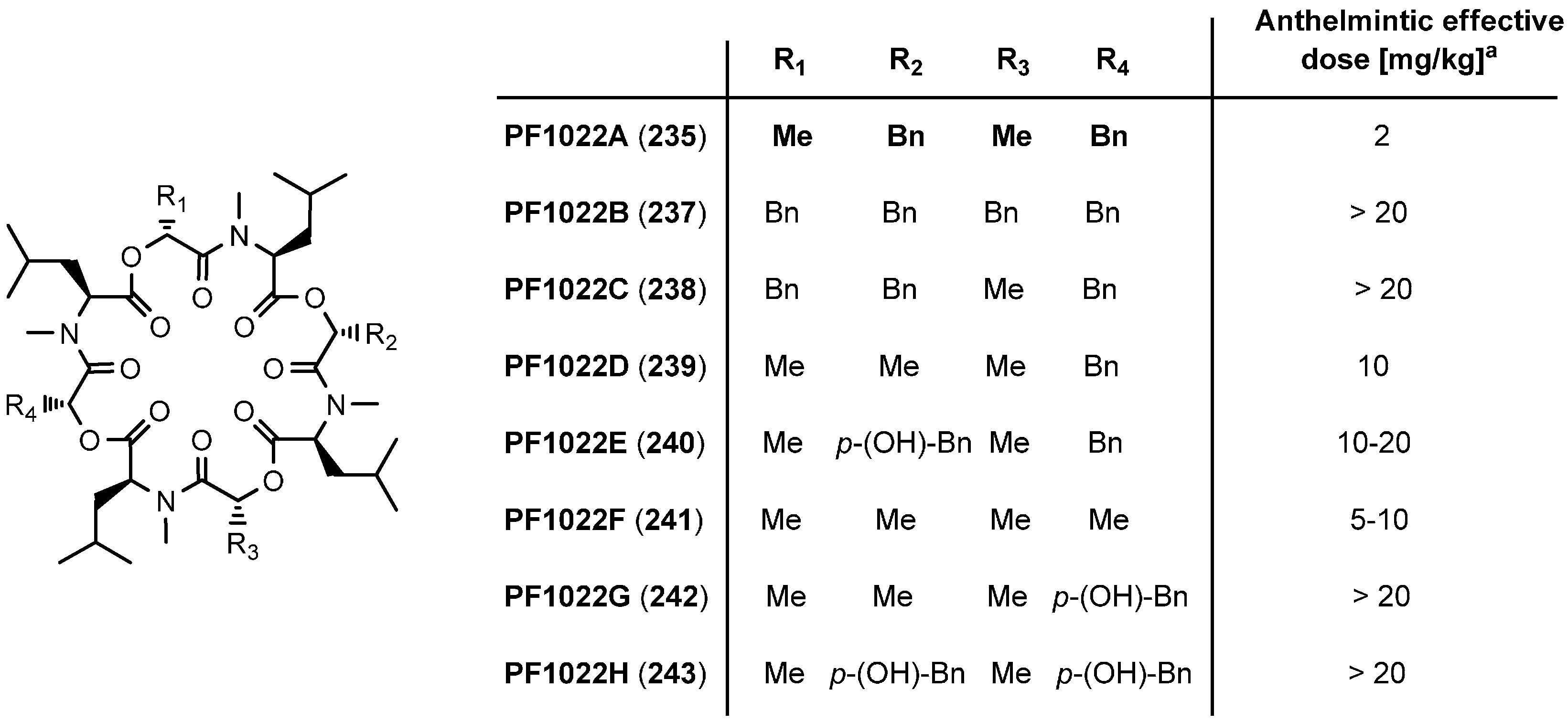
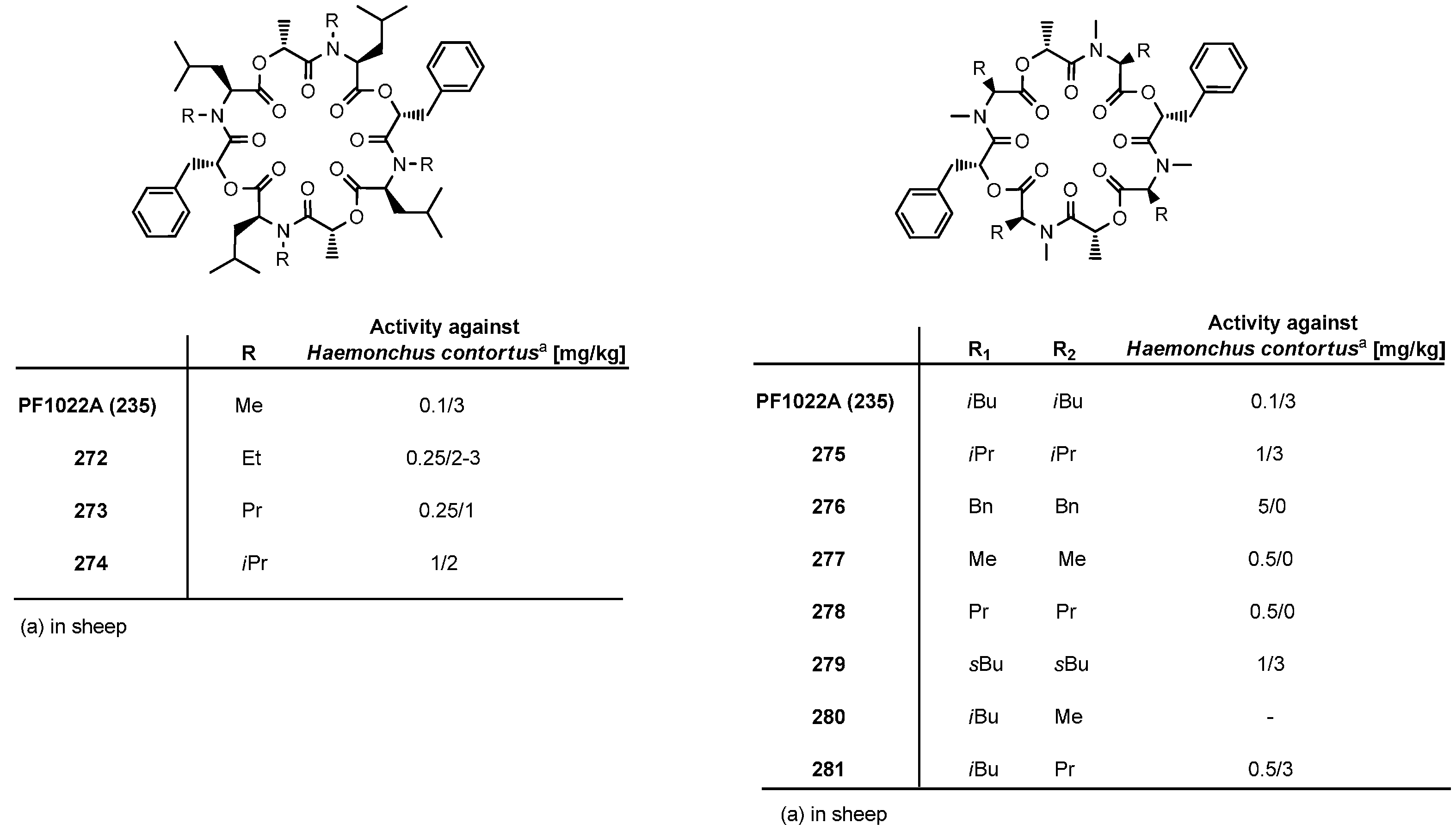
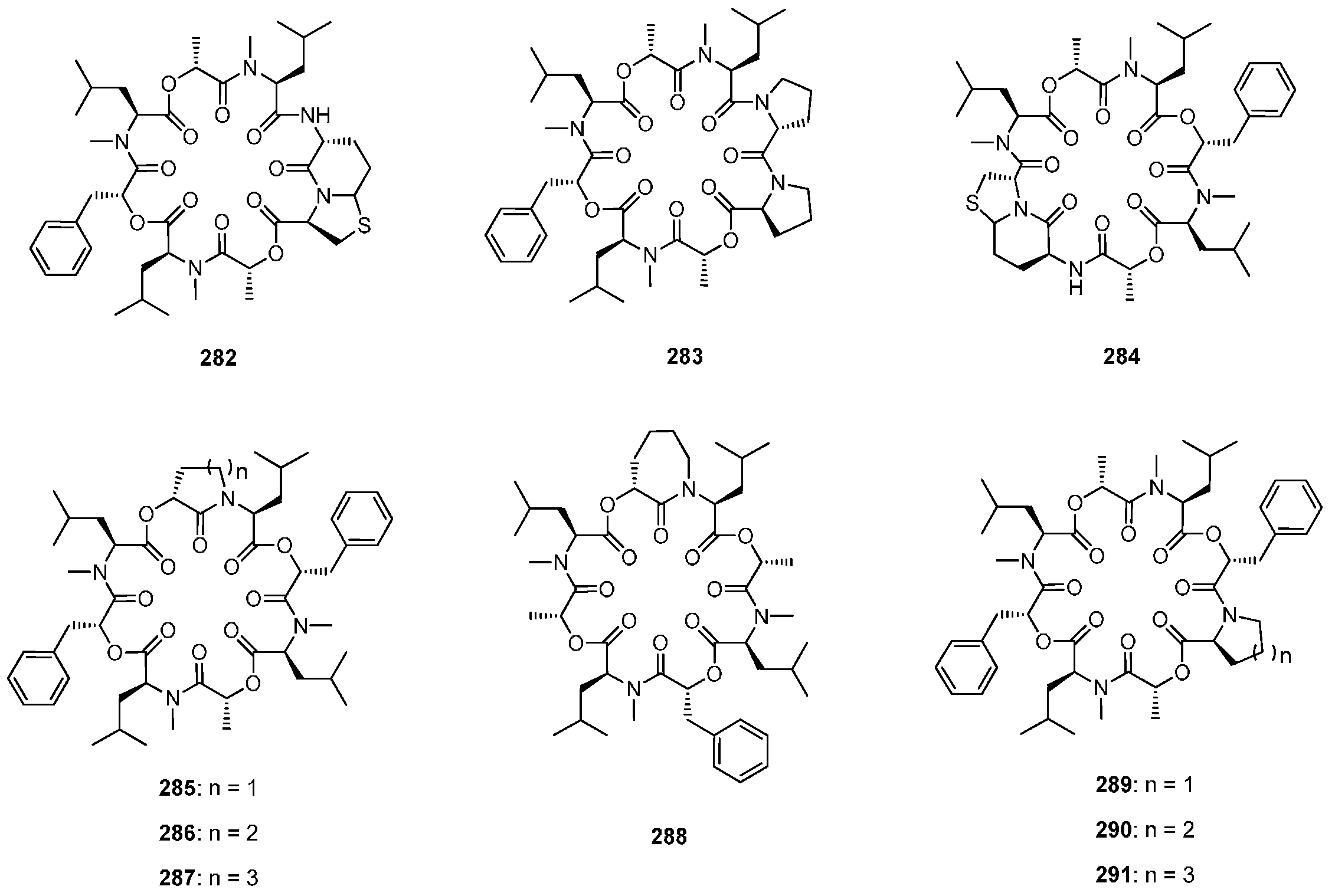
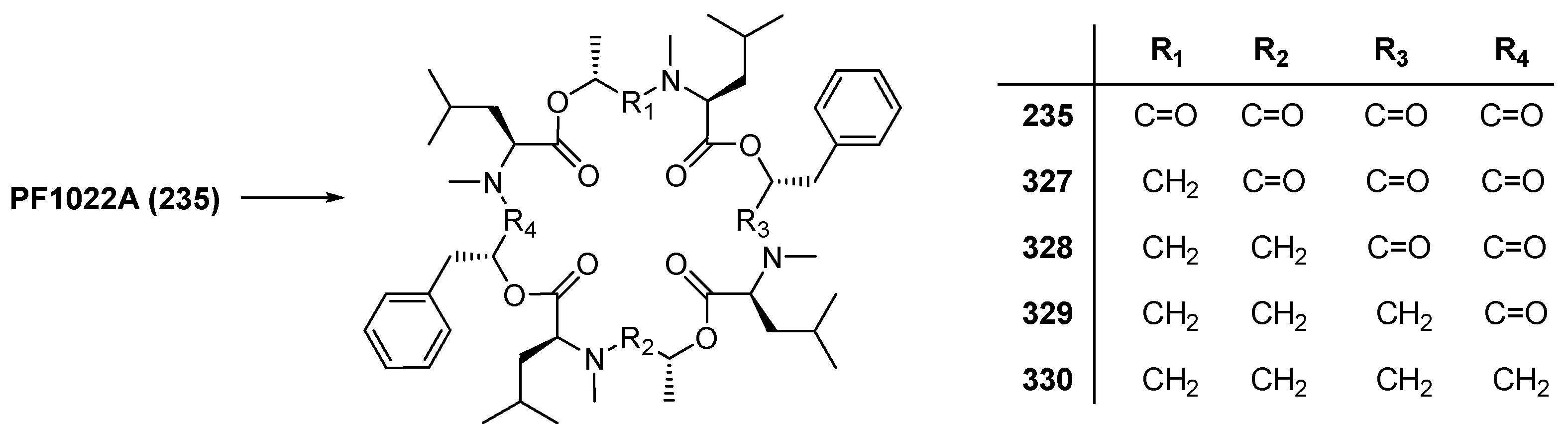
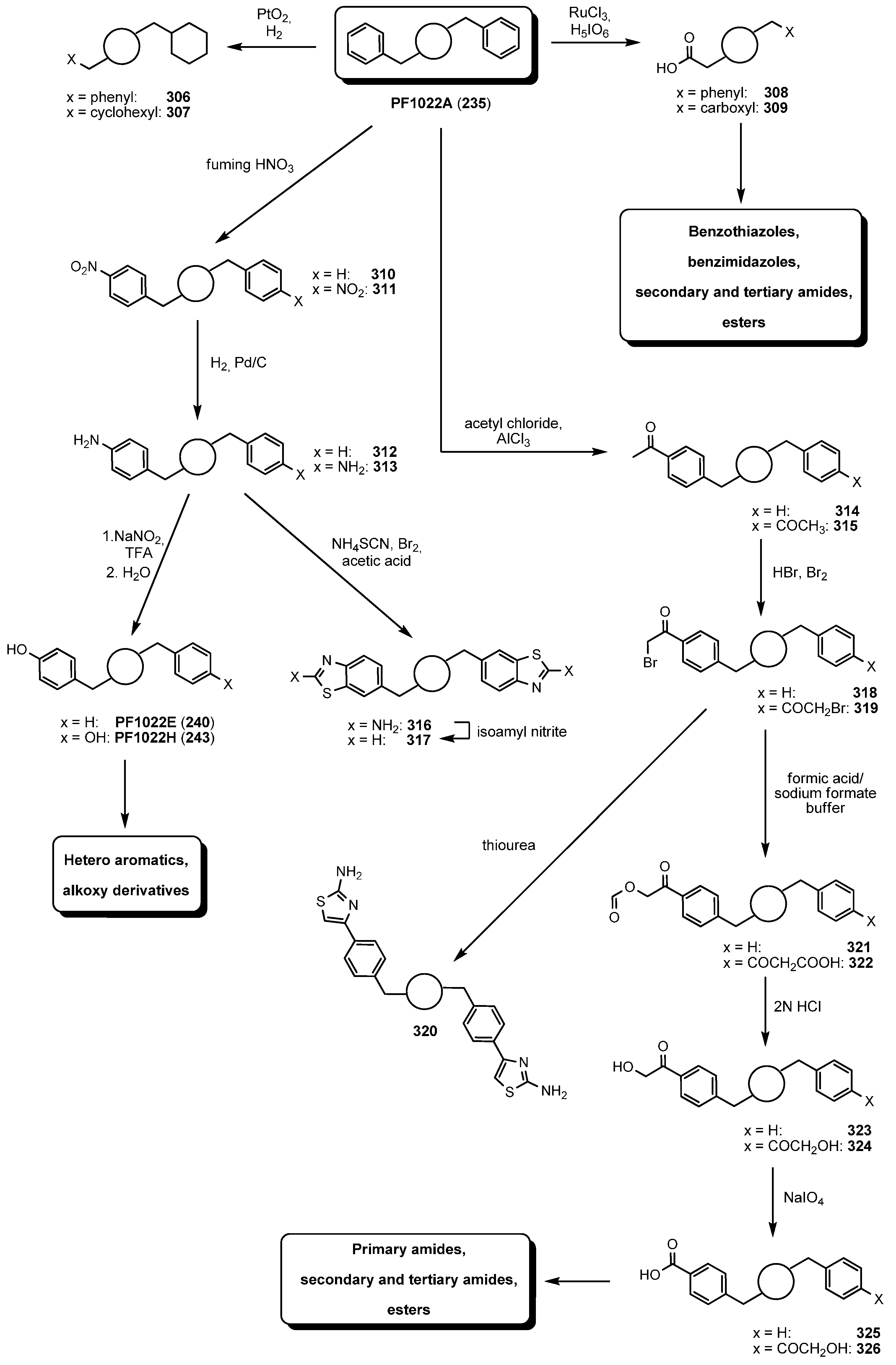
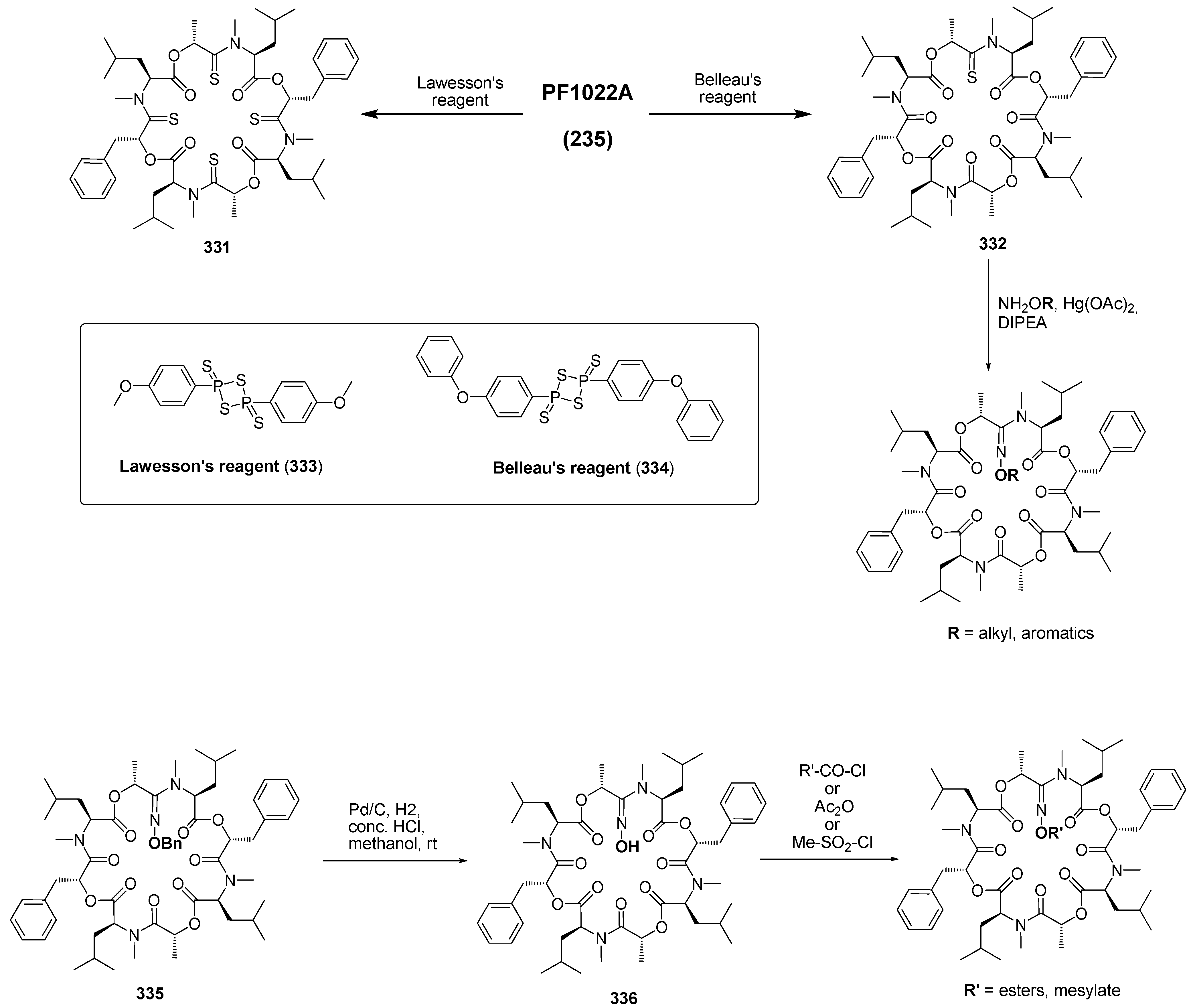
7.3.3. PF1022A Analogues by Direct Derivatization of the Natural Product


7.3.4. Biosynthesis of PF1022A
7.3.5. Mode of Action of PF1022A and Emodepside


8. Cyclononadepsipeptides
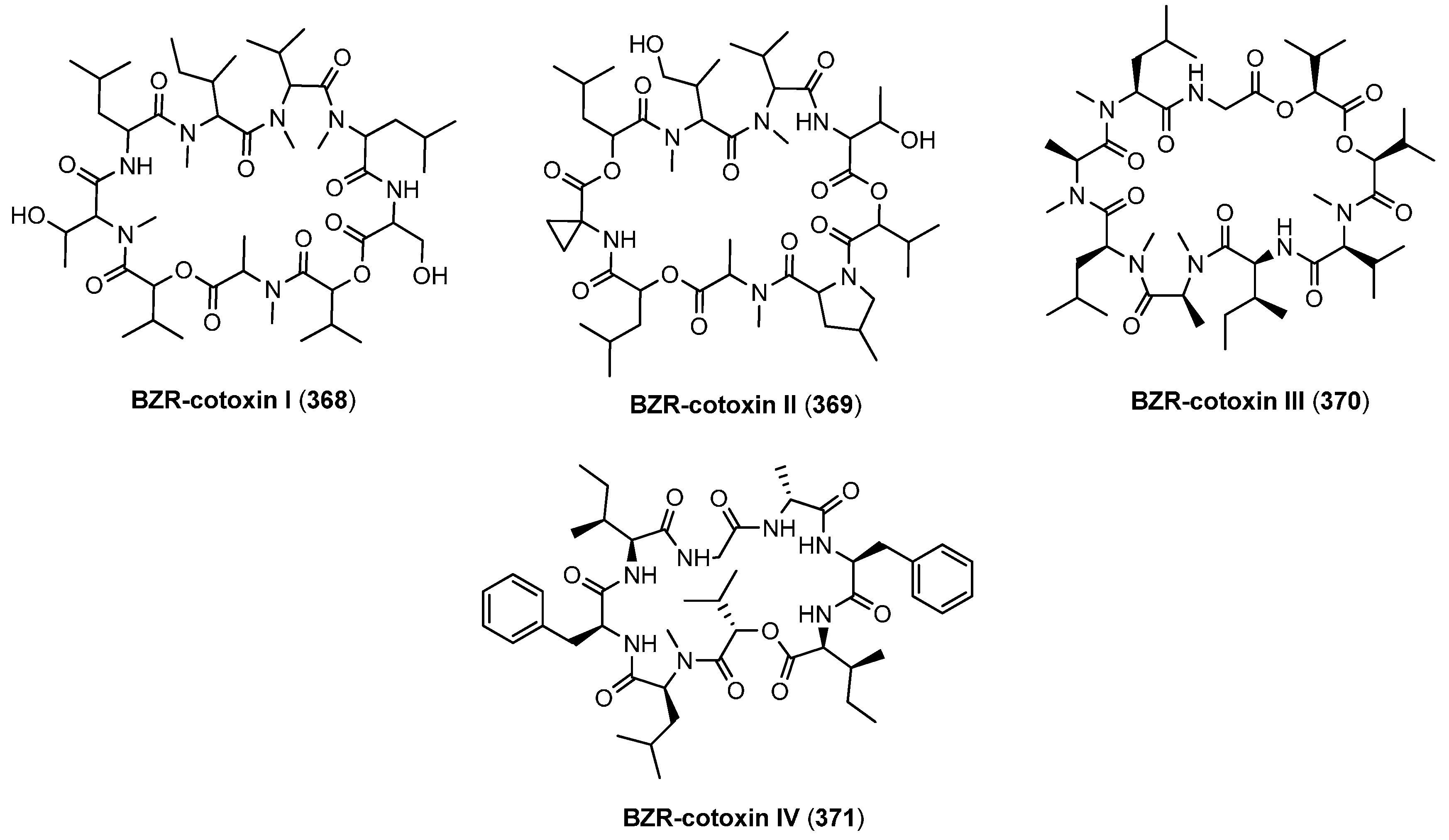
8.1. BZR-cotoxins I-IV

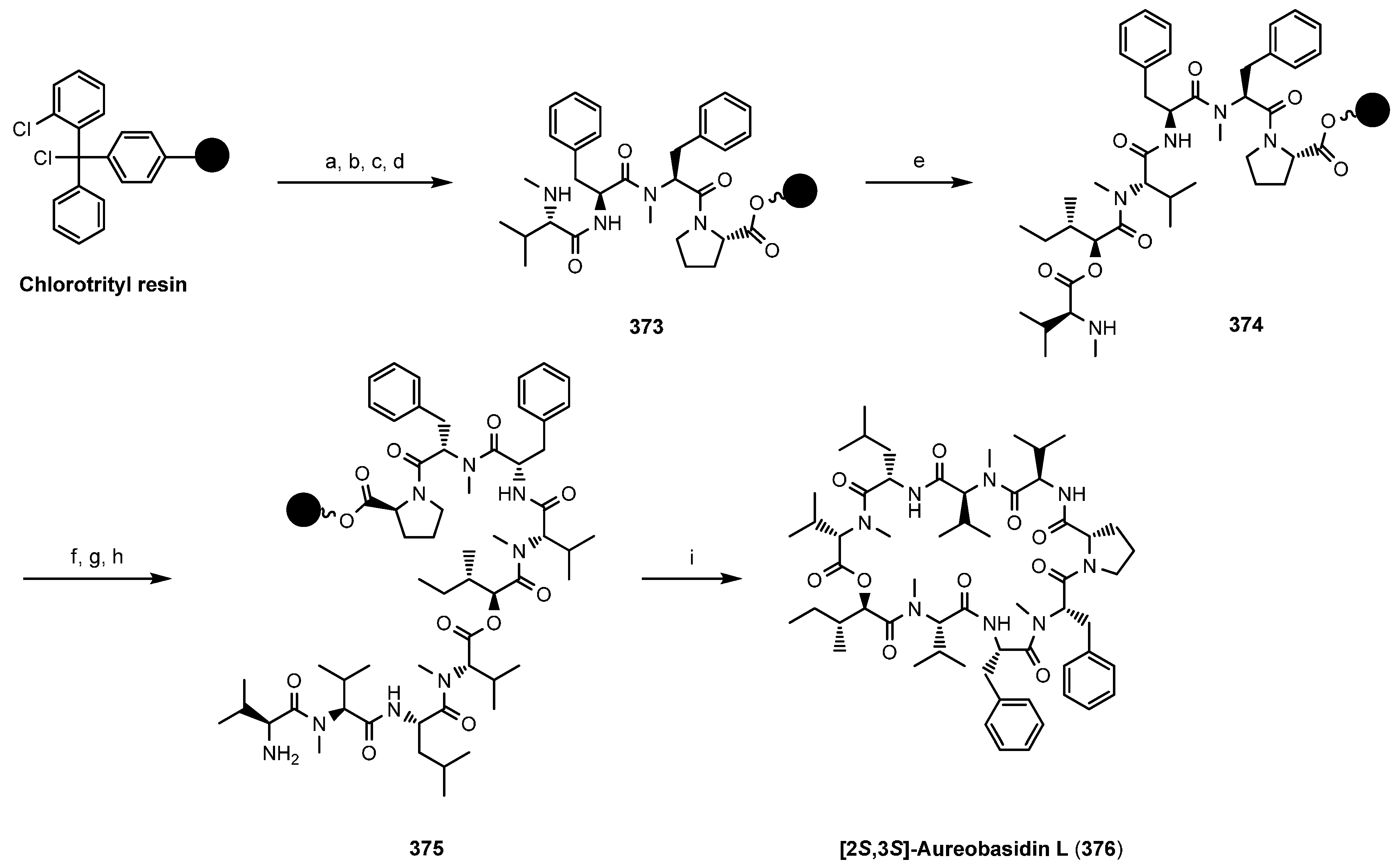
8.2. Aureobasidins


9. Cyclodecadepsipeptides
9.1. Clavariopsin A and B

10. Summary
Conflicts of Interest
References
- Dory, Y.L.; Mellor, J.M. Improved methods of synthesis of valinomycins. Tetrahedron Lett. 1989, 30, 1695–1698. [Google Scholar] [CrossRef]
- Hinaje, M.; Ford, M.; Banting, L.; Arkle, S.; Khambay, B. An investigation of the ionophoric characteristics of destruxin A. Arch. Biochem. 2002, 405, 73–77. [Google Scholar] [CrossRef] [PubMed]
- Davies, J.S. The cyclization of peptides and depsipeptides. J. Pept. Sci. 2003, 9, 471–501. [Google Scholar] [CrossRef] [PubMed]
- Davidson, B.S. Ascidians: procedure of amino acid-derived metabolites. Chem. Rev. 1993, 93, 1771–1791. [Google Scholar] [CrossRef]
- Moore, R.E. Cyclic peptides and depsipeptides from cyanobacteria: A review. J. Ind. Microbiol. 1996, 16, 134–143. [Google Scholar] [CrossRef] [PubMed]
- Kitir, B.; Baldry, M.; Ingmer, H.; Olsen, C.A. Total synthesis and structural validation of cyclodepsipeptides solonamide A and B. Tetrahedron 2014, in press. [Google Scholar]
- Li, W.; Schlecker, A.; Ma, D. Total synthesis of antimicrobial and antitumor cyclic depsipeptides. Chem. Commun. 2010, 46, 5403–5420. [Google Scholar] [CrossRef]
- Lüttenberg, S.; Sondermann, F.; Scherkenbeck, J. A practical method for the generation of Kaiser-oxime resin. Tetrahedron Lett. 2013, 54, 907–908. [Google Scholar]
- Stolze, S.C.; Kaiser, M. Case studies of the synthesis of bioactive cyclodepsipeptide natural products. Molecules 2013, 18, 1337–1367. [Google Scholar] [CrossRef] [PubMed]
- Humano, K.; Kinoshita, M.; Furuya, K.; Miyamoto, M.; Takamatsu, Y.; Hemmi, A.; Tanzawa, K. Leualacin, a novel calcium blocker from Hapsidospora irregularis. J. Antibiot. 1991, 45, 899–905. [Google Scholar] [CrossRef]
- Kikuchi, M.; Nosaka, K.; Akaji, K.; Konno, H. Solid phase total synthesis of callipeltin E isolated from marine sponge Latrunculia sp. Tetrahedron Lett. 2011, 52, 3872–3875. [Google Scholar] [CrossRef]
- Zhou, X.; Huang, H.; Li, J.; Song, Y.; Jiang, R.; Liu, J.; Zhang, S.; Hua, Y.; Ju, J. New anti-infective cycloheptadepsipeptide congeners and absolute stereochemistry from the deep sea-derived Streptomyces drozdowiczii SCIO 10141. Tetrahedron 2014, in press. [Google Scholar]
- Finking, R.; Marahiel, M.A. Biosynthesis of nonribosomal peptides. Annu. Rev. Microbiol. 2004, 58, 453–488. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Haynes, S.W.; Ames, B.D.; Wang, P.; Vien, L.P.; Walsh, C.T.; Tang, Y. Cyclization of fungal nonribosomal peptides by a terminal condensation-like domain. Nat. Chem. Biol. 2012, 8, 823–830. [Google Scholar] [CrossRef] [PubMed]
- Kopp, F.; Mahlert, C.; Grünewald, J.; Marahiel, M.A. Peptide macrocyclization: the reductase of the nostocyclopeptide synthetase triggers the self-assembly of a macrocyclic imine. J. Am. Chem. Soc. 2006, 128, 16478–16479. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Orozo, R.; Wijeratne, E.M.K.; Gunatilaka, A.A.L.; Stock, S.P.; Molnar, I. Biosynthesis of the cyclooligomer depsipeptide beauvericin, a virulence factor of the entomopathogenic fungus Beauveria bassiana. Chem. Biol. 2008, 15, 898–907. [Google Scholar] [CrossRef] [PubMed]
- Yu, D.; Xu, F.; Zi, J.; Wang, S.; Gage, D.; Zeng, J.; Zhan, J. Engineered production of fungal anticancer cyclooligomer depsipeptides in Saccharomyces cerevisae. Metab. Eng. 2013, 18, 60–68. [Google Scholar] [CrossRef] [PubMed]
