Different Fluorophore Labeling Strategies and Designs Affect Millisecond Kinetics of DNA Hairpins

Abstract
:
1. Introduction

2. Results and Discussion
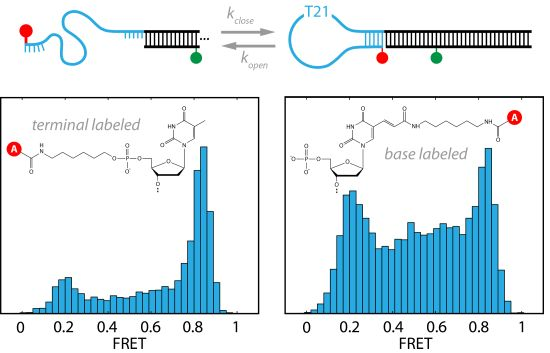
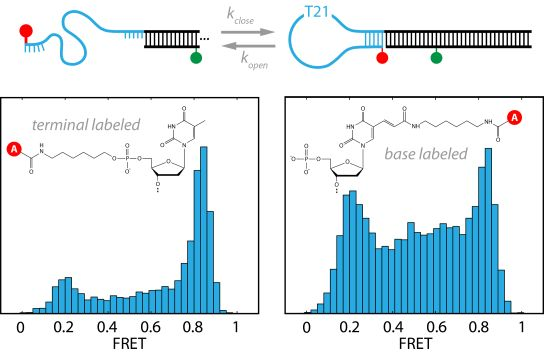
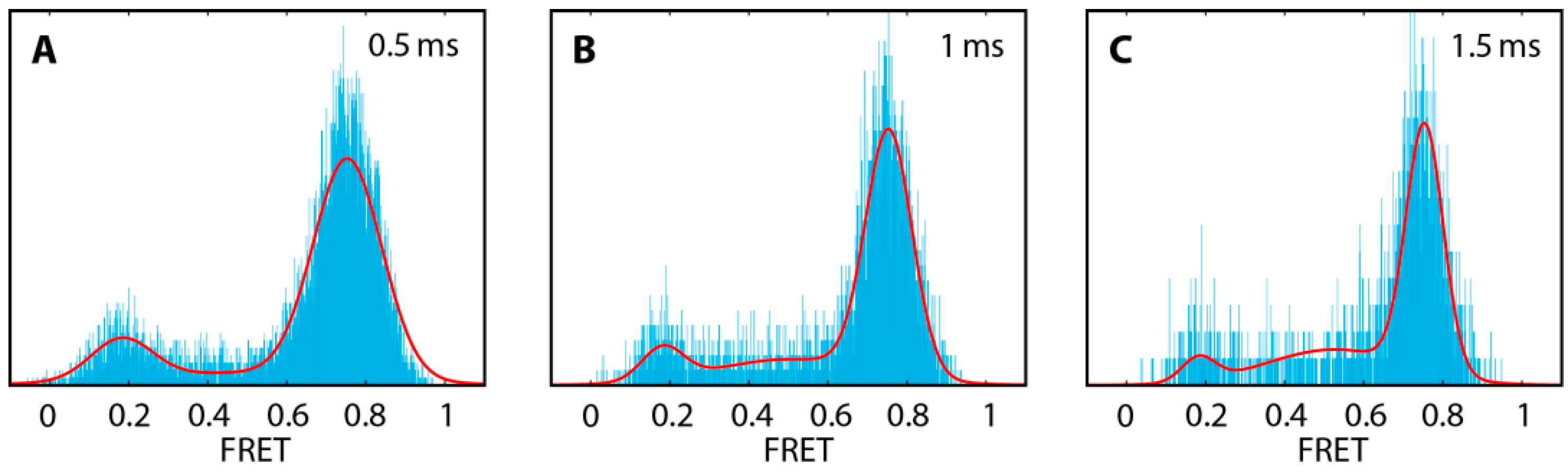
2.1. DNA Hairpins with Millisecond Kinetics



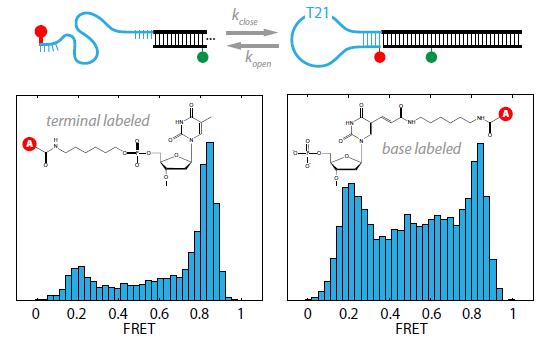{kind=link}
{kind=link}
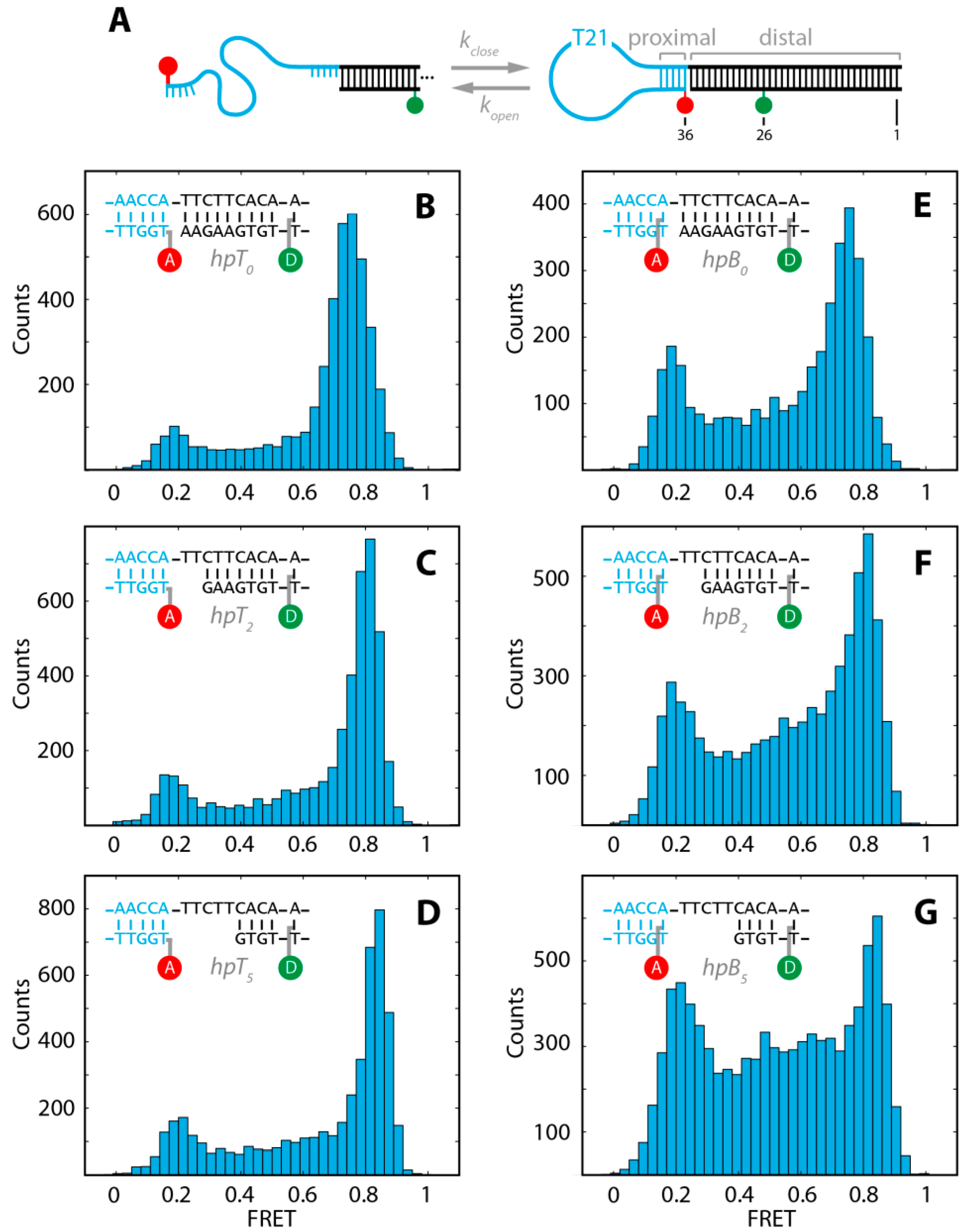{kind=link}
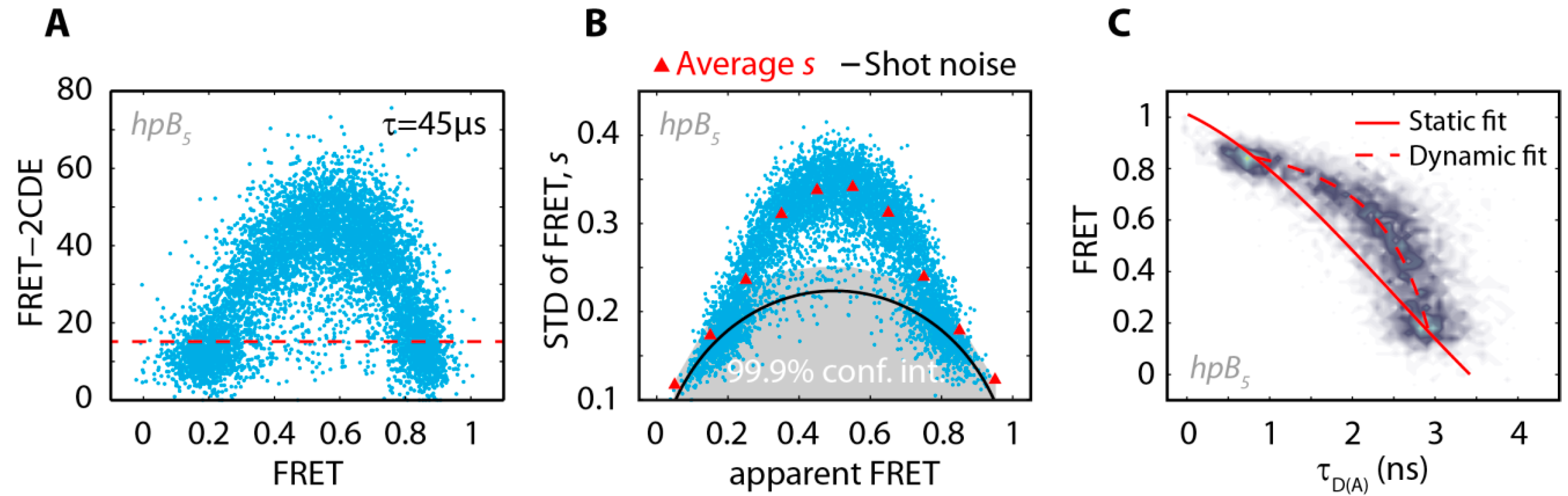{kind=link}
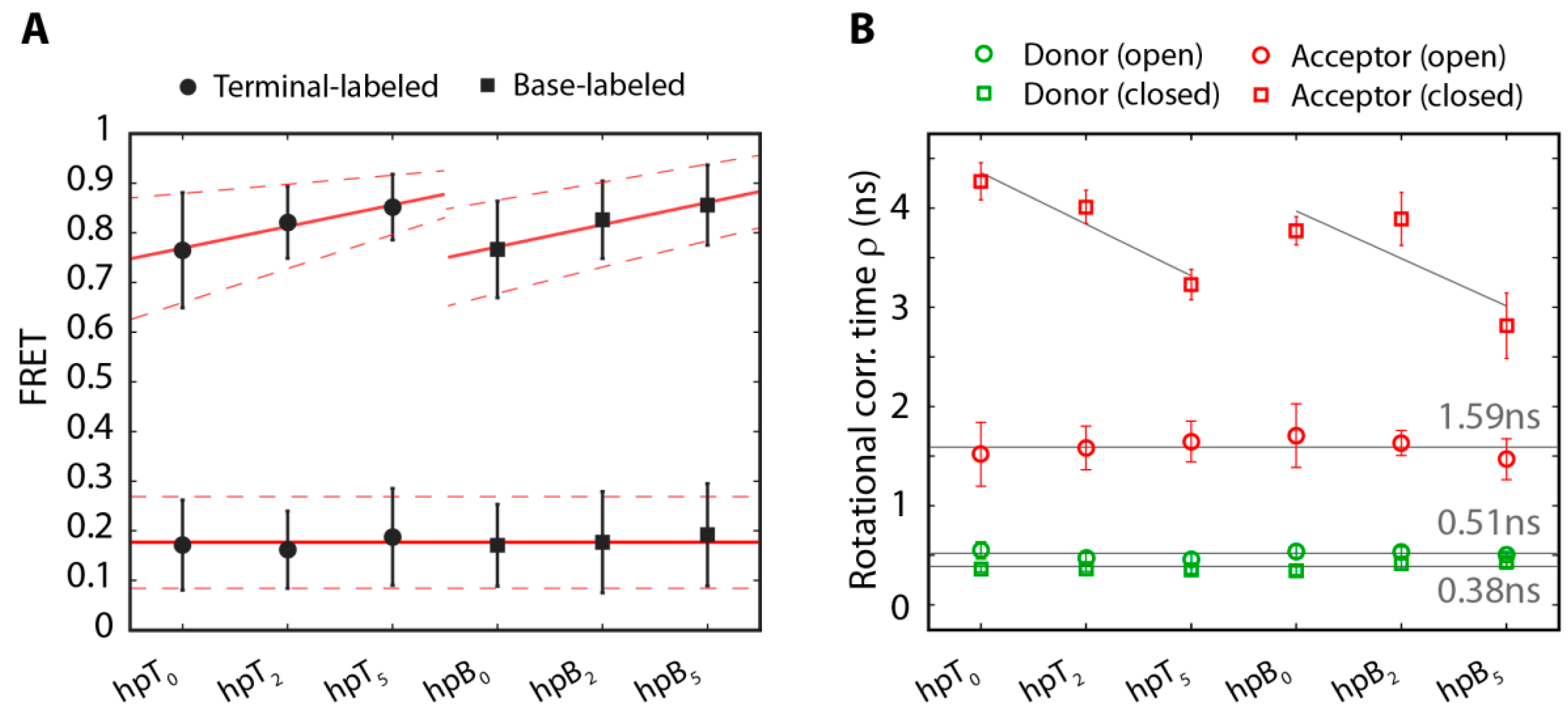{kind=link}
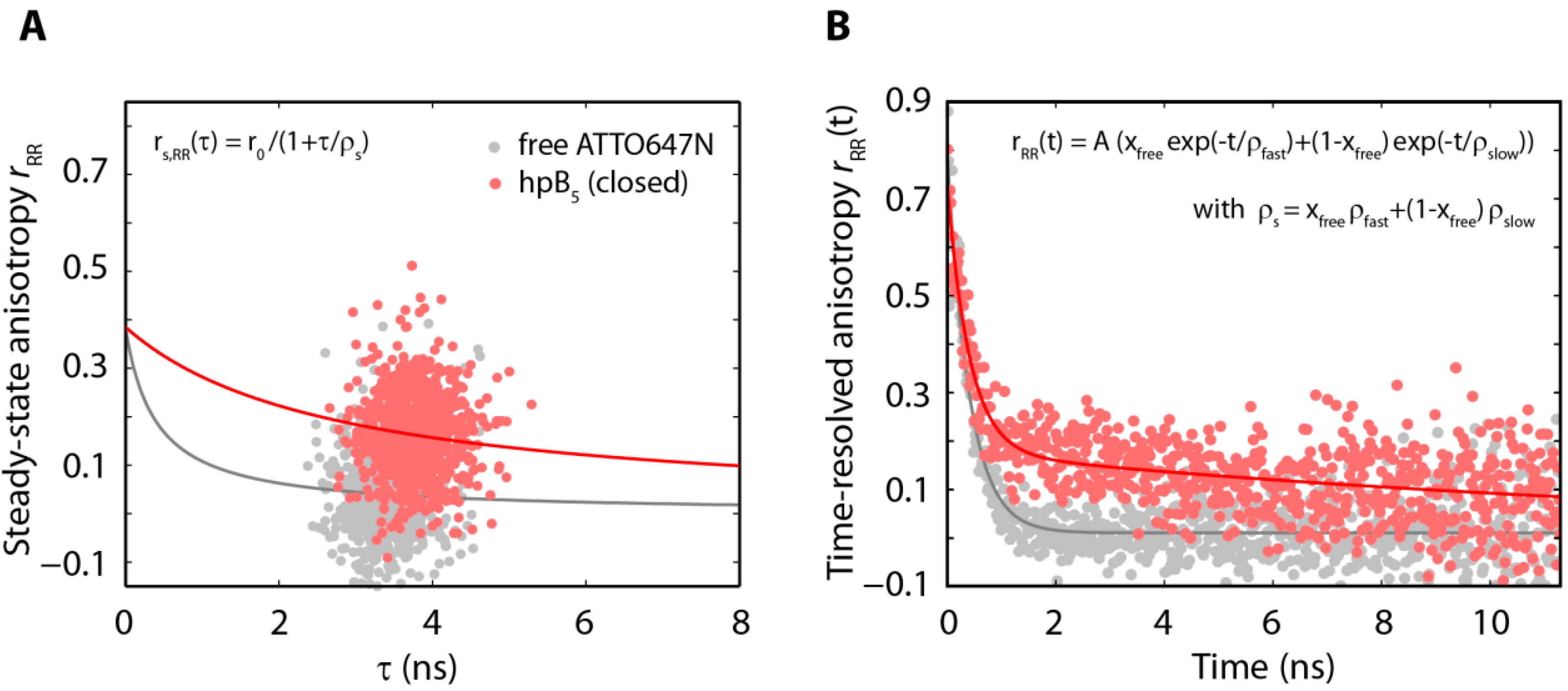{kind=link}
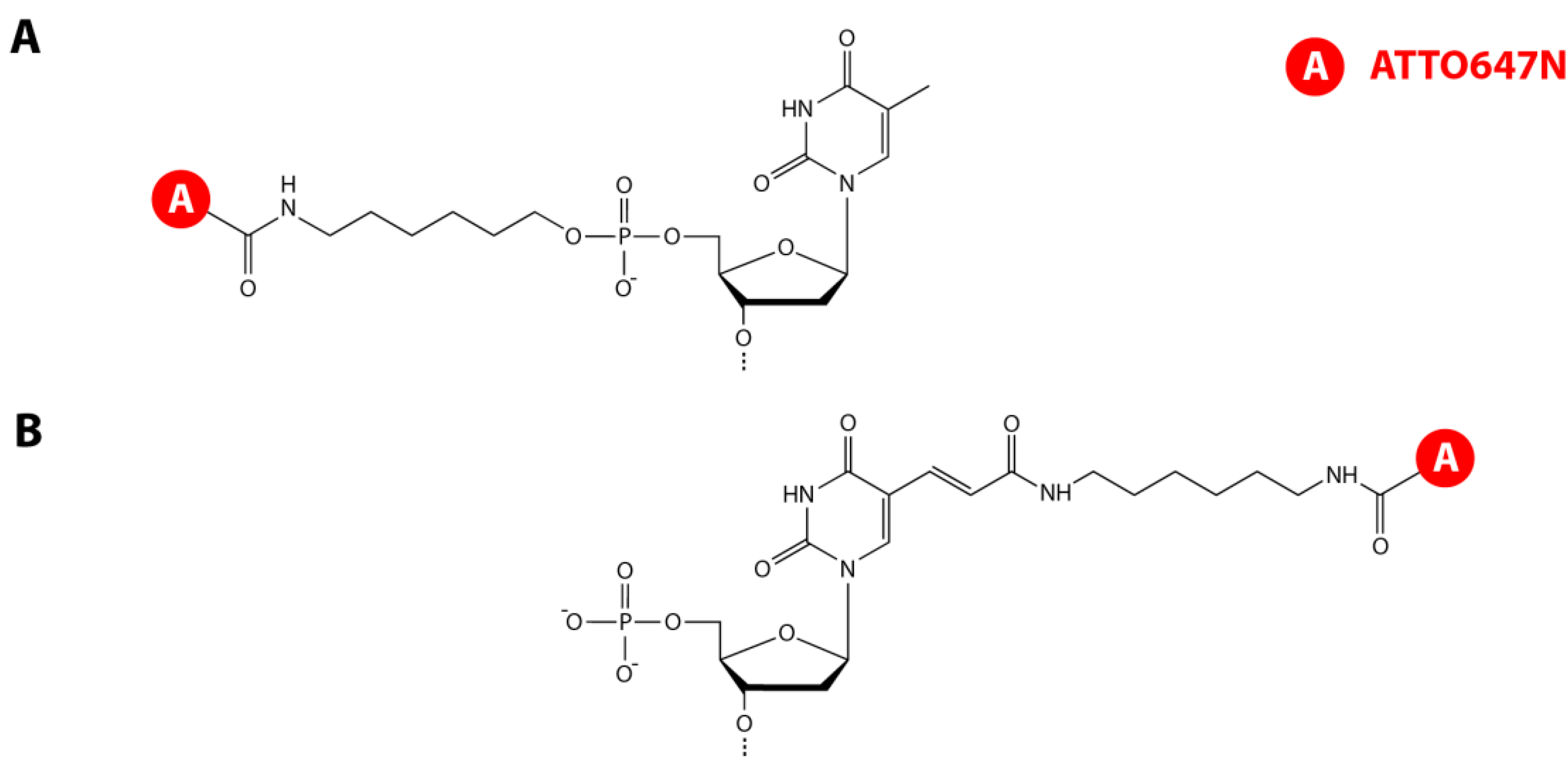{kind=link}
{kind=link}
{kind=link}
| Substrate | Eopen ± σopen | Eclose ± σclose | Nmol |
|---|---|---|---|
| hpT0 | 0.17 ± 0.05 | 0.77 ± 0.05 | 1622 |
| hpT2 | 0.16 ± 0.04 | 0.82 ± 0.04 | 1440 |
| hpT5 | 0.19 ± 0.05 | 0.85 ± 0.05 | 1589 |
| hpB0 | 0.17 ± 0.04 | 0.77 ± 0.04 | 1058 |
| hpB2 | 0.18 ± 0.05 | 0.83 ± 0.05 | 1267 |
| hpB5 | 0.19 ± 0.05 | 0.86 ± 0.05 | 1713 |
| Substrate | kclose (ms−1) | kopen (ms−1) | ∆G0 (kJ/mol) | ∆G‡close (kJ/mol) | ∆G‡open (kJ/mol) |
|---|---|---|---|---|---|
| hpT0 | 0.86 ± 0.05 | 0.20 ± 0.01 | −3.57 ± 0.26 | 19.98 ± 0.13 | 23.55 ± 0.11 |
| hpT2 | 0.92 ± 0.05 | 0.29 ± 0.01 | −2.83 ± 0.22 | 19.81 ± 0.13 | 22.64 ± 0.12 |
| hpT5 | 0.91 ± 0.02 | 0.35 ± 0.02 | −2.34 ± 0.19 | 19.84 ± 0.06 | 22.18 ± 0.12 |
| hpB0 | 0.66 ± 0.04 | 0.41 ± 0.03 | −1.17 ± 0.33 | 20.63 ± 0.15 | 21.79 ± 0.18 |
| hpB2 | 0.87 ± 0.02 | 0.55 ± 0.01 | −1.12 ± 0.10 | 19.95 ± 0.06 | 21.07 ± 0.05 |
| hpB5 | 0.90 ± 0.01 | 0.84 ± 0.01 | −0.17 ± 0.06 | 19.87 ± 0.03 | 20.04 ± 0.04 |
2.2. The Base-Labeled Hairpin is More Dynamic than the Terminal-Labeled Hairpin
2.3. The Acceptor Fluorophore on the Hairpin Interacts with the Double-Stranded Distal Stem


| Substrate | ρfast (ns) | ρslow (ns) | xfree | ρs,closed (ns) |
|---|---|---|---|---|
| hpT0 | 0.47 ± 0.11 | 15 ± 3 | 0.74 ± 0.07 | 4.27 ± 0.19 |
| hpT2 | 0.37 ± 0.10 | 14 ± 3 | 0.74 ± 0.06 | 4.01 ± 0.17 |
| hpT5 | 0.51 ± 0.08 | 13 ± 3 | 0.79 ± 0.06 | 3.23 ± 0.15 |
| hpB0 | 0.34 ± 0.09 | 14 ± 4 | 0.76 ± 0.08 | 3.77 ± 0.14 |
| hpB2 | 0.41 ± 0.12 | 19 ± 8 | 0.81 ± 0.08 | 3.89 ± 0.27 |
| hpB5 | 0.33 ± 0.16 | 15 ± 6 | 0.84 ± 0.08 | 2.82 ± 0.33 |
3. Experimental Section
3.1. DNA Hairpin Design and Labeling Strategies

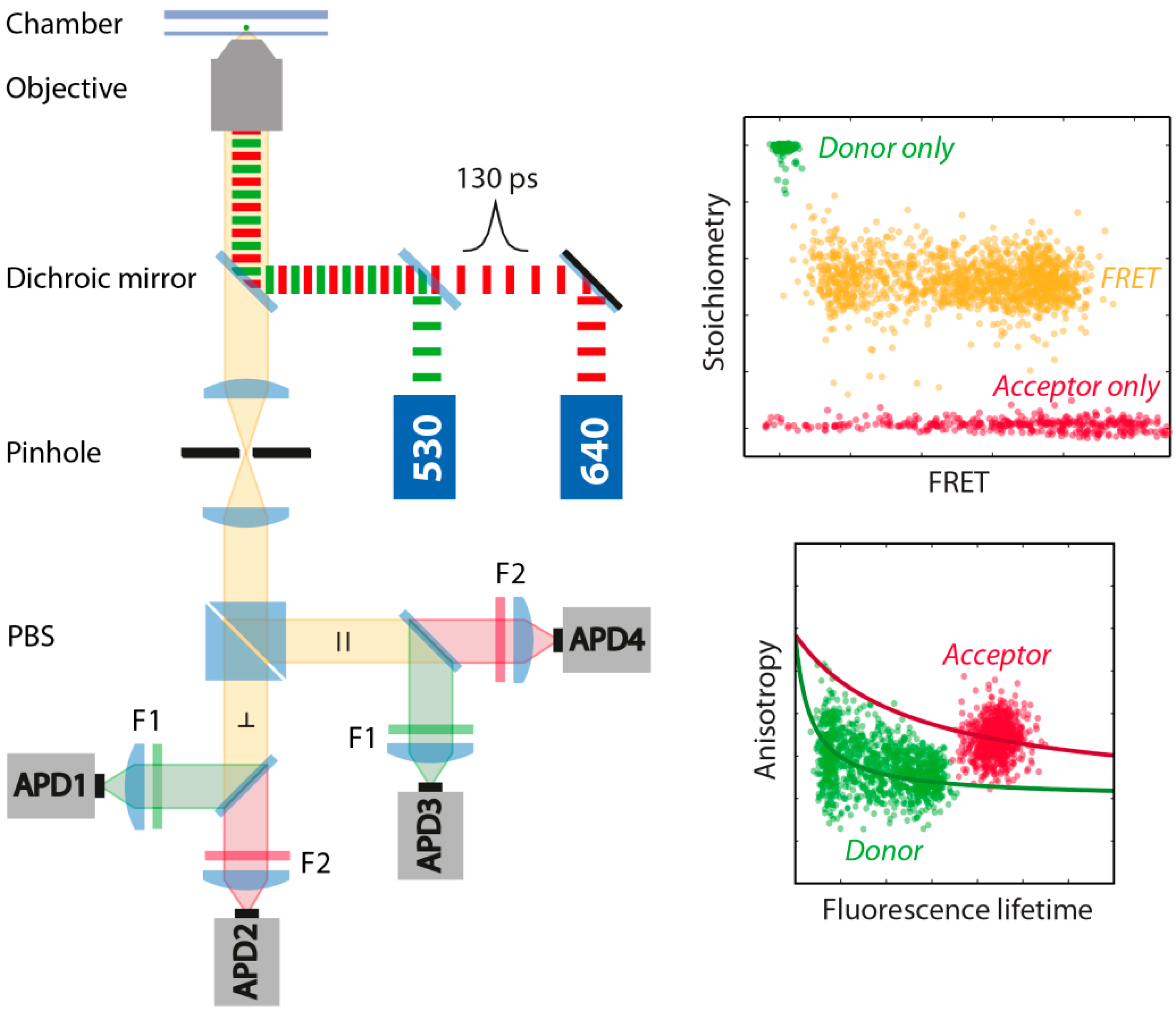
3.2. smFRET Measurements and Analysis Procedures
| Substrate | T (°C) |
|---|---|
| hpT0 | 21.34 ± 0.09 |
| hpT2 | 21.34 ± 0.07 |
| hpT5 | 21.41 ± 0.09 |
