3. Experimental Section
3.1. General Information
All melting points are corrected and determined on a stuart electric melting point apparatus (Microanalytical centre, ainshams university, Cairo, Egypt). Elemental analyses were carried out by Elementar Viro El-Microanalysis at the Micro-analytical Center, National Research Center, Egypt. IR spectra (KBr) were recorded on infrared spectrometer FT-IR 400D (New York, NY, USA) using OMNIC program and are reported frequency of absorption in terms of cm−1 and 1H-NMR spectra recorded on a Bruker spectrophotometer (Rheinstetten, Germany) at 400 MHz using TMS as internal standard and with residual signals of the deuterated solvent δ = 7.26 ppm for CDCl3 and δ 2.51 ppm for DMSO-d6. 13C-NMR spectra were recorded on the same spectrometer (Rheinstetten, Germany) at 100 MHz and referenced to solvent signals δ = 77 ppm for CDCl3 and δ 39.50 ppm for DMSO-d6. DEPT 135 NMR spectroscopy were used where appropriate to aid the assignment of signals in the 1H- and 13C-NMR spectra. The mass spectra were recorded on Shimadzu GCMS-QP-1000 EX mass spectrometer (Kyoto, Japan) used the electron ionization technique at 70 e.v. Homogeneity of all synthesized compounds was checked by TLC.
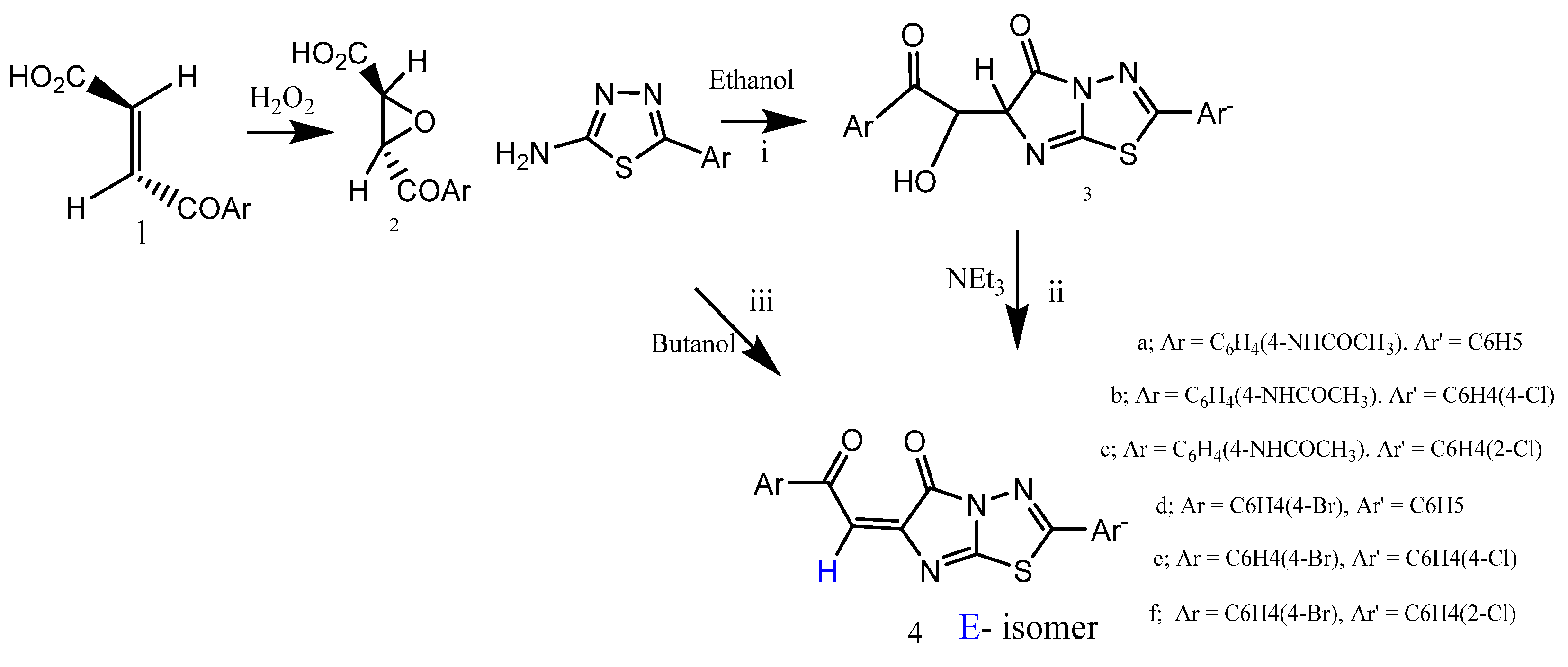
3.2. General Procedure for Synthesis of the Compounds 2, 3, 5a and 5c are in the Literature [19]
3.3. General Procedure for Synthesis of the Compounds 4a–f
Fuse the compounds 3a–f (0.01 mol) and 3–5 drops triethyl amine (TEA) in oil bath for 5 min, then refluxing in 50 mL of boiling aqueous ethanol for 5 h. The solid was separated after cooling and the pH of the solution was 6.5. The crude products were filtered, washed by petroleum ether (b.p. 40–60 °C), dried and then recrystallized from dioxane.
(E)-N-(4-(2-(5-Oxo-2-Phenylimidazo[2,1-b]1,3,4-thiadiazol-6(5H)ylidine)acetyl)phenyl)acetamide (4a). Yield 2.35 g (60%), light yellow finely crystalline, m.p. 176–178 °C. IR (KBr), υ, cm−1: 3245 (NH), 3055 (CH), 1706, 1670, 1650 (CO), 1613 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.54 (3H, s, CH3), 7.72 (1H, s, CH=, arylidine, 82% in form of E-configuration), 7.78 (1H, s, CH=, arylidine, 18% in form of Z-configuration), 7.48–7.86 (9ArH, m, aromatic protons), 13.2 (1H, s, acidic NH proton which exchanged in D2O), 13C-NMR δ, 40.0 (CH3CO), 105.4 (C2,6 Ph), 110.6 (C3,5 Ph), 125.5 (C4 Ar), 142.3 (C3,5 Ar), 144.8 (CH=), 147.4 (C2 Ar), 148 (C6 Ar), 148.5 (C4 Ph), 150.2 (C=CH), 152.4 (C1 Ph), 154.5 (C1 Ar), 156.0 (CNS), 162.2 (CN2S), 165.1 (CO imidaz.), 168.0 (CO amide), 190.2 (CO ketone), and found, %: C 61.50, H 3.59, N 14.30, S 8.19 for C20H14N4O3S. Calculated, %: C 61.53, H 3.61, N 14.35, S 8.21; MS: m/z 346 [M+ − CH2=C=O], 141 [imidazole thiadiazole moiety].
