
Synthesis and Preliminary Evaluation of N-Oxide Derivatives for the Prevention of Atherothrombotic Events


Abstract
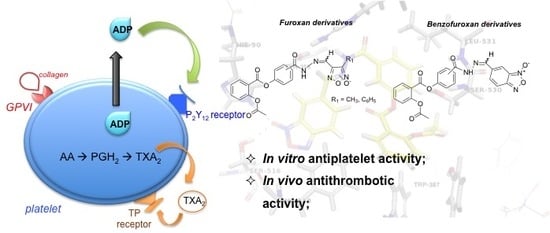
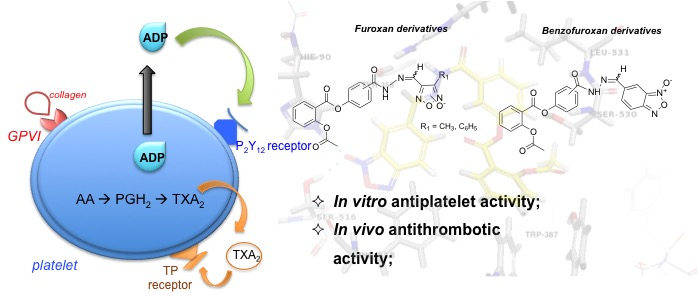
:
1. Introduction

2. Results and Discussion
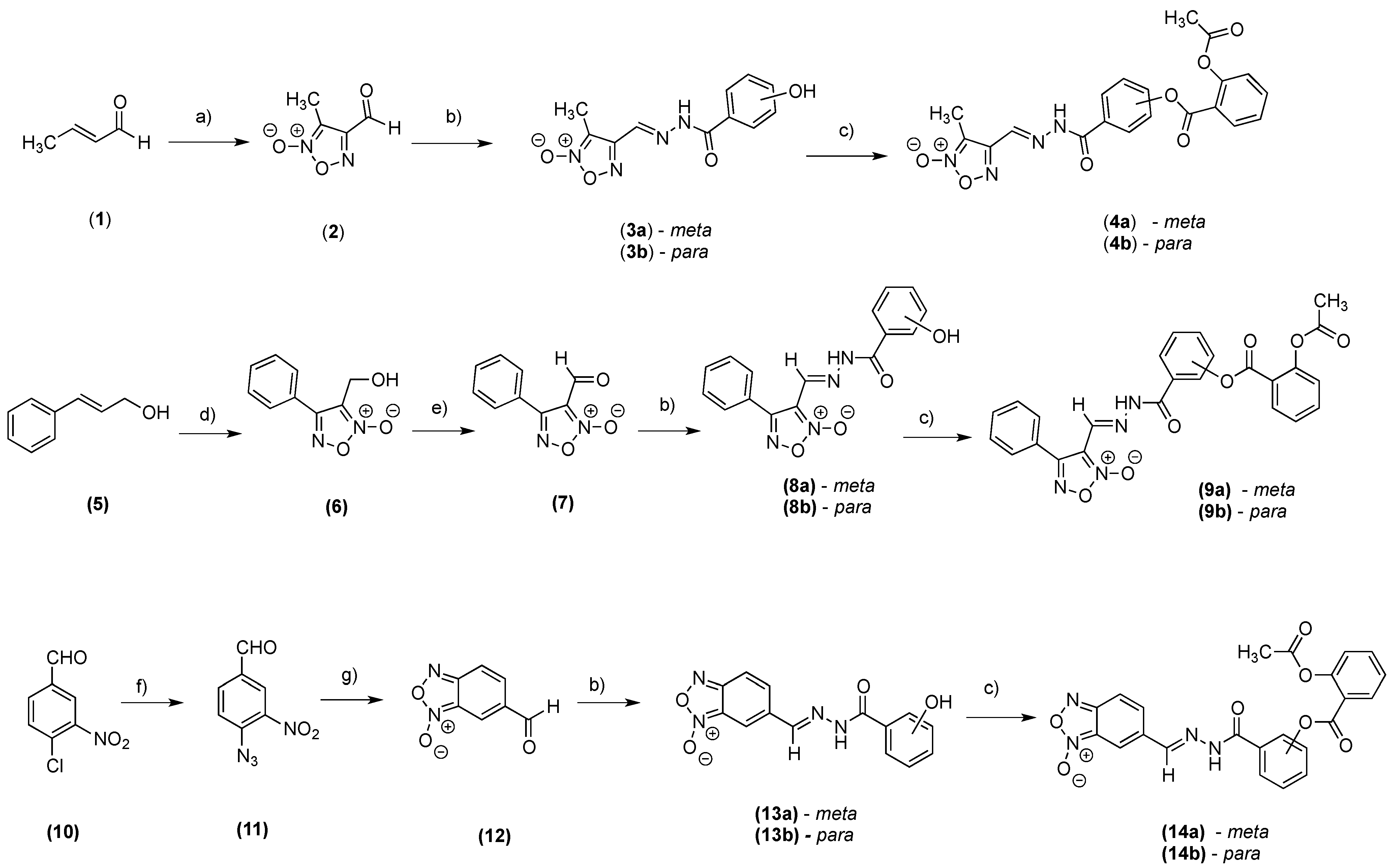
2.1. Chemistry

2.2. Pharmacological Evaluation
2.2.1. Antiplatelet Activity and Nitric Oxide Release

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | Platelet Inhibition (%) a | NO Release Data | ||
|---|---|---|---|---|
| ADP (10 μM) | Arachidonic Acid (100 μM) | Collagen (5 μg/mL) | %NO2− (mol/mol) b, 50 × 10−4 M l-Cys | |
| Control c | 0 | 0 | 0 | 0 |
| DNS | N.D. d | N.D. d | N.D. d | 10.5 ± 0.7 |
| ASA | 0 | 72.2 ± 3.7 * | 77.1 ± 3.7 * | 0 |
| 3a | 10.1 ± 3.3 * | 52.6 ± 4.1 * | 17.5 ± 2.7 * | 2.0 ± 0.7 |
| 3b | 16.8 ± 2.9 * | 47.8 ± 3.7 * | 10.5 ± 3.3 * | 1.9 ± 1.1 |
| 4a | 65.6 ± 2.4 †,* | 70.1 ± 2.9 * | 37.8 ± 4.1 * | 2.2 ± 0.4 |
| 4b | 75.6 ± 1.8 †,* | 87.5 ± 2.6 †,* | 34.5 ± 3.5 * | 1.8 ± 1.7 |
| 8a | 12.4 ± 3.7 †,* | 55.1 ± 4.3 * | 21.8 ± 5.1 * | 7.6 ± 1.2 |
| 8b | 9.2 ± 2.8 †,* | 31.6 ± 3.9 * | 8.8 ± 3.6 * | 7.5 ± 0.9 |
| 9a | 53.4 ± 2.1 †,* | 73.4 ± 3.3 * | 43.5 ± 4.7 * | 7.0 ± 0.8 |
| 9b | 68.8 ± 1.3 †,* | 82.7 ± 2.1 †,* | 47.2 ± 2.9 * | 7.9 ± 1.3 |
| 13a | 47.1 ± 2.5 * | 52.3 ± 3.8 * | 45.1 ± 3.8 * | 0 |
| 13b | 44.2 ± 5.1 †,* | 57.8 ± 4.9 * | 43.0 ± 3.1 * | 0 |
| 14a | 74.9 ± 2.2 †,* | 76.9 ± 3.1 * | 85.3 ± 3.7 * | 0 |