| hpB0 | 21.36 ± 0.15 |
| hpB2 | 21.34 ± 0.07 |
| hpB5 | 21.38 ± 0.14 |



 with n = 5, the number of photons.
with n = 5, the number of photons.


4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kapanidis, A.N.; Strick, T. Biology one molecule at a time. Trends Biochem. Sci. 2009, 34, 234–243. [Google Scholar] [CrossRef]
- Joo, C.; Balci, H.; Ishitsuka, Y.; Buranachai, C.; Ha, T. Advances in single-molecule fluorescence methods for molecular biology. Annu. Rev. Biochem. 2008, 77, 51–76. [Google Scholar] [CrossRef]
- Tan, E.; Wilson, T.J.; Nahas, M.K.; Clegg, R.M.; Lilley, D.M.J.; Ha, T. A four-way junction accelerates hairpin ribozyme folding via a discrete intermediate. Proc. Natl. Acad. Sci. USA 2003, 100, 9308–9313. [Google Scholar]
- Yeoman, J.A.; Orte, A.; Ashbridge, B.; Klenerman, D.; Balasubramanian, S. RNA conformation in catalytically active human telomerase. J. Am. Chem. Soc. 2010, 132, 2852–2853. [Google Scholar]
- Dupuis, N.F.; Holmstrom, E.D.; Nesbitt, D.J. Molecular-crowding effects on single-molecule RNA folding/unfolding thermodynamics and kinetics. Proc. Natl. Acad. Sci. USA 2014, 111, 8464–8469. [Google Scholar] [CrossRef]
- Zhuang, X.; Bartley, L.E.; Babcock, H.P.; Russell, R.; Ha, T.; Herschlag, D.; Chu, S. A single-molecule study of RNA catalysis and folding. Science 2000, 288, 2048–2051. [Google Scholar] [CrossRef]
- McKinney, S.A.; Freeman, A.D.J.; Lilley, D.M.J.; Ha, T. Observing spontaneous branch migration of Holliday junctions one step at a time. Proc. Natl. Acad. Sci. USA 2005, 102, 5715–5720. [Google Scholar] [CrossRef]
- Zhuang, X.; Kim, H.; Pereira, M.J.B.; Babcock, H.P.; Walter, N.G.; Chu, S. Correlating structural dynamics and function in single ribozyme molecules. Science 2002, 296, 1473–1476. [Google Scholar] [CrossRef]
- Wallace, M.I.; Ying, L.; Balasubramanian, S.; Klenerman, D. FRET Fluctuation spectroscopy: Exploring the conformational dynamics of a DNA hairpin loop. J. Phys. Chem. B 2000, 104, 11551–11555. [Google Scholar] [CrossRef]
- Grunwell, J.R.; Glass, J.L.; Lacoste, T.D.; Deniz, A.A.; Chemla, D.S.; Schultz, P.G. Monitoring the conformational fluctuations of DNA hairpins using single-pair fluorescence resonance energy transfer. J. Am. Chem. Soc. 2001, 123, 4295–4303. [Google Scholar] [CrossRef]
- Nir, E.; Michalet, X.; Hamadani, K.M.; Laurence, T.A.; Neuhauser, D.; Kovchegov, Y.; Weiss, S. Shot-noise limited single-molecule FRET histograms: Comparison between theory and experiments. J. Phys. Chem. B 2006, 110, 22103–22124. [Google Scholar]