- Süssmuth, R.; Müller, J.; von Döhren, H.; Molnar, I. Fungal cyclooligomer depsipeptides: From classical biochemistry to combinatorial biosynthesis. Nat. Prod. Rep. 2011, 28, 99–124. [Google Scholar]
- Miyashita, M.; Nakamori, T.; Miyagawa, H.; Akamatsu, M.; Ueno, T. Inhibitor activity of analogs of AM-toxin, a host-specific phytotoxin from the Alternaria alternate apple pathotype, on photosynthetic O2 evolution in apple leaves. Biosci. Biotechnol. Biochem. 2003, 67, 635–638. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.; Aoyagi, H.; Shimohigashi, Y.; Izumiya, N. Synthesis of cyclotetradepsipeptides, AM- toxin I and its analogs. Tetrahedon Lett. 1976, 11, 843–846. [Google Scholar] [CrossRef]
- Shimohigashi, Y.; Lee, S.; Kato, T.; Izumiya, N.; Ueno, T.; Fukami, H. Synthesis and necrotic activity of dihydro-AM-toxin I. Agric. Biol. Chem. 1977, 8, 1533–1534. [Google Scholar] [CrossRef]
- Ueda, K.; Waki, M.; Teruya, Y.; Izumiya, N. Facile synthesis of AM-toxin II. Bull. Chem. Soc. Jpn. 1989, 62, 1635–1638. [Google Scholar] [CrossRef]
- Hashimoto, K.; Sakai, M.; Okuno, T.; Shirahama, H. β-Phenylselenoalanine as a dehydroalanine precursor-efficient synthesis of alternariolide (AM-toxin I). Chem. Commun. 1996, 1139–1140. [Google Scholar] [CrossRef]
- Miyashita, M.; Nakamori, T.; Murai, T.; Miyagawa, H.; Akamatsu, M.; Ueno, T. Facile synthesis of AM-toxins and analogs as cyclic depsipeptides by the solid-phase method. Biosci. Biotechnol. Biochem. 1998, 9, 1799–1801. [Google Scholar] [CrossRef]
- Horikawa, E.; Kodaka, M.; Nakahara, Y.; Okuno, H.; Nakamura, K. Solid-phase synthesis of dehydropeptide, AM-toxin II, using a novel selenyl linker by side-chain tethered strategy. Tetrahedon Lett. 2001, 42, 8337–8339. [Google Scholar] [CrossRef]
- Konzono, T.; Kanmera, T.; Kato, T.; Ueno, T.; Izumiya, N. Synthesis of lactam analog of AM-toxin I. Agric. Biol. Chem. 1983, 47, 2631–2632. [Google Scholar] [CrossRef]
- Kunicki, J.B.; Peterson, M.N.; Alexander, L.D.; Ardi, V.C.; McConnell, J.R.; McAlpine, S.R. Synthesis and evaluation of biotinylated sansalvamide A analogs and their modulation of Hsp90. Bioorg. Med. Chem. Lett. 2011, 21, 4716–4719. [Google Scholar]
- Belofsky, G.N.; Jensen, P.R.; Fenical, W. Sansalvamide: A new cytotoxic cyclic depsipeptide produced by a marine fungus of the genus Fusarium. Tetrahedron Lett. 1999, 40, 2913–2916. [Google Scholar] [CrossRef]
- Cueto, M.; Jensen, P.R.; Fenical, W. N-methylsansalvamide, a cytotoxic cyclic depsipeptide from a marine fungus of the genus Fusarium. Phytochemistry 2000, 55, 223–226. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Lee, H.; Lee, C. A new cytotoxic cyclic pentadepsipeptide, neo-N-methylsansalvamide produced by Fusarium solani KCCM90040, isolated from potato. Food Chem. 2011, 126, 472–478. [Google Scholar] [CrossRef]
- Lee, Y.; Silverman, R.B. Rapid, high-yield, solid-phase synthesis of the antitumor antibiotic sansalvamide A using a side-chain-tethered phenylalanine building block. Org. Lett. 2000, 2, 3743–3746. [Google Scholar] [CrossRef] [PubMed]
- Gu, W.; Liu, S.; Silverman, R.B. Solid-phase, Pd-catalyzed silicon-aryl carbon bond formation. Synthesis of sansalvamide A peptide. Org. Lett. 2002, 4, 4171–4174. [Google Scholar]
- Carroll, C.L.; Johnston, J.V.C.; Kekec, A.; Brown, J.D.; Parry, E.P.; Cajica, J.; Medina, I.; Cook, K.M.; Corral, R.; Pan, P.; et al. Synthesis and cytotoxicity of novel sansalvamide A derivatives. Org. Lett. 2005, 7, 3481–3484. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Gu, W.; Lo, D.; Ding, X.Z.; Ujiki, M.; Adrian, T.E.; Stoff, G.A.; Silverman, R.B. N-Methylsansalvamide A peptide analogs. Potent antitumor agents. J. Med. Chem. 2005, 48, 3630–3638. [Google Scholar]
- Davis, M.R.; Styers, T.J.; Rodriguez, R.A.; Pan, P.; Vasko, R.C.; McAlpine, S.R. Synthesis and cytotoxicity of a new class of potent decapeptide macrocycles. Org. Lett. 2008, 10, 177–180. [Google Scholar]
- Alexander, L.D.; Sellers, R.P.; Davis, M.R.; Ardi, V.C.; Johnson, V.A.; Vasko, R.C.; MaAlpine, S.R. Evaluation of di-sansalvamide A derivatives: Synthesis, structure-activity relationship, and mechanism of action. J. Med. Chem. 2009, 52, 7927–7930. [Google Scholar] [CrossRef] [PubMed]