(E)-N-(4-(2-(2-(4-Chlorophenyl-5-oxo-imidazo[2,1-b]1,3,4-thiadiazol-6(5H)ylidine)acetyl)phenyl)acetamide (4b). Yield 2.85 g (67%), yellow finely crystalline, m.p. 210–212 °C. IR (KBr), υ, cm−1: 3245 (NH), 1710, 1691, 1655 (CO); 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.06 (3H, s, CH3), 7.73 (1H, s, CH=, arylidine, 78% in form of E-configuration), 7.81 (1H, s, CH=, arylidine, 22% in form of Z-configuration), 7.44–7.83 (8ArH, m, aromatic protons), 12.6 (1H, s, acidic NH proton which exchanged in D2O); 13C-NMR δ 22.5 (CH3CO), 121.2 (C2,6 Ph), 128.8 (C3,5 Ph), 129.5 (C4 Ar), 131.5 (C4 PhCl), 132.3 (C3,5 Ar), 134.6 (CH=), 137.1 (C2 Ar); 138.3 (C6 Ar), 140.2 (C=CH), 142.0 (C Ph), 142.4 (C1 Ar), 160.3 (CNS), 161.4 (CN2S), 166.5 (CO imidaz), 167.4 (CO amide), 191.0 (CO ketone) and found, %: C 56.53; H 3.06; N 13.16, Cl 8.30, S 7.53 for C20H13N4O3SCl. Calculated, %: C 56.54, H 3.08, N 13.19, Cl 8.34, S 7.55.
(E)-N-(4-(2-(2-(2-Chlorophenyl-5-oxo-imidazo[2,1-b]1,3,4-thiadiazol-6(5H)ylidine)acetyl)phenyl)acetamide (4c). Yield 2.68 g (63%), yellow finely crystalline, m.p. 198–200 °C. IR (KBr), υ, cm−1: 3245 (NH); 1710, 1691, 1655 (CO); 1630 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.06 (s, 3H, CH3), 7.69 (1H, s, CH=, arylidine, 80% in form of E-configuration), 7.77 (1H, s, CH=, arylidine, 20% in form of Z-configuration), 7.44–7.83 (8ArH, m, aromatic protons), 12.4 (1H, s, acidic NH proton which exchanged in D2O), and found, %: C 56.50, H 3.02, N 13.11, Cl 8.30, S 7.51 for C20H13N4SO3Cl. Calculated, %: C 56.54, H 3.08, N 13.19, Cl 8.34, S 7.55; MS: m/z 389 [M+ − Cl], 347 [389 − CH2CO].
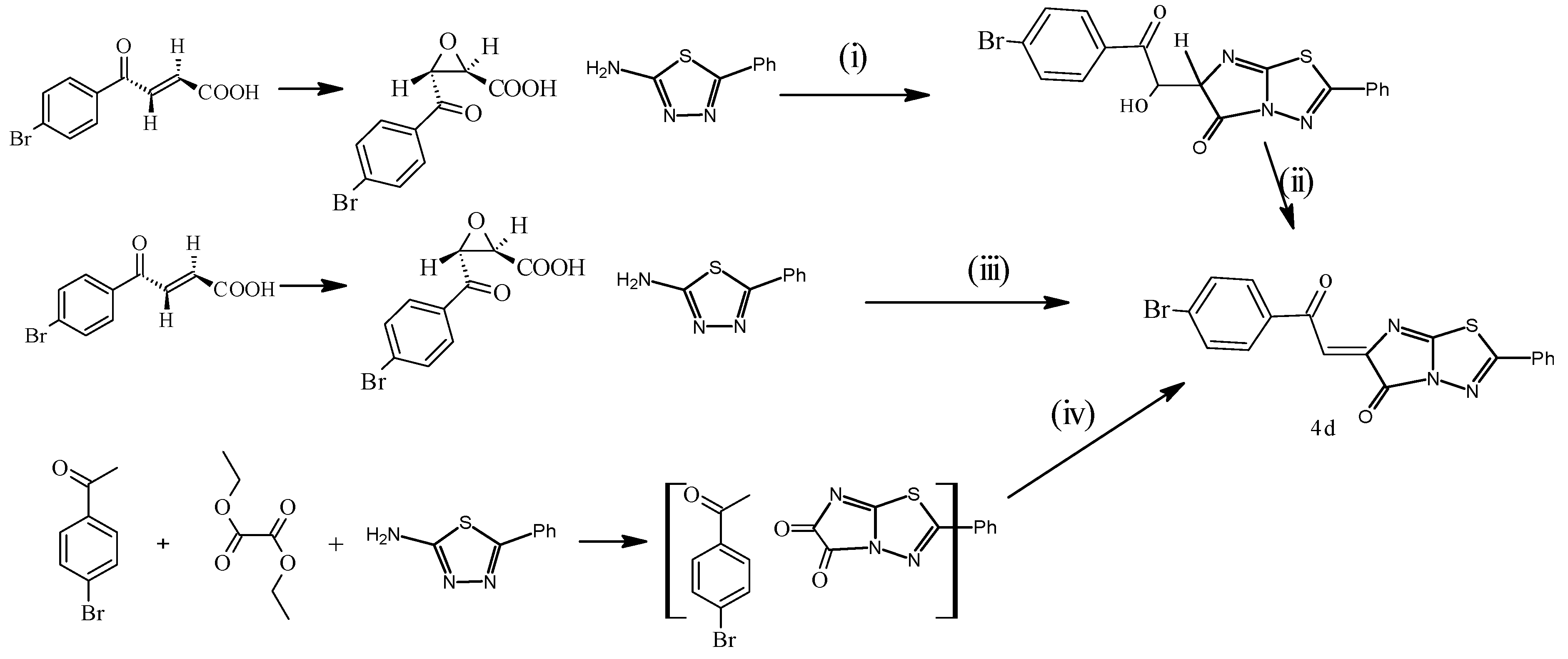
(E)-6-(4-Bromophenyl)-2-oxoethylidine)-2-phenylimidazo[2,1-b]1,3,4-thiadiazol-5(6H)-one (4d). Yield 2.72 g (66%), yellow powder, m.p. 152–154 °C. IR (KBr), υ, cm−1: 1694, 1672 (CO); 1613 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 7.70 (1H, s, CH=, arylidine, 86% in form of E-configuration), 7.79 (1H, s, CH=, arylidine, 14% in form of Z-configuration), 7.44–7.73 (9ArH, m, aromatic protons). 13C-NMR δ 119.6 (C2,6 Ph), 127.6 (C3,5 Ph), 130.5 (C4 Ph), 132.7 (C3,5 Ar), 135.3 (C2,6 Ar), 137.6 (C1 Ph), 139 (CH=), 141.2 (C4 Ar), 142.1 (C1 Ar), 145.5 (C=), 149.7 (CNS), 155.3 (CN2S), 166.8 (CO imidaz.), 179.2 (CO) and found, %: C 52.50; H 2.35; N 10.15. C18H10N3O2BrS. Calculated, %: C 52.42, H 2.42, N 10.19. MS: m/z 378 [M+ − CO], 335 [M+ − Ph], 263 [M+ − Ph(Br)], 138 [imidazolothiadiazole moiety].