- Santoso, Y.; Torella, J.P.; Kapanidis, A.N. Characterizing single-molecule FRET dynamics with probability distribution analysis. ChemPhysChem 2010, 11, 2209–2219. [Google Scholar] [CrossRef]
- Torella, J.P.; Holden, S.J.; Santoso, Y.; Hohlbein, J.; Kapanidis, A.N. Identifying molecular dynamics in single-molecule FRET experiments with burst variance analysis. Biophys. J. 2011, 100, 1568–1577. [Google Scholar] [CrossRef]
- Kügel, W.; Muschielok, A.; Michaelis, J. Bayesian-inference-based fluorescence correlation spectroscopy and single-molecule burst analysis reveal the influence of dye selection on DNA hairpin dynamics. ChemPhysChem 2012, 13, 1013–1022. [Google Scholar] [CrossRef]
- Tomov, T.E.; Tsukanov, R.; Masoud, R.; Liber, M.; Plavner, N.; Nir, E. Disentangling subpopulations in single-molecule FRET and ALEX experiments with photon distribution analysis. Biophys. J. 2012, 102, 1163–1173. [Google Scholar] [CrossRef]
- Tsukanov, R.; Tomov, T.E.; Berger, Y.; Liber, M.; Nir, E. Conformational dynamics of DNA hairpins at millisecond resolution obtained from analysis of single-molecule FRET histograms. J. Phys. Chem. B 2013, 117, 16105–16109. [Google Scholar] [CrossRef]
- Tsukanov, R.; Tomov, T.E.; Masoud, R.; Drory, H.; Plavner, N.; Liber, M.; Nir, E. Detailed study of DNA hairpin dynamics using single-molecule fluorescence assisted by DNA origami. J. Phys. Chem. B 2013, 117, 11932–11942. [Google Scholar] [CrossRef]
- Müller, B.K.; Reuter, A.; Simmel, F.C.; Lamb, D.C. Single-pair FRET characterization of DNA tweezers. Nano Lett. 2006, 6, 2814–2820. [Google Scholar] [CrossRef]
- Cisse, I.I.; Kim, H.; Ha, T. A rule of seven in Watson-Crick base-pairing of mismatched sequences. Nat. Struct. Mol. Biol. 2012, 19, 623–627. [Google Scholar] [CrossRef]
- Bokinsky, G.; Rueda, D.; Misra, V.K.; Rhodes, M.M.; Gordus, A.; Babcock, H.P.; Walter, N.G.; Zhuang, X. Single-molecule transition-state analysis of RNA folding. Proc. Natl. Acad. Sci. USA 2003, 100, 9302–9307. [Google Scholar] [CrossRef]
- Tomov, T.E.; Tsukanov, R.; Liber, M.; Masoud, R.; Plavner, N.; Nir, E. Rational design of DNA motors: Fuel optimization through single-molecule fluorescence. J. Am. Chem. Soc. 2013, 135, 11935–11941. [Google Scholar] [CrossRef]
- Nahas, M.K.; Wilson, T.J.; Hohng, S.; Jarvie, K.; Lilley, D.M.J.; Ha, T. Observation of internal cleavage and ligation reactions of a ribozyme. Nat. Struct. Mol. Biol. 2004, 11, 1107–1113. [Google Scholar] [CrossRef]