| 14b | 70.3 ± 1.7 †,* | 79.1 ± 1.9 †,* | 79.8 ± 3.4 * | 0 |
2.2.2. In Vivo Antithrombotic Activity
| Compounds | Paralyzed a or Died Animals/Total | % Protection |
|---|---|---|
| Control | 9/10 | 10 |
| ASA | 7/10 | 30 * |
| 3a | 6/10 | 40 * |
| 3b | 7/10 | 30 * |
| 4a | 6/10 | 40 * |
| 4b | 5/10 | 50 *,† |
| 8a | 6/10 | 40 * |
| 8b | 6/10 | 40 * |
| 9a | 7/10 | 30 * |
| 9b | 5/10 | 50 *,† |
| 13a | 3/10 | 70 *,† |
| 13b | 3/10 | 70 *,† |
| 14a | 2/10 | 80 *,† |
| 14b | 3/10 | 70 *,† |
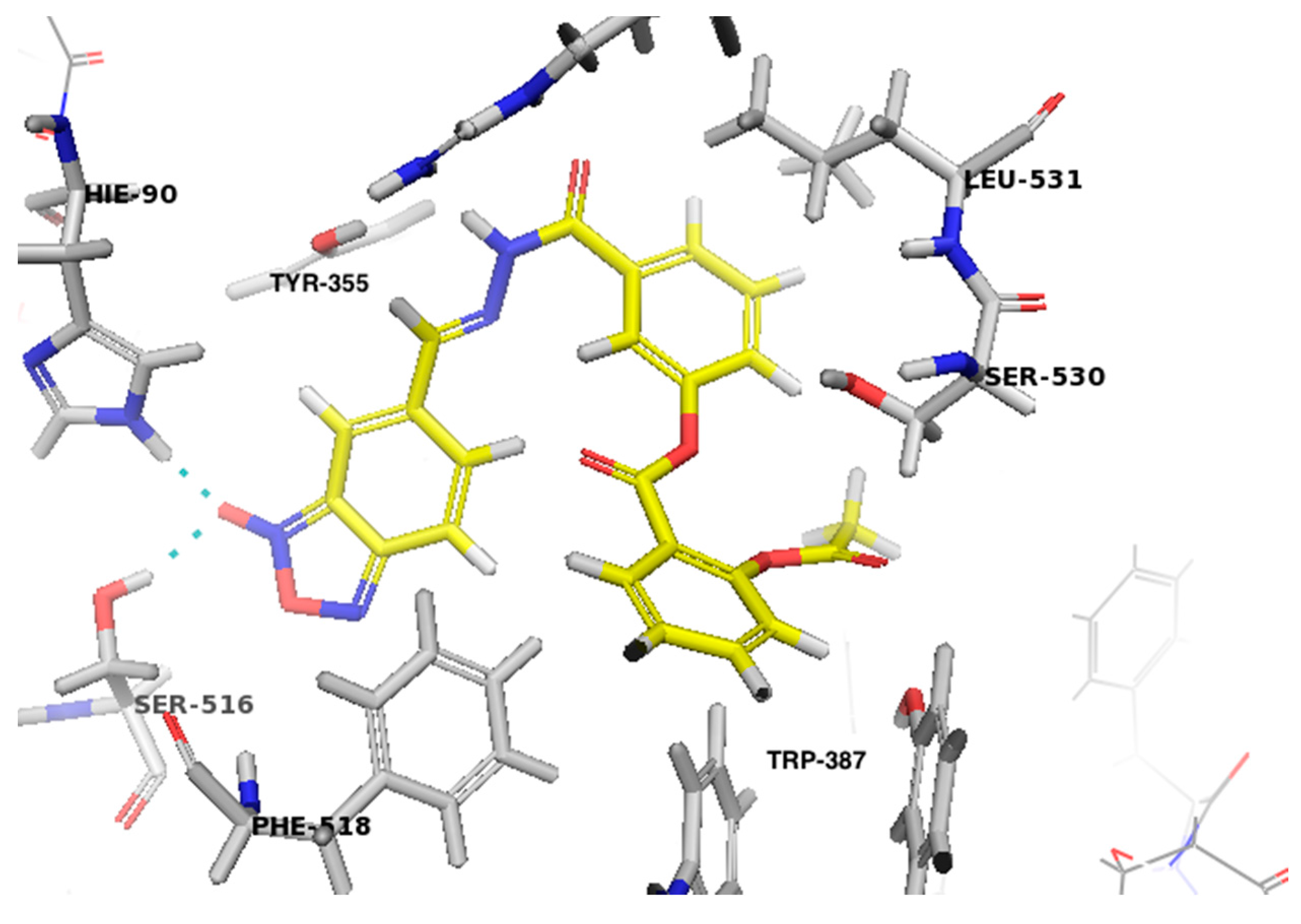
2.2.3. In Silico Studies

| Predicted properties | 14a |
|---|---|
| Physico-chemistry properties | Molecular weight: 460.1 H donors: 1 H acceptors: 9 LogP: 2.36 Number of rotatable bonds: 9 Surface area: 191.27 |
| Absorption | Water solubility: −4.71 log mol/L Caco2 permeability: −0.008 log Papp in 10−6 cm/s Intestinal absorption (humans): 86.54% absorbed |
| Distribution | VDss (humans): −0.869 log L/Kg |
| Metabolism | Substrate of CYP3A4 |
| Excretion | Total clearance: 0.386 log mL/min/Kg |
| Toxicity | AMES toxicity: No hERG I inhibitor: No |
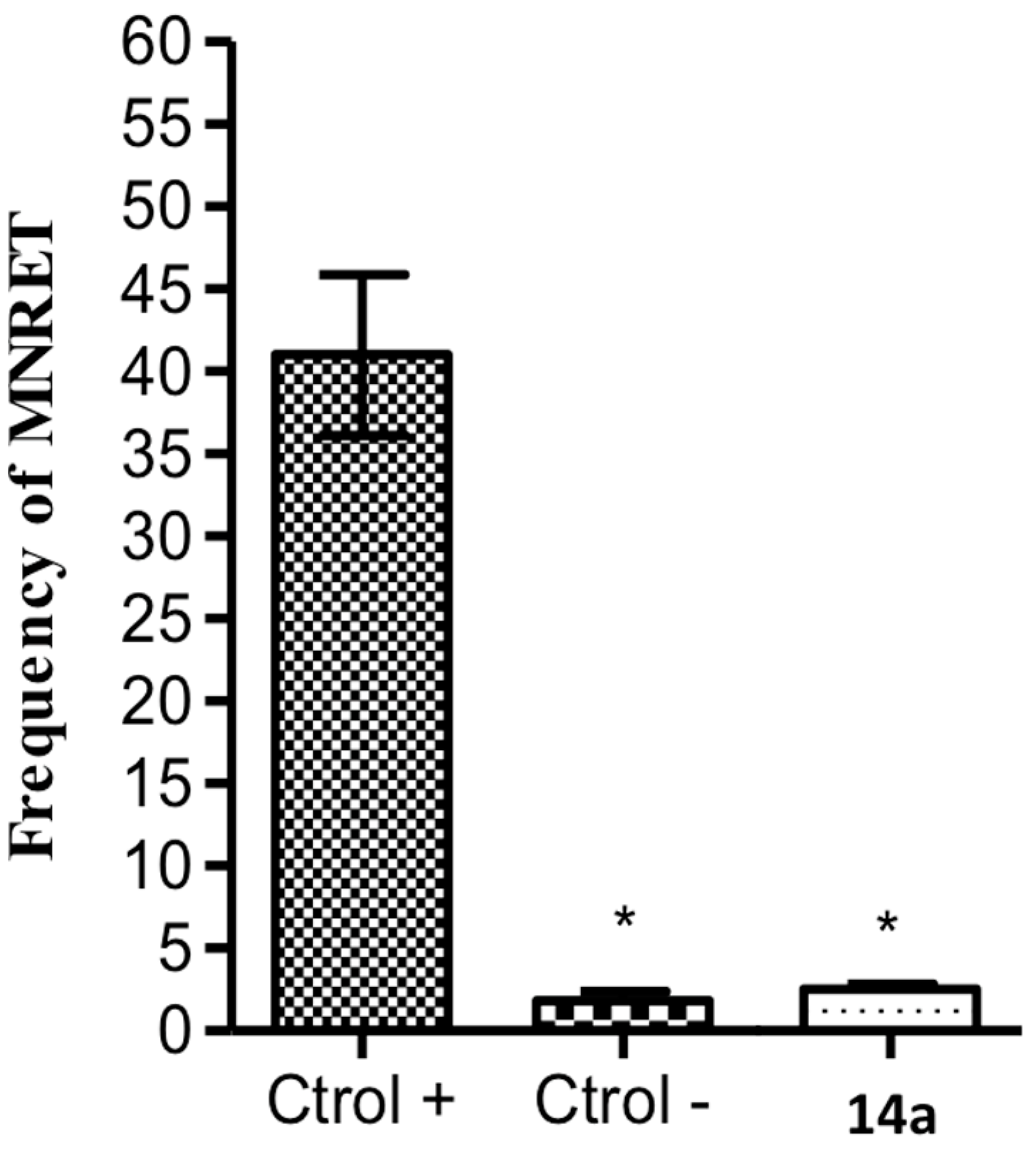
2.2.4. In Vivo Genotoxicity Studies

3. Experimental Section
3.1. General
3.2. Chemistry
3.2.1. General Procedures for the Preparation of Intermediates (3a,b, 8a,b and 13a,b)
3.2.2. General Procedures for the Preparation of Compounds (4a,b, 9a,b, and 14a,b)
3.3. Pharmacology
3.3.1. Animals
3.3.2. Antiplatelet Activity
3.3.3. In vitro Nitric Oxide Production