(E)-6-(4-Bromophenyl)-2-oxoethylidine)-2-(4-chlorophenyl)imidazo[2,1-b]1,3,4-thiadiazol-5(6H)-one (4e). Yield 3.17 g (71%), yellow finely crystalline, m.p. 168–170 °C. IR (KBr), υ, cm−1: 1710, 1691 (CO), 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm (J, Hz): 7.70 ( 1H, s, CH=, arylidine, 78% in form of E-configuration), 7.80 (1H, s, CH=, arylidine, 22% in form of Z-configuration), 7.60–7.83 (8ArH, m, aromatic protons), and found, %: C 48.30, H 2.10, N 9.33. C18H9N3O2BrClS. Calculated, %: C 48.37, H 2.15, N 9.40.
(E)-6-(4-Bromophenyl)-2-oxoethylidine)-2-(2-chlorophenyl)imidazo[2,1-b]1,3,4-thiadiazol-5(6H)-one (4f). Yield 3.04 g (68%), yellow finely crystalline, m.p. 180–182 °C. IR (KBr), υ, cm−1: 1708, 1684 (CO); 1630 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 7.73 (1H, s, CH=, arylidine, 73% in form of E-configuration), 7.84 (1H, s, CH=, arylidine, 27% in form of Z-configuration), 7.72–7.86 (8ArH, m, aromatic protons), and found, %: C 48.35, H 2.15, N 9.35 for C18H9N3O2BrClS. Calculated, %: C 48.37; H 2.15; N 9.40.
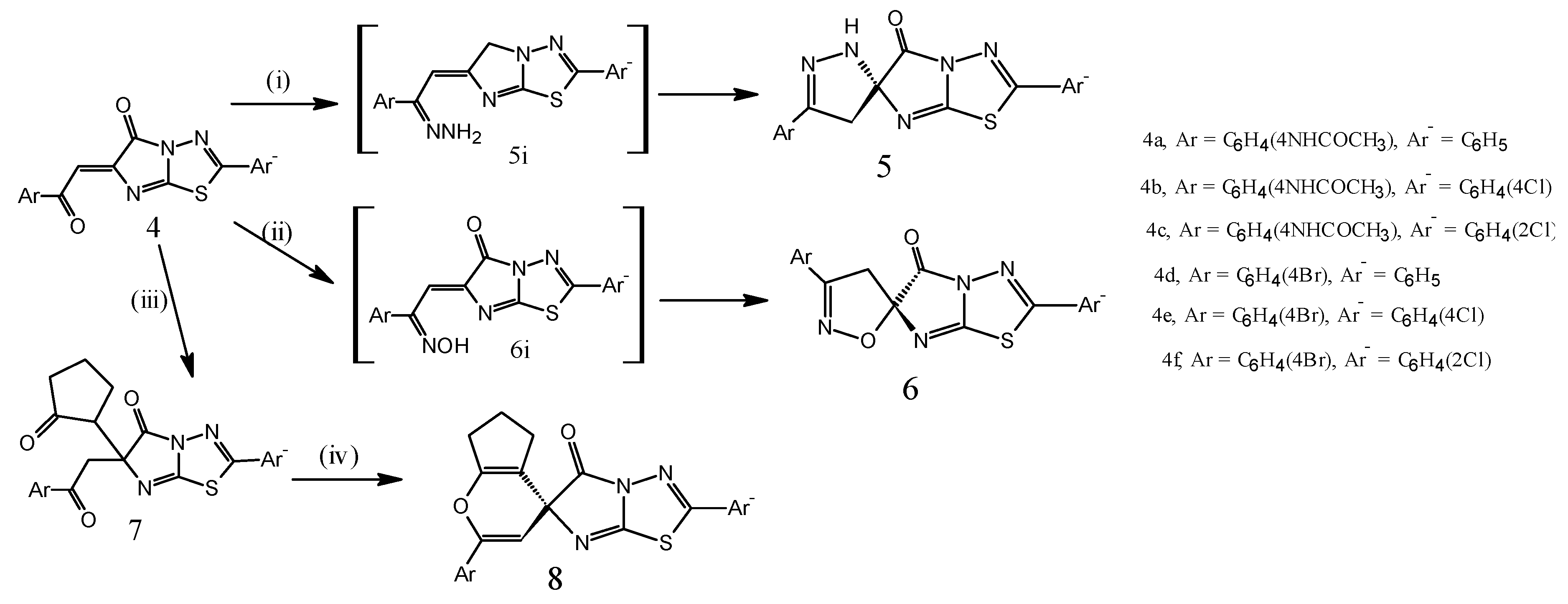
3.4. General Procedure for Synthesis of the Compounds 5b, 5d, and 5e
A mixture of chalcone derivatives 4b, 4d and 4e (5 mmol) and hydrazine hydrate (0.5 mL, 0.01 mol) in boiling ethanol (50 mL) was heated under reflux for 5 h. The reaction mixture was allowed to cool and the product was filtered, dried, and recrystallized from the benzene and/or ethanol.