- Schuler, B.; Lipman, E.A.; Eaton, W.A. Probing the free-energy surface for protein folding with single-molecule fluorescence spectroscopy. Nature 2002, 419, 743–747. [Google Scholar] [CrossRef]
- Margittai, M.; Widengren, J.; Schweinberger, E.; Schröder, G.F.; Felekyan, S.; Haustein, E.; König, M.; Fasshauer, D.; Grubmüller, H.; Jahn, R.; et al. Single-molecule fluorescence resonance energy transfer reveals a dynamic equilibrium between closed and open conformations of syntaxin 1. Proc. Natl. Acad. Sci. USA 2003, 100, 15516–15521. [Google Scholar]
- Diez, M.; Zimmermann, B.; Börsch, M.; König, M.; Schweinberger, E.; Steigmiller, S.; Reuter, R.; Felekyan, S.; Kudryavtsev, V.; Seidel, C.A.M.; et al. Proton-powered subunit rotation in single membrane-bound F0F1-ATP synthase. Nat. Struct. Mol. Biol. 2004, 11, 135–141. [Google Scholar] [CrossRef]
- Majumdar, D.S.; Smirnova, I.; Kasho, V.; Nir, E.; Kong, X.; Weiss, S.; Kaback, H.R. Single-molecule FRET reveals sugar-induced conformational dynamics in LacY. Proc. Natl. Acad. Sci. USA 2007, 104, 12640–12645. [Google Scholar]
- Santoso, Y.; Joyce, C.M.; Potapova, O.; le Reste, L.; Hohlbein, J.; Torella, J.P.; Grindley, N.D.F.; Kapanidis, A.N. Conformational transitions in DNA polymerase I revealed by single-molecule FRET. Proc. Natl. Acad. Sci. USA 2010, 107, 715–720. [Google Scholar] [CrossRef]
- Marcinowski, M.; Höller, M.; Feige, M.J.; Baerend, D.; Lamb, D.C.; Buchner, J. Substrate discrimination of the chaperone BiP by autonomous and cochaperone-regulated conformational transitions. Nat. Struct. Mol. Biol. 2011, 18, 150–158. [Google Scholar] [CrossRef]
- Borgia, A.; Wensley, B.G.; Soranno, A.; Nettels, D.; Borgia, M.B.; Hoffmann, A.; Pfeil, S.H.; Lipman, E.A.; Clarke, J.; Schuler, B. Localizing internal friction along the reaction coordinate of protein folding by combining ensemble and single-molecule fluorescence spectroscopy. Nat. Commun. 2012, 3. [Google Scholar] [CrossRef]
- Chung, H.S.; McHale, K.; Louis, J.M.; Eaton, W.A. Single-molecule fluorescence experiments determine protein folding transition path times. Science 2012, 335, 981–984. [Google Scholar] [CrossRef]
- Soranno, A.; Buchli, B.; Nettels, D.; Cheng, R.R.; Müller-Späth, S.; Pfeil, S.H.; Hoffmann, A.; Lipman, E.A.; Makarov, D.E.; Schuler, B. Quantifying internal friction in unfolded and intrinsically disordered proteins with single-molecule spectroscopy. Proc. Natl. Acad. Sci. USA 2012, 109, 17800–17806. [Google Scholar] [CrossRef]