- Davis, M.R.; Singh, E.K.; Wahyudi, H.; Alexander, L.D.; Kunicki, J.B.; Nazarova, L.A.; Fairweather, K.A.; Giltrap, A.M.; Jolliffe, K.A.; Mc Alpine, S.R. Synthesis of sansalvamide A peptidomimetics: Triazole, oxazole, thiazole and pseudoproline containing compounds. Tetrahedron 2012, 68, 1029–1051. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, D.M.; McConnel, J.R.; Alexander, L.D.; Tanaka, K.W.; Vera, C.M.; McAlpine, S.R. An Hsp90 modulator that exhibits a unique mechanistic profile. Bioorg. Med. Chem. Lett. 2012, 22, 3287–3290. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.; Yang, Y.; Zhao, C.; Huang, J.; Han, C.; Han, J. Effect of the 4'-substituted phenylalanine moiety of sansalvamide A peptide on antitumor activity. Med. Chem. Commun. 2014, 5, 463–467. [Google Scholar] [CrossRef]
- Kim, M.; Sohn, J.; Ahn, J.; Oh, H. Alternaramide, a cyclic depsipeptide from the marine-derived fungus Alternaria sp. SF-5016. J. Nat. Prod. 2009, 72, 2065–2068. [Google Scholar]
- Horton, A.E.; May, O.S.; Elsegood, M.R.J.; Kimber, M.C. Total synthesis of the marine-derived cyclic depsipeptide alternaramide. Synlett 2011, 22, 797–800. [Google Scholar]
- Oh, D.; Jensen, P.R.; Fenical, W. Zygosporamide, a cytotoxic cyclic depsipeptide from the marine-derived fungus Zygosporium masonii. Tetrahedron Lett. 2006, 47, 8625–8628. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, F.; Zhang, Y.; Liu, J.O.; Ma, D. Synthesis and antitumor activity of cyclodepsipeptide zygosporamide and its analogues. Bioorg. Med. Chem. Lett. 2008, 18, 4385–4387. [Google Scholar] [CrossRef] [PubMed]
- Sy-Cordero, A.A.; Pearce, C.J.; Oberlies, N.H. Revisting the enniatins: A review of their isolation, biosynthesis, structure determination, and biological activities. J. Antibiot. 2012, 65, 541–549. [Google Scholar]
- Krause, M.; Lindemann, A.; Glinski, M.; Hornbogen, T.; Bonse, G.; Jeschke, P.; Thielking, G.; Gau, W.; Kleinkauf, H.; Zocher, R. Directed biosynthesis of new enniatins. J. Antibiot. 2001, 54, 797–804. [Google Scholar] [CrossRef] [PubMed]
- Pieper, R.; Kleinkauf, H.; Zocher, R. Enniatin synthetases from different fusaria exhibiting distinct amino acid specificities. J. Antibiot. 1992, 45, 1273–1277. [Google Scholar] [CrossRef] [PubMed]
- Zhan, J.; Burns, A.M.; Liu, M.X.; Faeth, S.H.; Gunatilaka, A.A.L. Search for cell motility and angiogenesis inhibitors with potential anticancer activity: Beauvericin and other constitutions of two endophytic strains of Fusarium oxysporum. J. Nat. Prod. 2007, 70, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Xu, R.; Ouyang, Z.; Qian, C.; Shen, Y.; Wu, X.; Gu, Y.; Xu, Q.; Sun, Y. Beauvericin ameliorates experimental colitis by inhibiting activated T cells via downregulation of the PI3K/Akt signaling pathway. PLoS One 2013, 8, 1–11. [Google Scholar]
- Shemyakin, M.M.; Ovchinnikov, Y.A.; Kiryushkin, A.A.; Ivanov, V.T. The structure and total synthesis of enniatin B. Tetrahedron Lett. 1963, 14, 885–890. [Google Scholar]
- Hu, D.X.; Bielitza, M.; Koos, P.; Ley, V.S. A total synthesis of the ammonium ionophore, (−)-enniatin B. Tetrahedron Lett. 2012, 53, 4077–4079. [Google Scholar] [CrossRef]
- Jeschke, P.; Benet-Buchholz, J.; Harder, A.; Etzel, W.; Schindler, M.; Gau, W.; Weiss, H. Synthesis and anthelmintic activity of substituted (R)-phenyllactic acid containing cyclohexadepsipeptides. Bioorg. Med. Chem. Lett. 2006, 16, 4410–4415. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P.; Harder, A.; Etzel, W.; Schindler, M.; Thielking, G. Synthesis and anthelmintic activity of cyclohexadepsipeptides with cyclohexylmethyl side chains. Bioorg. Med. Chem. Lett. 2007, 17, 3690–3695. [Google Scholar] [CrossRef] [PubMed]
- Glinski, M.; Urbanke, C.; Hornbogen, T.; Zocher, R. Enniatin synthetase is a monomer with extended structure: evidence for an intermolecular mechanism. Arch. Microbiol. 2002, 178, 267–273. [Google Scholar] [CrossRef] [PubMed]