3.3.4. In vivo Antithrombotic Activity
3.3.5. In Silico Studies
3.3.6. In Vivo Genotoxicity Study
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- World Health Organization (WHO). Cardiovascular Diseases (CVDs). Fact sheet N 317. Available online: http://www.who.int/mediacentre/factsheets/fs317/en/ (accessed on 20 May 2015).
- Melnikova, I. The anticoagulants market. Nat. Rev. Drug Discov. 2009, 8, 353–354. [Google Scholar] [CrossRef] [PubMed]
- Chaudhari, K.; Hamad, B.; Sved, B.A. Antithrombotic drugs market. Nat. Rev. Drug Discov. 2014, 13, 571–572. [Google Scholar] [CrossRef] [PubMed]
- Pilgrim, T.; Windecker, S. Antiplatelet therapy for secondary prevention of coronary artery disease. Heart 2014, 100, 1750–1756. [Google Scholar] [CrossRef] [PubMed]
- Pusch, G.; Feher, G.; Kotai, K.; Tibold, A.; Gasztonyi, B.; Feher, A.; Papp, E.; Lupkovics, G.; Szapary, L.J. Aspirin resistance: Focus on clinical endpoints. Cardiovasc. Pharmacol. 2008, 52, 475–484. [Google Scholar] [CrossRef] [PubMed]
- Farré, A.J.L.; Tamargo, J.; Mateos-Cáceres, P.J.; Azcona, L.; Macaya, C. Old and new molecular mechanisms associated with platelet resistance to antithrombotics. Pharm. Res. 2010, 27, 2365–2373. [Google Scholar] [CrossRef] [PubMed]
- Michelson, A.D. Antiplatelet therapies for the treatment of cardiovascular disease. Nat. Rev. Drug Discov. 2010, 9, 154–169. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, T.A.; Diodati, J.G.; Pharand, C.J. Resistance to clopidogrel: A review of the evidence. Am. Coll. Cardiol. 2005, 45, 1157–1164. [Google Scholar] [CrossRef] [PubMed]