N-(4-(2-(2-Chlorophenyl)-5-oxo)-2′,4′-dihydro-5H-spiro[imidazo[2,1-b]1,3,4-thiadiazol-6,3′-pyrazol]-5′yl) phenyl)acetamide (5b). Yield 1.43 g (65%), off white crystal, m.p. 164–166 °C. IR (KBr), υ, cm−1: 3420 (NH); 1671, 1640 (CO); 1H-NMR (DMSO-d6), δ, ppm (J, Hz): 1.2 (d, 2H, CH2, J = 5.4); 2.5 (3H, s, CH3); 5.8 (1H, br. s, NH); 7.0–7.81 (8H, m, Ar-H); 12.40 (1H, br. s, NH of acetamide moiety), 13C-NMR δ, 22.3 (CH3CO); 69.6 (CH2C=N); 122.4 (C4 Ph), 126.8 (C3,5 Ph), 129.1 (C Spiro), 131.5 (C3 Ar), 131.8 (C5 Ar), 132.6 (C2,6 Ph), 137.4 (C2 Ar), 138 (C6 Ar), 140.2 (C4 Ar), 144.5 (C1 Ar), 145.1 (C1 Ph), 158.0 (C=N), 161.3 (CNS), 163.2 (CN2S), 165.3 (CO imidaz.), 167.8 (CO amide), and found, %: C 54.70, H 3.42, N 19.10, Cl 8.02, S 7.27 for C20H15N6O2ClS. Calculated, %: C 54.73, H 3.45, N 19.15, Cl 8.08, S 7.30.
5′-(4-Bromophenyl)-2-phenyl-2′,4′-dihydro-5H-spiro[imidazo[2,1-b]1,3,4-thiadiazol-6,3′-pyrazol]-5-one (5d). Yield 1.28 g (60%), White finely crystalline, m.p. 138–140 °C. IR (KBr), υ, cm−1: 3423, 3151 (NH), 1671, 1640 (CO), 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 1.2 (2H, d, CH2, J = 5.5), 2.5 (3H, s, CH3), 5.4 (1H, s, NH), 7.0–7.81 (9H, m, Ar-H); 13C-NMR δ, 69.4 (CH2C=N), 122.1 (C4 Ph), 125.9 (C3,5 Ph), 127.1 (C Spiro), 131.4 (C3 Ar), 131.8 (C5 Ar), 132.5 (C2,6 Ph), 136.8 (C2 Ar), 137.4 (C6 Ar), 140.2 (C4 Ar), 144.5 (C1 Ar); 145.1 (C1 Ph), 158.0 (C=N), 161.3 (CNS), 163.2 (CN2S); 165.3 (CO imidaz.), and found, %: C 50.70, H 2.81, N 16.40, Br 18.71, S 7.50 for C18H12N5OBrS. Calculated, %: C 50.72, H 2.84, N 16.43, Br 18.74, S 7.52.
5′-(4-Bromophenyl)-(2-(4-chlorophenyl)-2′,4′-dihydro-5H-spiro[imidazo[2,1-b]1,3,4-thiadiazol-6,3′-pyrazol]-5-one (5e). Obtained similarly to compound 5a from compound 4f (2.23 g, 5 mmol). Yield 1.50 g (65%), white finely crystalline, m.p. 150–152 °C. IR (KBr), υ, cm−1: 3423, 3333 (NH); 1680, 1648 (CO); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz); 1.4 (2H, d, CH2, J = 5.4), 2.5 (3H, s, CH3), 5.4 (1H, s, NH), 7.0–7.81 (8H, m, Ar-H); found, %: C 46.89, H 2.40, N 15.20, Br 17.32, Cl 7.67, S 6.92 for C18H11N5OBrClS. Calculated, %: C 46.92, H 2.41, N 15.20, Br 17.34, Cl 7.69, S 6.95; MS: m/z 464 [M+ + 2]; 461 [M+]; 418 [M+ − CH2=C=O]; 194 [spiro moiety].
3.5. General Procedure for Synthesis of the Compounds 6a–d
A mixture of 4a, 4b, 4d and/or 4f (5 mmol) and hydroxyl amine hydrochloride (0.52 g; 7.5 mmol) in boiling pyridine (25 mL) was heated under reflux for 6 h. The reaction mixture was allowed to cool, poured into ice/HCl until pH of the solution is 6.5, and the product was filtered, dried, and recrystallized from ethanol.
N-(4-(5-Oxo-2-phenyl-2′,4′-dihydro-5H-spiro[imidazo[2,1-b]1,3,4-thiadiazol-6,5′-isoxazol]-3′yl)phenyl)acetamide (6a). Yield 1.11g (55%), white finely crystalline. m.p. 197–200 °C. IR (KBr), υ, cm−1: 3425 (NH), 1647 (CO), 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.5 (3H, s, CH3), 3.59 (2H, d, CH2, J = 5.7), 7.53–7.96 (10H, m, Ar-H), and found, %: C 59.30, H 3.65, N 17.24. C20H15N5O3S. Calculated, %: C 59.26, H 3.70, N 17.28.
N-(4-(2-(4-Chlorophenyl-5-oxo-2′,4′-dihydro-5H-spiro[imidazo[2,1-b]1,3,4-thiadiazol-6,5′-isoxazol]-5′yl)phenyl)acetamide (6b). Yield 1.47 g (67%), white finely crystalline powder, m.p. 192–195 °C. IR (KBr) υ, cm−1: 3271 (NH), 1660 (CO), 1631 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.3 (3H, s, CH3), 3.20 (2H, d, CH2, J = 5.7), 7.53–7.96 (10H, m, Ar-H); 13C-NMR (DMSO), δ, ppm, 21.7 (CH3CO), 66.6 (CH2C=N), 119.7 (C spiro), 126.8 (C3,5 Ar), 128.3 (C3,5 PhCl), 130.5 (C2,6 PhCl), 134.5 (C2,6 Ar), 136.9 (C4 PhCl), 138.2 (C4 Ar), 140.2 (C1 Ar), 143.5 (C1 PhCl), 147.1 (C=N), 161.3 (CNS), 164.0 (CN2S), 166.4 (CO imidaz.), 168.0 (CO amide), and found, %: C 54.58, H 3.19, N 15.88, Cl 8.02, S 7.27 for C20H14N5O3ClS. Calculated, %: C 54.61, H 3.21, N 15.92, Cl 8.06, S 7.29.