- Madry, N.; Zocher, R.; Grodzki, K.; Kleinkauf, H. Selective synthesis of depsipeptides by the immobilized multienzyme enniatin synthetase. Appl. Microbiol. Biotechnol. 1984, 20, 83–86. [Google Scholar]
- Feifel, S.C.; Schmiederer, T.; Hornbogen, T.; Berg, H.; Süssmuth, R.D.; Zocher, R. In vitro synthesis of new enniatins: Probing the α-hydroxy carboxylic acid binding pocket of the multienzyme enniatin synthetase. ChemBioChem 2007, 8, 1767–1770. [Google Scholar]
- Peeters, H.; Zocher, R.; Kleinkauf, H. Synthesis of beauvericin by a multifunctional enzyme. J. Antibiot. 1988, 41, 352–359. [Google Scholar] [CrossRef] [PubMed]
- Peeters, H.; Zocher, R.; Madry, N.; Oelrichs, P.B.; Kleinkauf, H.; Kraepelin, G. Cell-free synthesis of the depsipeptide beauvericin. J. Antibiot. 1983, 36, 1762–1766. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Zhan, J.; Wijeratne, E.M.K.; Burns, A.M.; Gunatilaka, A.A.L.; Molnar, I. Cytotoxic and antihaptotactic beauvericin analogues from precursor-directed biosynthesis with the insect pathogen Beauveria bassiana ATCC 7159. J. Nat. Prod. 2007, 70, 1467–1471. [Google Scholar] [CrossRef] [PubMed]
- Nilanonta, C.; Isaka, M.; Kittakoop, P.; Trakulnaleamsai, S.; Tanticharoen, M.; Thebtaranonth, Y. Precursor-directed biosynthesis of beauvericin analogs by the insect pathogenic fungus Paecilomyces tenuipes BCC 1614. Tetrahedron 2002, 58, 3355–3360. [Google Scholar] [CrossRef]
- Matthes, D.; Richter, L.; Müller, J.; Denisiuk, A.; Feifel, S.C.; Xu, Y.; Espinosa-Artiles, P.; Süssmuth, R.D.; Molnar, I. In vitro chemoenzymatic and in vivo biocatalytic synthesis of new beauvericin analogues. Chem. Commun. 2012, 48, 5674–5676. [Google Scholar] [CrossRef]
- Isaka, M.; Yangchum, A.; Sappan, M.; Suvannakad, R.; Srikitikulchai, P. Cyclohexadepsipeptides from Acremonium sp. BCC 28424. Tetrahedron 2011, 67, 7929–7935. [Google Scholar] [CrossRef]
- Bunyapaiboonsri, T.; Vongvilai, P.; Auncharoen, P.; Isaka, M. Cyclohexadepsipeptides from the filamentous fungus Acremonium sp. BCC 2629. Helv. Chim. Acta 2012, 95, 963–972. [Google Scholar] [CrossRef]
- Vongvanich, N.; Kittakoop, P.; Isaka, M.; Trakulnaleamsai, S.; Vimuttipong, S.; Tanticharoen, M.; Thebtaranonth, Y.; Hirsutellide, A. a new antimycobacterial cyclohexadepsipeptide from the entomopathogenic fungus Hirsutella kobayasii. J. Nat. Prod. 2002, 65, 1346–1348. [Google Scholar]
- Xu, Y.; Chen, L.; Duan, X.; Meng, Y.; Jiang, L.; Li, M.; Zhao, G.; Li, Y. Total synthesis of hirsutellide A. Tetrahedron Lett. 2005, 46, 4377–4379. [Google Scholar] [CrossRef]
- Xu, Y.; Duan, X.; Li, M.; Jian, L.; Zhao, G.; Meng, Y.; Chen, L. Synthesis of the key precursor of hirsutellide A. Molecules 2005, 10, 259–264. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Chen, L.; Ma, Y.; Li, J.; Cao, X. Silver-ion mediated macrocyclization to form cyclohexadepsipeptide. Synlett 2007, 12, 1901–1904. [Google Scholar]
- Pohanka, A.; Menkis, A.; Levenfors, J.; Broberg, A. Low-abundance kutznerides from Kutznerica sp. 744. J. Nat. Prod. 2006, 69, 1776–1781. [Google Scholar] [CrossRef]
- Broberg, A.; Menkis, A.; Vasiliauskas, R. Kutznerides 1–4, depsipeptides from the actinomycete Kutzeria sp. 744 inhabiting mycorrhizal roots of Picea abies seedlings. J. Nat. Prod. 2006, 69, 97–102. [Google Scholar]
- Fujimori, D.G.; Hrvatin, S.; Neumann, C.S.; Strieker, M.; Marahiel, M.A.; Walsh, C.T. Cloning and characterization of the biosynthetic gene cluster for kutnerides. PNAS 2007, 104, 16498–16503. [Google Scholar] [CrossRef] [PubMed]
- Bevan, K.; Davies, J.S.; Hall, M.J.; Hassal, C.H.; Morton, R.B.; Phillips, D.A.S.; Ogihira, Y.; Thomas, W.A. The monamycins, a new family of cyclodepsipeptide antibiotics. Experientia 1970, 26, 122–123. [Google Scholar] [CrossRef] [PubMed]
- Hall, M.J. Mode of action of monamycin. Evidence for the formation of a complex between the monamycin cyclodepsipeptide antibiotics and some cations in solution. Biochem. Biophys. Res. Commun. 1970, 38, 590–598. [Google Scholar]