- Hankey, G.J.; Eikelboom, J.W. Aspirin resistance. Lancet 2006, 367, 606–617. [Google Scholar] [CrossRef]
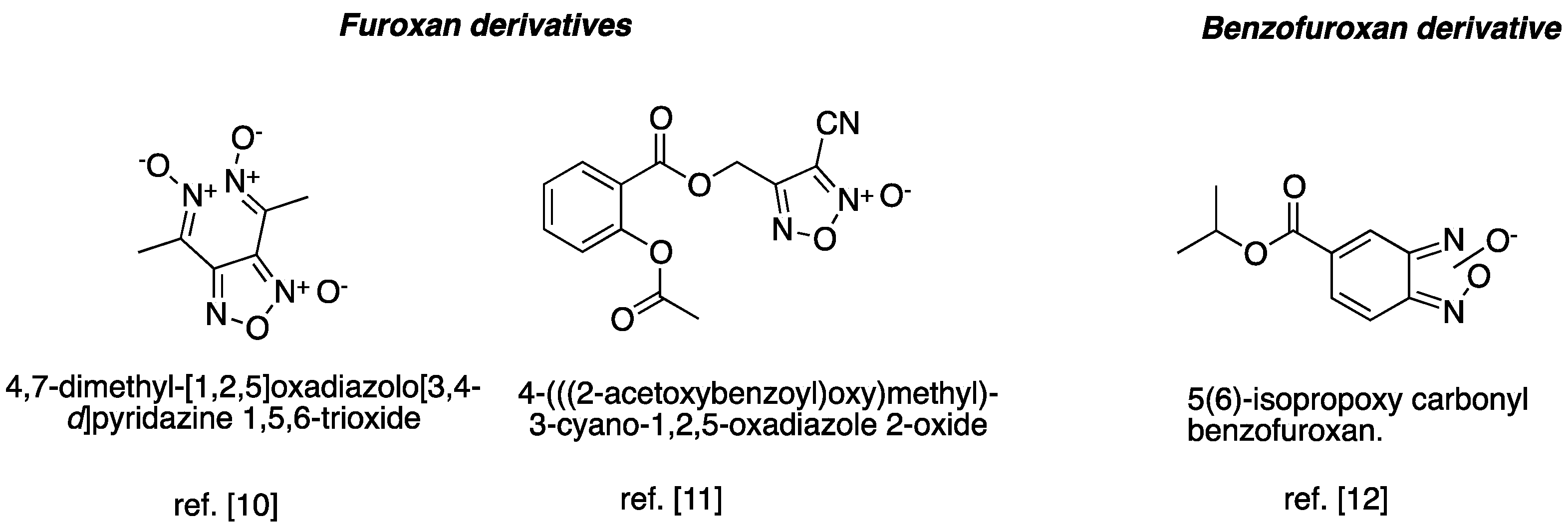
- Kots, A.Y.; Grafov, M.A.; Khropov, Y.V.; Betin, V.L.; Belushkina, N.N.; Busygina, O.G.; Yazykova, M.Y.; Ovchinnikov, I.V.; Kulikov, A.S.; Makhova, N.N.; et al. Vasorelaxant and antiplatelet activity of 4,7-dimethyl-1,2,5-oxadiazolo[3,4-d]pyridazine 1,5,6-trioxide: Role of soluble guanylate cyclase, nitric oxide and thiols. Br. J. Pharmacol. 2000, 129, 1163–1177. [Google Scholar] [CrossRef] [PubMed]
- Turnbull, C.M.; Cena, C.; Fruttero, R.; Gasco, A.; Rossi, A.G.; Megson, I.L. Mechanism of action of novel NO-releasing furoxan derivatives of aspirin in human platelets. Br. J. Pharmacol. 2006, 148, 517–526. [Google Scholar] [CrossRef] [PubMed]
- Sankaranarayanan, A. Benzofuroxan Derivatives, Their Therapeutic Uses and Pharmaceutical Compositions. U.S. Patent 6,232,331, 15 May 2001. [Google Scholar]
- Dutra, L.A.; de Almeida, L.; Passalacqua, T.G.; Reis, J.S.; Torres, F.A.E.; Martinez, I.; Peccinini, R.G.; Chung, M.C.; Chegaev, K.; Guglielmo, S.; et al. Leishmanicidal activities of novel synthetic furoxan and benzofuroxan derivatives. Antimicrob. Agents Chemother. 2014, 58, 4837–4847. [Google Scholar] [CrossRef] [PubMed]