- Sikor, M.; Mapa, K.; von Voithenberg, L.V.; Mokranjac, D.; Lamb, D.C. Real-time observation of the conformational dynamics of mitochondrial Hsp70 by spFRET. EMBO J. 2013, 32, 1639–1649. [Google Scholar] [CrossRef]
- Hohlbein, J.; Aigrain, L.; Craggs, T.D.; Bermek, O.; Potapova, O.; Shoolizadeh, P.; Grindley, N.D.F.; Joyce, C.M.; Kapanidis, A.N. Conformational landscapes of DNA polymerase I and mutator derivatives establish fidelity checkpoints for nucleotide insertion. Nat. Commun. 2013, 4. [Google Scholar] [CrossRef]
- Chung, H.S.; Eaton, W.A. Single-molecule fluorescence probes dynamics of barrier crossing. Nature 2013, 502, 685–688. [Google Scholar] [CrossRef]
- Ratzke, C.; Hellenkamp, B.; Hugel, T. Four-colour FRET reveals directionality in the Hsp90 multicomponent machinery. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- Roy, R.; Hohng, S.; Ha, T. A practical guide to single-molecule FRET. Nat. Methods 2008, 5, 507–516. [Google Scholar] [CrossRef]
- Schuler, B.; Hofmann, H. Single-molecule spectroscopy of protein folding dynamics—Expanding scope and timescales. Curr. Opin. Struct. Biol. 2013, 23, 36–47. [Google Scholar] [CrossRef]
- Bikard, D.; Loot, C.; Baharoglu, Z.; Mazel, D. Folded DNA in action: Hairpin formation and biological functions in prokaryotes. Microbiol. Mol. Biol. Rev. 2010, 74, 570–588. [Google Scholar] [CrossRef]
- Bevilacqua, P.C.; Blose, J.M. Structures, kinetics, thermodynamics, and biological functions of RNA hairpins. Annu. Rev. Phys. Chem. 2008, 59, 79–103. [Google Scholar] [CrossRef]
- Kortmann, J.; Narberhaus, F. Bacterial RNA thermometers: Molecular zippers and switches. Nat. Rev. Microbiol. 2012, 10, 255–265. [Google Scholar] [CrossRef]
- Tan, W.; Wang, K.; Drake, T.J. Molecular beacons. Curr. Opin. Chem. Biol. 2004, 8, 547–553. [Google Scholar] [CrossRef]
- Tyagi, S.; Kramer, F.R. Molecular beacons: Probes that fluoresce upon hybridization. Nat. Biotechnol. 1996, 14, 303–308. [Google Scholar] [CrossRef]
- Bonnet, G.; Krichevsky, O.; Libchaber, A. Kinetics of conformational fluctuations in DNA hairpin-loops. Proc. Natl. Acad. Sci. USA 1998, 95, 8602–8606. [Google Scholar] [CrossRef]
- Bonnet, G.; Tyagi, S.; Libchaber, A.; Kramer, F.R. Thermodynamic basis of the enhanced specificity of structured DNA probes. Proc. Natl. Acad. Sci. USA 1999, 96, 6171–6176. [Google Scholar] [CrossRef]
- Wallace, M.I.; Ying, L.; Balasubramanian, S.; Klenerman, D. Non-Arrhenius kinetics for the loop closure of a DNA hairpin. Proc. Natl. Acad. Sci. USA 2001, 98, 5584–5589. [Google Scholar] [CrossRef]