- Hale, K.J.; Jogiya, N.; Manaviazar, S. Monamycin synthetic studies. Pt 1. An enantiospecific total synthesis of (3S,5S)-5-hydroxypiperazic acid from d-Mannitol. Tetrahedron Lett. 1998, 39, 7163–7166. [Google Scholar]
- Hall, M.J.; Handford, B.O.; Hassyl, C.H.; Phillips, D.A.S.; Rees, A.V. Bromomonamycins, unnatural analogues of the monamycin cyclodepsipeptide antibiotics: Production, isolation, and biological activity. Antimircob. Agents Chemother. 1973, 3, 380–383. [Google Scholar] [CrossRef]
- Lam, K.S.; Heslter, G.A.; Mattel, J.M.; Mamber, S.W.; Forenza, S. Himastatin, a new antitumor antibiotic from Streptomyces hygroscopius. I. Taxonomy of producing organism, fermentation and biological activity. J. Antibiot. 1990, 8, 956–960. [Google Scholar]
- Left, J.E.; Schröder, D.R.; Krishan, B.S.; Matron, J.A. Himastatin, a new antitumor antibiotic from Streptomyces hygroscopicus. II. Isolation and characterization. J. Antibiot. 1990, 8, 961–966. [Google Scholar]
- Left, J.E.; Schroeder, D.R.; Golik, J.; Matson, J.A.; Doyle, T.W.; Lam, K.S.; Hill, S.E.; Lee, M.S.; Whitney, J.L.; Krishnan, B.S. Himastatin, a new antitumor antibiotic from Streptomyces hygroscopicus. III. Structural elucidation. J. Antibiot. 1996, 49, 299–311. [Google Scholar]
- Ruiz-Sanchis, R.; Savina, S.A.; Albericio, F.; Alvarez, M. Structure, bioactivity and synthesis of natural products with hexahydropyrrolo[2,3-b]indole. Chem. Eur. J. 2011, 17, 1388–1408. [Google Scholar]
- Kamenecka, T.M.; Danishefsky, S.J. Discovery through total synthesis: A retrospective on the himastatin problem. Chem. Eur. J. 2001, 7, 41–63. [Google Scholar] [CrossRef] [PubMed]
- Kamenecka, T.M.; Danishefsky, S.J. Studies in the total synthesis of himastatin: A revision of the stereochemical assignment. Angew. Chem. Int. Ed. 1998, 37, 2993–2995. [Google Scholar] [CrossRef]
- Kamenecka, T.M.; Danishefsky, S.J. Total synthesis of himastatin: Confirmation of the revised stereostructure. Angew. Chem. Int. Ed. 1998, 37, 2995–2998. [Google Scholar] [CrossRef]
- Ma, J.; Wang, Z.; Huang, H.; Luo, M.; Zuo, D.; Wang, B.; Sun, A.; Cheng, Yi.; Zhang, C.; Ju, J. Biosynthesis of himastatin: assembly line and characterization of three cytrochrome P450 enzymes involved in the post-tailoring oxidative steps. Angew. Chem. Ind. Ed. 2011, 50, 7797–7802. [Google Scholar] [CrossRef]
- Isaka, M.; Palasarn, S.; Supothina, S.; Komwijit, S.; Luangsa-ard, J. Bioactive compounds from the scale insect pathogenic fungus Conoideocrella tenius BCC 18627. J. Nat. Prod. 2011, 74, 782–789. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Palasarn, S.; Lapanun, S.; Striklung, K. Peacilodepsipeptide A, an antimalarial and antitumor cyclohexadepsipeptide from the insect pathogenic fungus Peacilomyces cinnamomeus BCC 9616. J. Nat. Prod. 2007, 70, 675–678. [Google Scholar] [CrossRef] [PubMed]
- Yang, M.J.; Wu, J.; Yang, Z.D.; Zhang, Y.M. First total synthesis of peacilodepsipeptide A. Chin. Chem. Lett. 2009, 20, 527–530. [Google Scholar] [CrossRef]
- Isaka, M.; Berkaew, P.; Intereya, K.; Komwijit, S.; Sathitkunanon, T. Antiplasmodial and antiviral cyclohexadepsipeptides from the endophytic fungus Pullularia sp. BCC 8613. Tetrahedron 2007, 63, 6855–6860. [Google Scholar]
- Ebrahim, W.; Kjer, J.; El Amrani, M.; Wray, V.; Lin, W.; Ebel, R.; Lai, D.; Proksch, P. Pullularins E and F, two new peptides from the endophytic fungus Bionecteria ochroleuca isolated from the mangrove plant Sonneratia caseolaris. Mar. Drugs 2012, 10, 1081–1091. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Palasarn, S.; Sriklung, K.; Kocharin, K. Cyclohexadepsipeptides from the insect pathogenic fungus Hirsutella nivea BB 2594. J. Nat. Prod. 2005, 68, 1680–1682. [Google Scholar] [CrossRef] [PubMed]
- Isaka, M.; Rugseree, N.; Maithip, P.; Kongsaeree, P.; Prabpai, S.; Thebtaranonth, Y. Hirsutellones A-E, antimycobacterial alkaloids from the insect pathogenic fungus Hirsutella nivea BCC 2594. Tetrahedron 2005, 61, 5577–5583. [Google Scholar] [CrossRef]