- Monge, A.; López, D.C.A.; Ezpeleta, O.; Cerecetto, H.; Dias, E.; Di Maio, R.; González, M.; Onetto, S.; Seoane, G.; Suescun, L.; Mariezcurrena, R. Synthesis and biological evaluation of 1,2,5-oxadiazole N-oxide derivatives as hypoxia-selective cytotoxins. Pharmazie 1998, 53, 758–764. [Google Scholar] [PubMed]
- Chelucci, R.C.; Dutra, L.A.; Lopes Pires, M.E.; de Melo, T.R.; Bosquesi, P.L.; Chung, M.C.; dos Santos, J.L. Antiplatelet and antithrombotic activities of non-steroidal anti-inflammatory drugs containing an N-acyl hydrazone subunit. Molecules 2014, 19, 2089–2099. [Google Scholar] [CrossRef] [PubMed]
- Born, G.V.R.; Cross, M.J. The aggregation of blood platelets. J. Physiol. 1963, 168, 178–195. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.L.; Lanaro, C.; Lima, L.M.; Gambero, S.; Franco-Penteado, C.F.; Alexandre-Moreira, M.S.; Wade, M.; Yerigenahally, S.; Kutlar, A.; Meiler, S.E.; et al. Design, synthesis, and pharmacological evaluation of novel hybrid compounds to treat sickle cell disease symptoms. J. Med. Chem. 2011, 54, 5811–5819. [Google Scholar] [CrossRef] [PubMed]
- DiMinno, G.; Silver, M.J. Mouse antithrombotic assay: A simple method for the evaluation of antithrombotic agents in vivo. Potentiation of antithrombotic activity by ethyl alcohol. J. Pharmacol. Exp. Ther. 1983, 225, 57–60. [Google Scholar] [PubMed]
- Lima, L.M.; Frattani, F.S.; dos Santos, J.L.; Castro, H.C.; Fraga, C.A.; Zingali, R.B.; Barreiro, E.J. Synthesis and anti-platelet activity of novel arylsulfonate—acylhydrazone derivatives, designed as antithrombotic candidates. Eur. J. Med. Chem. 2008, 43, 348–356. [Google Scholar] [CrossRef] [PubMed]