- Jung, J.; van Orden, A. Folding and unfolding kinetics of DNA hairpins in flowing solution by multiparameter fluorescence correlation spectroscopy. J. Phys. Chem. B 2005, 109, 3648–3657. [Google Scholar] [CrossRef]
- Kim, J.; Doose, S.; Neuweiler, H.; Sauer, M. The initial step of DNA hairpin folding: A kinetic analysis using fluorescence correlation spectroscopy. Nucleic Acids Res. 2006, 34, 2516–2527. [Google Scholar] [CrossRef]
- Torres, T.; Levitus, M. Measuring conformational dynamics: A new FCS-FRET approach. J. Phys. Chem. B 2007, 111, 7392–7400. [Google Scholar] [CrossRef]
- Jung, J.; Ihly, R.; Scott, E.; Yu, M.; van Orden, A. Probing the complete folding trajectory of a DNA hairpin using dual beam fluorescence fluctuation spectroscopy. J. Phys. Chem. B 2008, 112, 127–133. [Google Scholar] [CrossRef]
- Li, H.; Ren, X.; Ying, L.; Balasubramanian, S.; Klenerman, D. Measuring single-molecule nucleic acid dynamics in solution by two-color filtered ratiometric fluorescence correlation spectroscopy. Proc. Natl. Acad. Sci. USA 2004, 101, 14425–14430. [Google Scholar] [CrossRef]
- Jung, J.; van Orden, A. A three-state mechanism for DNA hairpin folding characterized by multiparameter fluorescence fluctuation spectroscopy. J. Am. Chem. Soc. 2006, 128, 1240–1249. [Google Scholar] [CrossRef]
- Di Fiori, N.; Meller, A. The Effect of dye-dye interactions on the spatial resolution of single-molecule FRET measurements in nucleic acids. Biophys. J. 2010, 98, 2265–2272. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Tynan, C.J.; Rolfe, D.J.; Clarke, D.T.; Martin-Fernandez, M. Hydrophobic fluorescent probes introduce artifacts into single molecule tracking experiments due to non-specific binding. PLoS One 2013, 8, e74200. [Google Scholar]
- Hwang, H.; Kim, H.; Myong, S. Protein induced fluorescence enhancement as a single molecule assay with short distance sensitivity. Proc. Natl. Acad. Sci. USA 2011, 108, 7414–7418. [Google Scholar] [CrossRef]
- Lee, W.; von Hippel, P.H.; Marcus, A.H. Internally labeled Cy3/Cy5 DNA constructs show greatly enhanced photo-stability in single-molecule FRET experiments. Nucleic Acids Res. 2014, 42, 5967–5977. [Google Scholar] [CrossRef]
- Iqbal, A.; Arslan, S.; Okumus, B.; Wilson, T.J.; Giraud, G.; Norman, D.G.; Ha, T.; Lilley, D.M.J. Orientation dependence in fluorescent energy transfer between Cy3 and Cy5 terminally attached to double-stranded nucleic acids. Proc. Natl. Acad. Sci. USA 2008, 105, 11176–11181. [Google Scholar]