- Chen, Y.; Bilban, M.; Foster, C.A.; Boger, D.L. Solution-phase parallel synthesis of a pharmacophore library of HUN-7293 analogues: A general chemical mutagenesis approach to defining structure-function properties of naturally occurring cyclic (depsi)peptides. J. Am. Chem. Soc. 2002, 124, 5431–5440. [Google Scholar] [CrossRef] [PubMed]
- Boger, D.L.; Keim, H.; Oberhauser, B.; Schreiner, E.P.; Foster, C.A. Total synthesis of HUN-7293. J. Am. Chem. Soc. 1999, 121, 6197–6205. [Google Scholar] [CrossRef]
- Boger, D.L.; Chen, Y.; Foster, C.A. Synthesis and evaluation of aza HUN-7293. Bioorg. Med. Chem. Lett. 2000, 10, 1741–1744. [Google Scholar] [CrossRef] [PubMed]
- Schreiner, E.P.; Kern, M.; Steck, A. A convenient protocol for selective cleavage of 2-hydroxy acid amides. Application to semisynthesis of the cyclic heptapeptide aza HUN-7293. J. Org. Chem. 2002, 67, 8299–8304. [Google Scholar]
- Kanaoka, M.; Isogai, A.; Murakoshi, S.; Ichinoe, M.; Suzuki, A.; Tamura, S. Bassianolide, a new insecticidal cyclodepsipeptide from Beauveria bassiana and Verticillium lecanii. Agric. Biol. Chem. 1978, 42, 629–635. [Google Scholar] [CrossRef]
- Nakajyo, S.; Shimizu, K.; Kometani, A.; Suzuki, A.; Ozaki, H.; Urakawa, N. On the inhibitory mechanism of bassianolide, a cyclodepsipeptide, in acetylcholine-induced contraction in guinea-pig taenia coli. Jpn. J. Pharmacol. 1983, 33, 573–582. [Google Scholar] [CrossRef] [PubMed]
- Jirakkakul, J.; Punya, J.; Pongpattanakitshote, S.; Paungmoung, P.; Vorapreeda, N.; Tachaleat, A.; Klomnara, C.; Tanticharoen, M.; Cheevadhanarak, S. Identification of the nonribosomal peptide synthetase gene responsible for bassianolide synthesis in wood-decaying fungus Xylaria sp. BCC1067. Microbiology 2008, 154, 995–1006. [Google Scholar] [CrossRef]
- Kanaoka, M.; Isogai, A.; Suzuki, A. Synthesis of bassianolide. Tetrahedron Lett. 1977, 46, 4049–4050. [Google Scholar] [CrossRef]
- Shiomi, K.; Matsui, R.; Kakei, A.; Yamaguchi, Y.; Masuma, R.; Hatano, H.; Arai, N.; Isozaki, M.; Tanaka, H.; Kobayashi, S.; et al. Verticilide, a new ryanodine-binding inhibitor, produced by Verticilium sp. FKI-1033. J. Antibiot. 2010, 63, 77–82. [Google Scholar]
- Ohshiro, T.; Matsudo, D.; Kazuhiro, T.; Uchida, R.; Nonaka, K.; Masuma, R.; Tomada, H. New verticilides, inhibitors of acyl-CoA: Cholesterol acyltransferase, produced by Verticillium sp. FKI-2679. J. Antibiot. 2012, 65, 255–262. [Google Scholar]
- Monma, S.; Sunazuka, T.; Nagai, K.; Arai, T.; Shiomi, K.; Matsui, R.; Omura, S. Verticilide: Elucidation of absolute configuration and total synthesis. Org. Lett. 2006, 8, 5601–5604. [Google Scholar] [CrossRef] [PubMed]
- Sasaki, T.; Takagi, M.; Yaguchi, T.; Miyadoh, S.; Okada, T.; Koyama, M. A new anthelmintic cyclodepsipeptide, PF1022A. J. Antibiot. 1992, 45, 692–697. [Google Scholar] [CrossRef] [PubMed]
- Sakanaka, O.; Okada, Y.; Ohyama, M.; Matsumoto, M.; Takahashi, M.; Murai, Y.; Iinuma, K.; Harder, A.; Mencke, N. Novel Cyclic Depsipeptide PF1022A Derivatives. Patent WO97/11064, 1997. [Google Scholar]
- Dutton, F.E.; Nelson, S.J. Synthesis of PF1022A, an anthelmintic cyclodepsipeptide. J. Antibiot. 1994, 47, 1322–1327. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, M.; Iinuma, K.; Isogai, A.; Suzuki, A. Total synthesis of the anthelmintic cyclodepsipeptide, PF1022A. Biosci. Biotechnol. Biochem. 1994, 58, 1193–1194. [Google Scholar] [CrossRef]
- Scherkenbeck, J.; Plant, A.; Harder, A.; Mencke, N. A highly efficient synthesis of the anthelmintic cyclooctadepsipeptide PF1022A. Tetrahedron 1995, 51, 8459–8470. [Google Scholar] [CrossRef]
- Kobayashi, M.; Nanba, T.; Toyama, T.; Saito, A. Synthesis and anthelmintic activity of the cyclodepsipeptide PF1022A. Annu. Rep. Sankyo Res. Lab. 1994, 46, 67–75. [Google Scholar]
- Lee, B.H. Solid-phase synthesis of cyclooctadepsipeptide PF1022A analogs using a cyclization-cleavage method with oxime-resin. Tetrahedron Lett. 1997, 38, 757–760. [Google Scholar] [CrossRef]