- Hernández, P.; Cabrera, M.; Lavaggi, M.L.; Celano, L.; Tiscornia, I.; Rodrigues da Costa, T.; Thomson, L.; Bollati-Fogolín, M.; Miranda, A.L.P.; Lima, L.M.; et al. Discovery of new orally effective analgesic and anti-inflammatory hybrid furoxanyl N-acylhydrazone derivatives. Bioorg. Med. Chem. 2012, 20, 2158–2171. [Google Scholar] [CrossRef] [PubMed]
- Kalgutkar, A.S.; Crews, B.C.; Rowlinson, S.W.; Garner, C.; Seibert, K.; Marnett, L.J. Aspirin-like molecules that covalently inactivate cyclooxygenase-2. Science 1998, 280, 1268–1270. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.; Blundell, T.L.; Ascher, D.B. pkCSM: Predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Hayashi, M.; MacGregor, J.T.; Gatehouse, D.G.; Blakey, D.H.; Dertinger, S.D.; Abramsson-Zetterberg, L.; Krishna, G.; Morita, T.; Russo, A.; Asano, N.; et al. In vivo erythrocyte micronucleus assay. III. Validation and regulatory acceptance of automated scoring and the use of rat peripheral blood reticulocytes, with discussion of non-hematopoietic target cells and a single dose-level limit test. Mutat. Res. 2007, 627, 10–30. [Google Scholar] [CrossRef] [PubMed]