3′-(4-Bromophenyl)-2-phenyl-4′H,5H-spiro[imidazo[2,1-b]1,3,4-thiadiazol-6,5′-isoxazol]-5-one (6c). Yield 1.56 g (70%), white finely crystalline, m.p. 197–200 °C. IR (KBr) υ, cm−1: 3425 (NH); 1630 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 3.99 (2H, s, CH2), 7.53–7.96 (10H, m, Ar-H). 13C-NMR (DMSO), δ, ppm, 65.8 (CH2C=N); 111.2 (C spiro); 116.2 (C3,5 Ar); 120.9 (C3,5 Ph); 124.6 (C2,6 Ar); 132.7 (C2,6 Ph); 134.3 (C4 Ph); 136.3 (C4 PhBr), 139.2 (C1, PhBr); 140.6 (C1, Ph); 153.0 (C=N); 157.2 (CNS); 162.3 (CN2S); 166.0 (CO imidaz.) and found, %: C 50.60, H 2.58, N 13.12, Br 18.68, S 7.48 for C18H11N4O2BrS. Calculated, %: C 50.60, H 2.60, N 13.11, Br 18.70, S 7.50.
3′-(4-Bromophenyl)-2-(2-chlorophenyl)-4′H,5H-spiro[imidazo[2,1-b]1,3,4-thiadiazol-6,5′-isoxazol]-5-one (6d). Yield 1.39 g (58%), white finely crystalline powder, m.p. 192–195 °C. IR (KBr) υ, cm−1: 3271 (NH), 1680 (CO), 1631 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 3.49 (2H, br. s, CH2), 7.53–7.96 (10H, m, Ar-H), found %: C 46.85, H 2.16, N 12.11, Br 17.29, Cl 7.65, S 6.92 for C18H10N4O2BrClS. Calculated, %: C 46.82, H 2.18, N 12.13, Br 17.31, Cl 7.68, S 6.94.
3.6. General Procedure for Synthesis of the Compounds 7a–c
A mixture of 4a, 4b and/or 4d (5 mmol), cyclopentanone (0.45 mL, 5 mmol), (50%) NaOH (5 mL) and ethanol (25 mL) was refluxed for 3 h, and left overnight for 3 days. The reaction mixture was poured into ice/HCl, until pH of the solution becomes 6.5. The crude product was filtered, washed by petroleum ether (b.p. 40–60 °C), and then crystalized from benzene.
N-(4-(2-(5-Oxo-6-(3-oxocyclopentyl)-2-phenyl-5,6-dihydroimidazo[2,1-b][1,3,4]thiadiazol-6-yl)acetyl)phenyl)acetamide (7a). Yield 1.57 g (65%), white finely crystalline powder, m.p. 176–178 °C. IR (KBr) υ, cm−1: 3245 (NH); 1685, 1670, 1650 (CO), 1613 (C=N); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 1.12 (6H, m, 3CH2), 2.02 (1H, dd, H-cyclopent.); 2.36 (3H, s, CH3); 2.50 (2H, s, CH2CO); 7.44–7.73 (9ArH, m, aromatic protons); 13.2 (1H, s, acidic NH proton which exchanged in D2O) and found, %: C 63.25; H 4.65, N 11.79, S 6.77 for C25H22N4SO4. Calculated, %: C 63.28, H 4.67, N 11.81, S 6.76. MS: m/z 474; 432 [M+ − CH2=C=O]; 141 [imidazolothiadiazole moiety].
N-(4-(2-(2-(4-Chlorophenyl)-5-oxo-6-(3-oxocyclopentyl)-5,6-dihydroimidazo[2,1-b][1,3,4]thiadiazol-6-yl)acetyl)phenyl)acetamide (7b). Yield 1.88 g (74%), white finely crystalline. m.p. 210–212 °C. IR (KBr), υ, cm−1: 3245 (NH); 1710, 1691, 1655 (CO); 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm (J, Hz): 1.2 (6H, m, 3CH2), 2.02 (1H, dd, H-cyclopent.); 2.47 (3H, s, CH3); 2.51(2H, s, CH2CO); 7.44–7.83 (8ArH, m, aromatic protons; 8.2 (1H, s, acidic NH proton which exchanged in D2O); 13C-NMR (DMSO), δ, ppm, 22.4 (CH3CO); 31.8 (C3,4 cyclopent); 57.2 (C5 cyclopent); 67.8 (CH2CO spiro); 109.3 (C2, CH, cyclopent); 119.6 (C3,5 Ar−); 122.2 (C spiro); 124.3 (C3,5 Ar); 131.7 (C2,6 Ar−); 135.5 (C2,6 Ar); 141.5 (C4 Ar−), 143.4 (C4 Ar), 144.1 (C1 Ar−), 152.6 (C1 Ar), 161.0 (CNS), 164.0 (CN2S), 167.0 (CO imidaz), 168.0 (CO amide), 190.2 (CO ketone) 192.0 (CO cyclopent) and found, %: C 59.05, H 4.15, N 11.03, Cl 6.94, S 6.28 for C25H21N4O4ClS. Calculated, %: C 59.00, H 4.16, N 11.01, Cl 6.96, S 6.30.
6-(2-(4-Bromophenyl)-2-oxoethyl)-2-(4-chlorophenyl)-6-(3-oxocyclopentyl)imidazo[2,1-b][1,3,4]thiadiazol-5(6H)-one (7c). Yield 1.83 g (70%), white finely crystalline. m.p. 196–198 °C. IR (KBr), υ, cm−1: 3245 (NH); 1710, 1691, 1655 (CO); 1630 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 1.2 (6H, m, 3CH2), 2.08 (1H, dd, H-cyclohex.), 2.32 (3H, s, CH3), 2.53 (2H, s, CH2CO), 7.62–7.83 (8ArH, m, aromatic protons), 8.2 (1H, s, acidic NH proton which exchanged in D2O); And found, %: C 52.05; H 3.25; N 7.93, Br 15.03, Cl 6.66, S 6.00 for C23H17N3O3BrClS. Calculated, %: C 52.04, H 3.23; N 7.92, Br 15.05, Cl 6.68, S 6.03; MS: m/z 531 [M+ + 2], 529 [M+] 170; 139.
3.7. General Procedure for Synthesis of the Compounds 8a–c
A mixture of 7a (1.2 g, 2.5 mmol) and acetic anhydride (5 mL, 50 mmol) was refluxed in water bath for 2 h. The excess acetic anhydride was removed by fraction distillation and the separated product was filtered, dried and recrystallized from a mixture of toluene–ethanol.