- Nishino, N.; Xu, M.; Mihara, H.; Fujimoto, T. Sequence dependent cyclization-cleavage of dipeptides from the oxime resin and its prevention. Bull. Chem. Soc. Jpn. 1991, 65, 991–994. [Google Scholar] [CrossRef]
- Kumagai, H. Efficient preparation of cyclic peptide mixtures by solid phase synthesis and cyclization cleavage with oxime resin. Tetrahedron Lett. 1995, 36, 4837–4840. [Google Scholar] [CrossRef]
- Dutton, F.E.; Lee, B.H. Epsilon-lactam analogs of the anthelmintic cyclodepsipeptide PF1022A. Tetrahedron Lett. 1998, 39, 5313–5316. [Google Scholar] [CrossRef]
- Lee, B.H.; Dutton, F.E.; Thompson, D.P.; Thomas, E.M. Generation of a small library of cyclodepsipeptides PF1022A analogues using a cyclization-cleavage method with oxime resin. Bioorg. Med. Chem. Lett. 2002, 12, 353–356. [Google Scholar] [CrossRef] [PubMed]
- Lüttenberg, S.; Sondermann, F.; Scherkenbeck, J. Anthelmintic PF1022A: Stepwise solid-phase synthesis of a cyclodepsipeptide containing N-methyl amino acids. Tetrahedron 2012, 68, 2068–2073. [Google Scholar]
- Scherkenbeck, J.; Lüttenberg, S.; Ludwig, M.; Brüchner, K.; Kotthaus, A. Segment solid-phase total synthesis of the anthelmintic cyclooctadepsipeptides PF1022A and emodepside. Eur. J. Org. Chem. 2012, 1546–1553. [Google Scholar] [CrossRef]
- Scherkenbeck, J.; Jeschke, P.; Harder, A. PF1022A and related cyclodepsipeptides—A novel class of anthelmintics. Curr. Top. Med. Chem. 2002, 2, 759–777. [Google Scholar] [CrossRef] [PubMed]
- Scherkenbeck, J.; Dyker, H.; Gondol, D.; Harder, A.; Plant, A.; Reichel, F. Synthesis, conformational studies and anthelmintic activity of a constrained PF1022A analogue. Pest. Sci. 1999, 55, 457–461. [Google Scholar] [CrossRef]
- Dyker, H.; Harder, A.; Scherkenbeck, J. Chimeric cyclodepsipeptides as mimetics for the anthelmintic PF1022A. Bioorg. Med. Chem. Lett. 2004, 14, 6129–6132. [Google Scholar] [CrossRef] [PubMed]
- Dutton, F.E.; Lee, B.H.; Johnson, S.S.; Coscarelli, E.M.; Lee, P.H. Restricted conformation analogues of an anthelmintic cyclodepsipeptide. J. Med. Chem. 2003, 46, 2057–2073. [Google Scholar] [CrossRef] [PubMed]
- Dyker, H.; Scherkenbeck, J.; Gondol, D.; Goehrt, A.; Harder, A. Azadepsipeptides: Synthesis and evaluation of a novel class of peptidomimetics. J. Org. Chem. 2001, 66, 3760–3766. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, M.; Okada, Y.; Takahashi, M.; Sakanaka, O.; Matsumoto, M.; Atsumi, K. Structure-activity relationship of anthelmintic cyclooctadepsipeptides. Biosci. Biotechnol. Biochem. 2011, 75, 1354–1363. [Google Scholar] [CrossRef] [PubMed]
- Ohyama, M. Chemistry of anthelmintic cyclodepsipeptides. Sci. Report of Meiji Seika Kaisha 2006, 45, 8–34. [Google Scholar]
- Dyker, H.; Harder, A.; Mencke, N.; Scherkenbeck, J.; Georg von, S. Desoxycyclodepsipeptides and Their Use for Combatting Endoparasites. Patent WO19980554469A1, 1998. [Google Scholar]
- Jeschke, P.; Harder, A.; Etzel, W.; Gau, W.; Thielking, G.; Bonse, G.; Iinuma, K. Synthesis and anthelmintic activity of thioamide analogues of cyclic octadepsipeptides such as PF1022A. Pest. Manag. Sci. 2001, 57, 1000–1006. [Google Scholar] [CrossRef] [PubMed]
- Jeschke, P.; Harder, A.; Samson-Himmelstjerna, G.V.; Etzel, W.; Gau, W.; Thielking, G.; Bonse, G. Synthesis of anthelmintically active N-methylated amidoxime analogues of the cyclic octadepsipeptide PF1022A. Pest. Manag. Sci. 2002, 58, 1205–1215. [Google Scholar] [CrossRef] [PubMed]
- Weckwerth, W.; Miyamoto, K.; Iinuma, K.; Krause, M.; Glinski, M.; Storm, T.; Bonse, G.; Kleinkauf, H.; Zocher, R. Biosynthesis of PF1022A and related cyclooctadepsipeptides. J. Biol. Chem. 2000, 275, 17909–17915. [Google Scholar] [CrossRef] [PubMed]
- Müller, J.; Feifel, S.C.; Schmiederer, T.; Zocher, R.; Süssmuth, R.D. In vitro synthesis of new cyclodepsipeptides of the PF1022A-type: Probing the α-d-hydroxy acid tolerance of PF1022 synthetase. ChemBioChem 2009, 10, 323–328. [Google Scholar]