- Cabrera, M.; Lavaggi, M.L.; Hernández, P.; Merlino, A.; Gerpe, A.; Porcal, W.; Boiani, M.; Ferreira, A.; Monge, A.; de Cerain, A.L.; et al. Cytotoxic, mutagenic and genotoxic effects of new anti-T. cruzi 5-phenylethenylbenzofuroxans. Contribution of phase I metabolites on the mutagenicity induction. Toxicol. Lett. 2009, 190, 140–149. [Google Scholar] [CrossRef] [PubMed]
- Gabay, M.; Cabrera, M.; Maio, R.D.; Paez, J.A.; Campillo, N.; Lavaggi, M.L.; Cerecetto, H.; González, M. Mutagenicity of N-oxide containing heterocycles and related compounds: experimental and theoretical studies. Curr. Top Med. Chem. 2014, 14, 1374–1387. [Google Scholar] [CrossRef] [PubMed]
- Dos Santos, J.L.; Lanaro, C.; Chelucci, R.C.; Gambero, S.; Bosquesi, P.L.; Reis, J.S.; Lima, L.M.; Cerecetto, H.; González, M.; Costa, F.F.; et al. Design, synthesis, and pharmacological evaluation of novel hybrid compounds to treat sickle cell disease symptoms. Part II: Furoxan derivatives. J. Med. Chem. 2012, 55, 7583–7592. [Google Scholar] [CrossRef] [PubMed]
- Maestro, version 9.1; Software for docking studies; Schrodinger: New York, NY, USA, 2012.
- Rimon, G.; Sidhu, R.S.; Lauver, D.A.; Lee, J.Y.; Sharma, N.P.; Yuan, C.; Frieler, R.A.; Trievel, R.C.; Lucchesi, B.R.; Smith, W.L. Coxibs interfere with the action of aspirin by binding tightly to one monomer of cyclooxygenase-1. Proc. Natl. Acad. Sci. USA 2010, 107, 28–33. [Google Scholar] [CrossRef] [PubMed]
- LigPrep, version 2.5; Software for molecular energy minimization; Schrodinger: New York, NY, USA, 2012.