N-(4-(5′-Oxo-2′-phenyl-6,7-dihydro-5H,5′H-spiro[cyclopenta[b]pyran-4,6′-imidazo[2,1-b][1,3,4]thiadiazol]-2-yl)phenyl)acetamide (8a). Yield 502 mg (45%), white powder. m.p. 140–142 °C. IR (KBr), υ, cm−1: 3245 (NH); 1646, 1668 (CO); 1613 (C=N). 1H-NMR (DMSO-d6), δ, ppm (J, Hz): 1.43 (6H, m, 3CH2), 2.5 (3H, s, CH3), 6.7 (1H, s, pyrane-H), 7.44–7.73 (9ArH, m, aromatic protons); 13.2 (1H, s, acidic NH proton which exchanged in D2O), and found, %: C 65.75, H 4.40, N 12.25, S 7.00 for C25H20N4O3S. Calculated, %: C 65.77, H 4.42, N 12.27, S 7.02; MS: m/z 456 [M+], 337 [M+ − PhNCO], 141 [imidazolothiadiazole moiety].
N-(4-(2′-(4-Chlorophenyl)-5′-oxo-6,7-dihydro-5H,5′H-spiro[cyclopenta[b]pyran-4,6′-imidazo[2,1-b][1,3,4]thiadiazol]-2-yl)phenyl)acetamide (8b). Yield 516 mg (39%), white powder. m.p. 172–174 °C. IR (KBr) υ, cm−1: 1630 (C=N), 1645, 1670 (CO), 3245 (NH). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 1.37 (6H, m, 3CH2); 2.06 (3H, s, CH3), 6.6–6.7 (s,1H, pyrane), 7.44–7.83 (8ArH, m, aromatic protons), 8.2 (1H, s, acidic NH proton which exchanged in D2O), found, %: C 61.13, H 3.88, N 11.43, Cl 7.20, S 6.50 for C25H19N4O3ClS. Calculated, %: C 61.16, H 3.90, N 11.41, Cl 7.22, S 6.53; MS: m/z 490 [M+], 377 [M+ − PhCl], 170, 140.
2-(4-Bromophenyl)-2′-(4-chlorophenyl)-5′-oxo-6,7-dihydro-5H,5′H-spiro[cyclopenta[b]pyran-4,6′-imidazo[2,1-b][1,3,4]thiadiazol]-5′-one (8c). Yield 420 mg (35%), white, finely crystalline powder; m.p. 236–238 °C. IR (KBr), υ, cm−1: 3245 (NH), 1672 (CO), 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm (J, Hz): 1.28 (8H, m, 4CH2), 6.6–6.7 (1H, s, pyrane), 7.44–7.83 (8ArH , m, aromatic protons), and found, %: C 53.85, H 2.95; N 8.19, Br 15.56, Cl 6.90, S 6.23 for C23H15N3O2BrClS. Calculated, %: C 53.87, H 2.95, N 8.19, Br 15.58, Cl 6.91, S 6.26; MS: m/z 511 [M+].
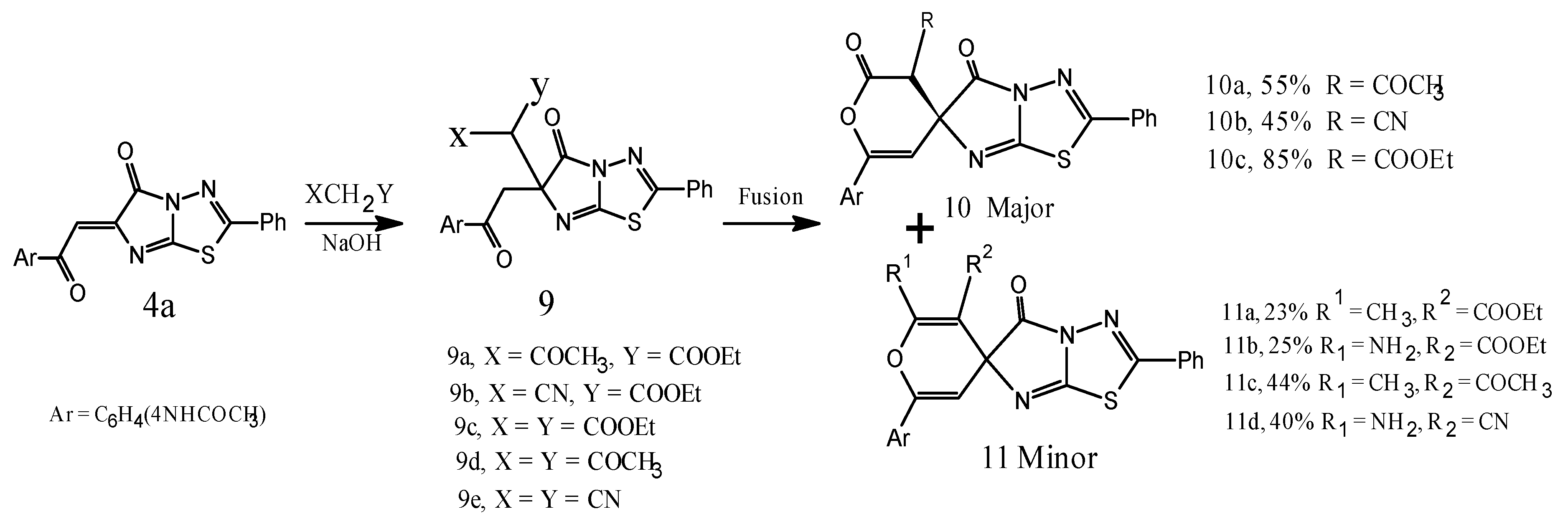
3.8. General Procedure for Synthesis of the Compounds 9a–c
A mixture of 4a (3.91 g, 0.01mol), and carbon acids e.g. ethylacetoacetate, ethylcyanoacetate, diethylmalonate, acetylacetone, malononitrile (0.01 mol), (50%) NaOH (8 mL) and ethanol (50 mL), was made and left overnight for 3 days. The reaction mixture was poured into ice/HCl, the crude product was filtered and washed by petroleum ether (b.p. 40–60 °C), and then crystallized from ethanol.
Ethyl 2-(6-(2-(4-acetamidophenyl)-2-oxoethyl)-5-oxo-2-phenyl-5,6-dihydroimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-3-oxobutanoate (9a). Yield 2.08 g (40%), white finely crystalline, m.p. 180–182 °C. IR (KBr), υ, cm−1: 3245 (NH), 1742, 1671, 1655 (CO), 1630 (C=N). 1HNMR (DMSO-d6), δ, ppm, (J, Hz): 1.21 (3H, t, CH3), 2.32–2.34 (6H, s, 2CH3), 2.47 (2H, s, CH2CO), 4.23 (2H, q, CH2CO), 5.3 (1H, s, methine), 7.62–7.83 (9ArH, m, aromatic protons), 11.2 (1H, s, acidic NH proton which exchanged in D2O); and found, %: C 59.93, H 4.62, N 10.72, S 6.14 for C26H24N4O6S. Calculated, %: C 59.99, H 4.65, N 10.76, S 6.16; MS: m/z 520 [M+], 170, 139.