- Sanborn, M.E.; Connolly, B.K.; Gurunathan, K.; Levitus, M. Fluorescence properties and photophysics of the sulfoindocyanine Cy3 linked covalently to DNA. J. Phys. Chem. B 2007, 111, 11064–11074. [Google Scholar] [CrossRef]
- Ha, T.; Tinnefeld, P. Photophysics of fluorescent probes for single-molecule biophysics and super-resolution imaging. Annu. Rev. Phys. Chem. 2012, 63, 595–617. [Google Scholar] [CrossRef]
- Luo, G.; Wang, M.; Konigsberg, W.H.; Xie, X.S. Single-molecule and ensemble fluorescence assays for a functionally important conformational change in T7 DNA polymerase. Proc. Natl. Acad. Sci. USA 2007, 104, 12610–12615. [Google Scholar] [CrossRef]
- Müller, B.K.; Zaychikov, E.; Bräuchle, C.; Lamb, D.C. Pulsed interleaved excitation. Biophys. J. 2005, 89, 3508–3522. [Google Scholar] [CrossRef]
- Kapanidis, A.N.; Lee, N.K.; Laurence, T.A.; Doose, S.; Margeat, E.; Weiss, S. Fluorescence-aided molecule sorting: Analysis of structure and interactions by alternating-laser excitation of single molecules. Proc. Natl. Acad. Sci. USA 2004, 101, 8936–8941. [Google Scholar] [CrossRef]
- Kudryavtsev, V.; Sikor, M.; Kalinin, S.; Mokranjac, D.; Seidel, C.A.M.; Lamb, D.C. Combining MFD and PIE for accurate single-pair Förster resonance energy transfer measurements. ChemPhysChem 2012, 13, 1060–1078. [Google Scholar] [CrossRef]
- Sisamakis, E.; Valeri, A.; Kalinin, S.; Rothwell, P.J.; Seidel, C.A.M. Accurate single-molecule FRET studies using multiparameter fluorescence detection. Methods Enzymol. 2010, 475, 455–514. [Google Scholar] [CrossRef]
- Chung, H.S.; Gopich, I.V.; McHale, K.; Cellmer, T.; Louis, J.M.; Eaton, W.A. Extracting rate coefficients from single-molecule photon trajectories and FRET efficiency histograms for a fast-folding protein. J. Phys. Chem. A 2011, 115, 3642–3656. [Google Scholar]
- Gopich, I.V; Szabo, A. FRET efficiency distributions of multistate single molecules. J. Phys. Chem. B 2010, 114, 15221–15226. [Google Scholar] [CrossRef]
- Woodside, M.T.; Behnke-Parks, W.M.; Larizadeh, K.; Travers, K.; Herschlag, D.; Block, S.M. Nanomechanical measurements of the sequence-dependent folding landscapes of single nucleic acid hairpins. Proc. Natl. Acad. Sci. USA 2006, 103, 6190–6195. [Google Scholar]
- SantaLucia, J. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef]
- Zuker, M. Mfold web server for nucleic acid folding and hybridization prediction. Nucleic Acids Res. 2003, 31, 3406–3415. [Google Scholar] [CrossRef]