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shelley, M.; Perry, J.K.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds (3a,b, 4a,b, 8a,b, 9a,b, 13a,b, and 14a,b) are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rosseto, L.A.; Pires, M.E.L.; Melchior, A.C.B.; Bosquesi, P.L.; Pavan, A.R.; Marcondes, S.; Chung, M.C.; Santos, J.L.d. Synthesis and Preliminary Evaluation of N-Oxide Derivatives for the Prevention of Atherothrombotic Events. Molecules 2015, 20, 18185-18200. https://doi.org/10.3390/molecules201018185
Rosseto LA, Pires MEL, Melchior ACB, Bosquesi PL, Pavan AR, Marcondes S, Chung MC, Santos JLd. Synthesis and Preliminary Evaluation of N-Oxide Derivatives for the Prevention of Atherothrombotic Events. Molecules. 2015; 20(10):18185-18200. https://doi.org/10.3390/molecules201018185
Chicago/Turabian StyleRosseto, Leandro Augusto, Maria Elisa Lopes Pires, Aylime Castanho Bolognesi Melchior, Priscila Longhin Bosquesi, Aline Renata Pavan, Sisi Marcondes, Man Chin Chung, and Jean Leandro dos Santos. 2015. "Synthesis and Preliminary Evaluation of N-Oxide Derivatives for the Prevention of Atherothrombotic Events" Molecules 20, no. 10: 18185-18200. https://doi.org/10.3390/molecules201018185
APA StyleRosseto, L. A., Pires, M. E. L., Melchior, A. C. B., Bosquesi, P. L., Pavan, A. R., Marcondes, S., Chung, M. C., & Santos, J. L. d. (2015). Synthesis and Preliminary Evaluation of N-Oxide Derivatives for the Prevention of Atherothrombotic Events. Molecules, 20(10), 18185-18200. https://doi.org/10.3390/molecules201018185