- Harder, A.; Schmitt-Wrede, H.; Krücken, J.; Marinovski, P.; Wunderlich, F.; Willson, J.; Amliwala, K.; Holden-Dye, L.; Walker, R. Cyclooctadepsipeptides—An anthelmintically active class of compounds exhibiting a novel mode of action. Int. J. Antimicrob. Ag. 2003, 22, 318–331. [Google Scholar] [CrossRef]
- Guest, M.; Bull, K.; Walker, R.J.; Amliwala, K.; O’Connor, V.; Harder, A.; Holden-Dye, L.; Hopper, A.N. The calcium-activated potassium channel, SLO-1, is required for the action of the novel cyclo-octadepsipeptide anthelmintic, emodepside, in Caenorhabditis elegans. Int. J. Parasitol. 2007, 37, 1577–1588. [Google Scholar] [CrossRef] [PubMed]
- Ueda, K.; Xiao, J.; Doke, N.; Nakatsuka, S. Isolation and structure of BZR-cotoxin IV produced by Bipolaris zeicola race 3, the cause of leaf spot disease in corn. Tetrahedron Lett. 1995, 36, 741–744. [Google Scholar] [CrossRef]
- Ueda, K.; Xiao, J.; Doke, N.; Nakatsuka, S. Structure of BZR-cotoxin I produced by Bipolaris zeicola race 3, the cause of leaf spot disease in corn. Tetrahedron Lett. 1994, 35, 7033–7036. [Google Scholar] [CrossRef]
- Ueda, K.; Xiao, J.; Doke, N.; Nakatsuka, S. Structure of BZR-cotoxin II produced by Bipolaris zeicola race 3, the cause of leaf spot disease in corn. Tetrahedron Lett. 1994, 33, 5377–5380. [Google Scholar] [CrossRef]
- Ueda, K.; Xiao, J.; Doke, N.; Nakatsuka, S. Structure of BZR-cotoxin III produced by Bipolaris zeicola race 3, the cause of leaf spot desease in corn. Nat. Prod. Lett. 1994, 6, 43–48. [Google Scholar] [CrossRef]
- Ikai, K.; Shiomi, K.; Takesako, K.; Mizutani, S.; Yamamoto, J.; Ogawa, Y.; Ueno, M.; Kato, I. Structures of aureobasidins B to R. J. Antibiot. 1991, 44, 1187–1198. [Google Scholar] [CrossRef] [PubMed]
- Yoshikawa, Y.; Ikai, K.; Umeda, Y.; Ogawa, A.; Takesako, K.; Kato, I. Isolation, structures, and antifungal activities of new aureobasidins. J. Antibiot. 1993, 46, 1347–1354. [Google Scholar] [CrossRef] [PubMed]
- Kurome, T.; Inami, K.; Inoue, T.; Ikai, K.; Takesako, K.; Kato, I.; Shiba, T. Total synthesis of an antifungal cyclic depsipeptide aureobasidin A. Tetrahedron 1996, 52, 4327–4346. [Google Scholar] [CrossRef]
- Jao, E.; Cooper, A.B.; Rane, D.F.; Saksena, A.K.; Desai, J.; Wang, J.; Girijavallabhan, V.M.; Ganguly, A.K. Total synthesis of the antifungal cyclic depsipeptides sch 57697 and aureobasidin A. Tetrahedron Lett. 1996, 37, 5661–5664. [Google Scholar] [CrossRef]
- Maharani, R.; Brownlee, R.T.C.; Hughes, A.B.; Abbott, B.M. A total synthesis of a highly N-methylated cyclodepsipeptide [2S,3S]-aureobasidin L using solid-phase methods. Tetrahedron 2014, 70, 2351–2358. [Google Scholar] [CrossRef]
- Suzuki, Y.; Ojika, M.; Sakagami, Y.; Kajda, K.; Fudou, R.; Kameyama, T. New cyclic depsipeptide antibiotics, clavariopsin A and B, produced by an aquatic hyphomycetes, Clavariopsis aquatica. 2. Structure analysis. J. Antibiot. 2001, 54, 22–28. [Google Scholar] [CrossRef]
- Kaika, K.; Fudou, R.; Kameyama, T.; Tubaki, K.; Suzuki, Y.; Ojika, M.; Sakagami, Y. New cyclic depsipeptide antibiotics, clavariopsins A and B, produced by an aquatic hyphomycetes, Clavariopsis aquatica. 1. Taxonomy, fermentation, isolation, and biological properties. J. Antibiot. 2001, 54, 17–21. [Google Scholar]
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Sivanathan, S.; Scherkenbeck, J. Cyclodepsipeptides: A Rich Source of Biologically Active Compounds for Drug Research. Molecules 2014, 19, 12368-12420. https://doi.org/10.3390/molecules190812368
Sivanathan S, Scherkenbeck J. Cyclodepsipeptides: A Rich Source of Biologically Active Compounds for Drug Research. Molecules. 2014; 19(8):12368-12420. https://doi.org/10.3390/molecules190812368
Chicago/Turabian StyleSivanathan, Sivatharushan, and Jürgen Scherkenbeck. 2014. "Cyclodepsipeptides: A Rich Source of Biologically Active Compounds for Drug Research" Molecules 19, no. 8: 12368-12420. https://doi.org/10.3390/molecules190812368
APA StyleSivanathan, S., & Scherkenbeck, J. (2014). Cyclodepsipeptides: A Rich Source of Biologically Active Compounds for Drug Research. Molecules, 19(8), 12368-12420. https://doi.org/10.3390/molecules190812368