Ethyl 2-(6-(2-(4-acetamidophenyl)-2-oxoethyl)-5-oxo-2-phenyl-5,6-dihydroimidazo[2,1-b][1,3,4]thiadiazol-6-yl)-2-cyanoacetate (9b). Yield 1.87 g (37%), white finely crystalline, m.p. 164–166 °C. IR (KBr), υ, cm−1: 3331, 3245 (NH), 1734, 1670, 1645 (CO), 1630 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 1.23 (3H, t, CH3), 2.32 (3H, s, CH3), 2.47 (2H, s, CH2CO), 4.09 (2H, q, CH2CO), 5.1 (1H, s, methine), 7.35–7.72 (9ArH, m, aromatic protons), 12.1 (1H, s, acidic NH proton which exchanged in D2O), found, %: C 59.60, H 4.17, N 13.88, S 6.35 for C25H21N5O5S. Calculated, %: C 59.63, H 4.20, N 13.91, S 6.37; MS: m/z 503 [M+], 430 [M+ − COOEt], 170, 140.
Diethyl 2-(6-(2-(4-acetamidophenyl)-2-oxoethyl)-5-oxo-2-phenyl-5,6-dihydroimidazo[2,1-b][1,3,4]thiadiazol-6-yl)malonate (9c). Yield 3.86 g (70%), white finely crystalline. m.p. 144–146 °C. IR (KBr), υ, cm−1: 3245 (NH), 1671, 1742 (CO), 1630 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 1.22 (6H, t, 2CH3), 2.32–2.35 (6H, s, 2CH3), 2.47 (2H, s, CH2CO), 4.45 (4H, q, CH2CO), 5.6 (1H, s, methine), 7.62–7.83 (9ArH, m, aromatic protons), 11.2 (1H, s, acidic NH proton which exchanged in D2O). 13C-NMR (DMSO), δ, 14.5 (2CH3CH2), 22.4 (CH3CO Ar), 60.5 (2CH3CH2CO), 64.7 (CH2CO spiro), 92.6 (CH(CO)2), 114.6 (C4 Ph), 122.1 (C3,5 Ph), 127.5 (C spiro), 128.2 (C3,5 Ar), 129.6 (C2,6 Ph), 131.6 (C2,6 Ar), 134.2 (C4 Ar), 137.9 (C1 Ph), 140.2 (C1 Ar), 149.5 (CNS), 154.1 (CN2S), 163.4 (CO imidaz), 168.3 (CO amide), 176.6 (2CO ester), 193.0 (CO ketone), and found, %: C 58.86; H 4.72; N 10.15, S 5.79 for C27H26N4O7S. Calculated, %: C 58.90, H 4.76, N 10.18, S 5.82; MS: m/z 550 [M+], 170, 139.
N-(4-(2-(6-(2,4-Dioxopentan-3-yl)-5-oxo-2-phenyl-5,6-dihydroimidazo[2,1-b][1,3,4]thiadiazol-6-yl)acetyl)phenyl)acetamide (9d). Obtained similarly to compound 9a, from compound 4a (3.91 g, 0.01 mol) and acetylacetone (1.05 mL, 0.01 mol). Crystalised from benz-ethanol. Yield 2.94 g (60%), white powder. m.p. 158–160 °C. IR (KBr) υ, cm−1: 3245 (NH); 1670, 1645 (CO), 1630 (C=N). 1H-NMR (DMSO-d6), δ, ppm (J, Hz): 2.32–2.46 (9H, br. s, 3CH3), 2.47 (2H, s, CH2CO), 5.2 (1H, s, methine), 7.35–7.72 (9ArH, m, aromatic protons), 12.1 (1H, s, acidic NH proton which exchanged in D2O), and found, %: C 61.22, H 4.49, N 11.40, S 6.52 for C25H22N4O5S. Calculated, %: C 61.21; H 4.52; N 11.42, S 6.54; MS: m/z 490 [M+]; 447 [M+ − COCH3], 170, 140.
N-(4-(2-(6-(Dicyanomethyl)-5-oxo-2-phenyl-5,6-dihydroimidazo[2,1-b][1,3,4]thiadiazol-6-yl)acetyl)phenyl)acetamide (9e). Crystalized from ethanol. Yield 3.29 g (72%), white powder, m.p. 122–124 °C. IR (KBr), υ, cm−1: 3320 (NH); 1655 (CO); 1630 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.32 (3H, s, CH3), 2.47 (2H, s, CH2CO), 5.0(1H, s, methine), 7.35–7.72 (9ArH, m, aromatic protons), 12.1 (1H, s, acidic NH proton which exchanged in D2O). 13C-NMR, δ, 21.8 (CH3CO Ar), 62.5 (CH2CO spiro), 95.1 (CH(CN)2), 112.6 (C4 Ph), 119.3 (C3,5 Ph), 124.5 (C spiro), 125.4 (C3,5 Ar), 128.6 (C2,6 Ph), 131.2 (C2,6 Ar), 133.4 (C4 Ar), 135.6 (C1 Ph), 139.2 (C1 Ar), 147.7 (CNS), 152.1 (CN2S), 163.2 (CO imidaz), 167.3 (CO amide), 170.6 (2CN), 189.4 (CO ketone); found, %: C 60.49, H 3.50, N 18.38, S 7.00 for C23H16N6O3S: C 60.52, H 3.53, N 18.41, S 7.02; and MS: m/z 456 [M+], 380 [M+ − Ph], 170, 140.
3.9. General Procedure for Synthesis of the Compounds 10a–c and 11a–d
The adduct 9 (1.9 mmol) was fused in oil bath for 1 h. The reaction mixture was poured into ice, the crude product filtered and washed by petroleum ether (b.p. 40–60 °C), and then crystalized.