- Markham, N.R.; Zuker, M. DINAMelt web server for nucleic acid melting prediction. Nucleic Acids Res. 2005, 33, (Suppl. 2). W577–W581. [Google Scholar] [CrossRef]
- Sindbert, S.; Kalinin, S.; Nguyen, H.; Kienzler, A.; Clima, L.; Bannwarth, W.; Appel, B.; Müller, S.; Seidel, C.A.M. Accurate distance determination of nucleic acids via Förster resonance energy transfer: Implications of dye linker length and rigidity. J. Am. Chem. Soc. 2011, 133, 2463–2480. [Google Scholar] [CrossRef]
- Spiriti, J.; Binder, J.K.; Levitus, M.; van der Vaart, A. Cy3-DNA stacking interactions strongly depend on the identity of the terminal basepair. Biophys. J. 2011, 100, 1049–1057. [Google Scholar] [CrossRef]
- Unruh, J.R.; Gokulrangan, G.; Lushington, G.H.; Johnson, C.K.; Wilson, G.S. Orientational dynamics and dye-DNA interactions in a dye-labeled DNA aptamer. Biophys. J. 2005, 88, 3455–3465. [Google Scholar] [CrossRef]
- Gokulrangan, G.; Unruh, J.R.; Holub, D.F.; Ingram, B.; Johnson, C.K.; Wilson, G.S. DNA aptamer-based bioanalysis of IgE by fluorescence anisotropy. Anal. Chem. 2005, 77, 1963–1970. [Google Scholar] [CrossRef]
- Cordes, T.; Vogelsang, J.; Tinnefeld, P. On the mechanism of Trolox as antiblinking and antibleaching reagent. J. Am. Chem. Soc. 2009, 131, 5018–5019. [Google Scholar] [CrossRef]
- Swoboda, M.; Henig, J.; Cheng, H.-M.; Brugger, D.; Haltrich, D.; Plumeré, N.; Schlierf, M. Enzymatic oxygen scavenging for photostability without pH drop in single-molecule experiments. ACS Nano 2012, 6, 6364–6369. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hartmann, A.; Krainer, G.; Schlierf, M. Different Fluorophore Labeling Strategies and Designs Affect Millisecond Kinetics of DNA Hairpins. Molecules 2014, 19, 13735-13754. https://doi.org/10.3390/molecules190913735
Hartmann A, Krainer G, Schlierf M. Different Fluorophore Labeling Strategies and Designs Affect Millisecond Kinetics of DNA Hairpins. Molecules. 2014; 19(9):13735-13754. https://doi.org/10.3390/molecules190913735
Chicago/Turabian StyleHartmann, Andreas, Georg Krainer, and Michael Schlierf. 2014. "Different Fluorophore Labeling Strategies and Designs Affect Millisecond Kinetics of DNA Hairpins" Molecules 19, no. 9: 13735-13754. https://doi.org/10.3390/molecules190913735
APA StyleHartmann, A., Krainer, G., & Schlierf, M. (2014). Different Fluorophore Labeling Strategies and Designs Affect Millisecond Kinetics of DNA Hairpins. Molecules, 19(9), 13735-13754. https://doi.org/10.3390/molecules190913735