N-(4-(3′-Acetyl-2′,5-dioxo-2-phenyl-2′,3′-dihydro-5H-spiro[imidazo[2,1-b][1,3,4]thiadiazole-6,4′-pyran]-6′-yl)phenyl)acetamide (10a). Crystalized from dioxane. Yield 495 mg (55%), white finely crystalline, m.p. 210–212 °C. IR (KBr), υ, cm−1: 3245 (NH), 1743, 1685, 1670, 1650 (CO), 1613 (C=N); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.36 (6H, s, 2CH3), 4.50 (1H, s, CH(CO)2), 6.2 (1H, s, PyH), 7.44–7.73 (9ArH, m, aromatic protons), 13.2 (1H, s, acidic NH proton which exchanged in D2O); found: %: C 60.70, H 3.79, N 11.75, S 6.76 for C24H18N4O5S. Calculated, %: C 60.75, H 3.82, N 11.81, S 6.76; MS: m/z 474 [M+], 141 [imidazolothiadiazole moiety].
N-(4-(3′-Cyano-2′,5-dioxo-2-phenyl-2′,3′-dihydro-5H-spiro[imidazo[2,1-b][1,3,4]thiadiazole-6,4′-pyran]-6′-yl)phenyl)acetamide (10b). Crystalized from ethanol. Yield 409 mg (45%), white finely crystalline, m.p. 198–200 °C. IR (KBr), υ, cm−1: 3245 (NH), 2220 (CN), 1740, 1691, 1672, 1655, (CO), 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.47 (3H, s, CH3), 4.51 (1H, s, CH(CO)(CN)), 6.1 (1H, s, PyH), 7.44–7.83 (9ArH, m, aromatic protons), 8.2 (1H, s, acidic NH); found, %: C 60.35, H 3.28, N 15.31, S 7.00 for C23H15N5O4S. Calculated, %: C 60.39; H 3.31; N 15.31, S 7.01.
Ethyl 6′-(4-acetamidophenyl)-2′,5-dioxo-2-phenyl-2′,3′-dihydro-5H-spiro[imidazo[2,1-b][1,3,4] thiadiazole-6,4′-pyran]-3′-carboxylate (10c). Yield 1.17 g (85%), white finely crystalline, m.p. 162–164 °C. IR (KBr), υ, cm−1: 3245 (NH), 1752, 1738, 1670, 1650 (CO), 1613 (C=N). 1H-NMR (DMSO-d6), δ, ppm (J, Hz): 1.2 (2H, t, CH2), 2.5 (3H, s, CH3), 4.11 (2H, q, CH2O), 4.4 (1H, s, CH(CO)2), 6.2(1H, s, PyH), 7.44–7.73 (9ArH, m, aromatic protons); 13.2 (1H, s, acidic NH proton which exchanged in D2O); found, %: C 59.50, H 3.95, N 11.08, S 6.32 for C25H20N4O6S. Calculated, %: C 59.52, H 4.00, N 11.11, S 6.35; MS: m/z 504 [M+], 462 [M+ − CH2=C=O], 141.
Ethyl 6′-(4-acetamidophenyl)-2′-methyl-5-oxo-2-phenyl-5H-spiro[imidazo[2,1-b][1,3,4]thiadiazole-6,4′-pyran]-3′-carboxylate (11a). Crystalized from benzene. Yield 219 mg (23%), white powder, m.p. 180–182 °C. IR (KBr), υ, cm−1: 3245 (NH), 1742, 1671, 1655, (CO), 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 1.21 (3H, t, CH3), 2.32 (6H, s, 2CH3), 4.23 (2H, q, CH2CO), 6.3 (s, 1H, PyH), 7.62–7.83 (9ArH, m, aromatic protons), 11.2 (1H, s, acidic NH proton which exchanged in D2O); found, %: C 62.15, H 4.38, N 11.13, S 6.35 for C26H22N4O5S. Calculated, %: C 62.14, H 4.41, N 11.15, S 6.38; MS: m/z 502 [M+], 170, 139.
Ethyl 6′-(4-acetamidophenyl)-2′-amino-5-oxo-2-phenyl-5H-spiro[imidazo[2,1-b][1,3,4]thiadiazole-6,4′-pyran]-3′-carboxylate (11b). Crystalized from benzene. Yield 249 mg (25%), white powder, m.p. 164–166 °C. IR (KBr), υ, cm−1: 3331, 3245 (NH), 1734, 1670, 1645 (CO), 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 1.23 (3H, t, CH3), 2.32 (6H, s, 2CH3), 4.09 (2H, q, CH2CO), 5.2(2H, br. s, NH2), 6.1 (1H, s, PyH), 7.35–7.72 (9ArH, m, aromatic protons), 12.1 (1H, s, acidic NH proton which exchanged in D2O), and found, %: C 59.60, H 4.18, N 13.90, S 6.35 for C25H21N5O5S. Calculated, %: C 59.63, H 4.20, N 13.91, S 6.37; MS: m/z 503 [M+], 430 [M+ − COOEt]; 170.
N-(4-(3′-Acetyl-2′-methyl-5-oxo-2-phenyl-5H-spiro[imidazo[2,1-b][1,3,4]thiadiazole-6,4′-pyran]-6′-yl)phenyl)acetamide (11c). Yield 810 mg (64%), white powder; m.p. 194–196 °C. IR (KBr), υ, cm−1: 3245(NH), 1687, 1670, 1650, (CO), 1613 (C=N). 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.52 (9H, br. s, 3CH3), 6.3 (1H, s, PyH), 7.35–7.80 (9ArH, m, aromatic protons), 11.6 (1H, s, acidic NH proton which exchanged in D2O); found, %: C 63.52, H 4.25, N 11.85, S 6.75 for C25H20N4O4S. Calculated, %: C 63.55, H 4.27, N 11.86, S 6.78; MS: m/z 472 [M+], 353 [M+ − PhN=C=O], 141 [imidazolothiadiazole moiety].
N-(4-(2′-Amino-3′-cyano-5-oxo-2-phenyl-5H-spiro[imidazo[2,1-b][1,3,4]thiadiazole-6,4′-pyran]-6′-yl)phenyl)acetamide (11d). Yield 600 mg (60%), white finely crystalline, m.p. 222–224 °C. IR (KBr), υ, cm−1: 3285, 3245 (NH), 2220 (CN), 1672, 1655 (CO), 1630 (C=N); 1H-NMR (DMSO-d6), δ, ppm, (J, Hz): 2.46 (3H, s, CH3), 5.2 (2H, br. s, NH2), 6.3 (1H, s, PyH), 7.25–7.66 (9ArH, m, aromatic protons), 11.3 (1H, s, acidic NH proton which exchanged in D2O); found, %: C 60.50; H 3.51; N 18.40, S 7.00 for C23H16N6O3S. Calculated, %: C 60.52, H 3.53, N 18.41, S 7.02. MS: m/z 456 [M+].








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}









