
Acid Catalyzed Alcoholysis of Sulfinamides: Unusual Stereochemistry, Kinetics and a Question of Mechanism Involving Sulfurane Intermediates and Their Pseudorotation



Abstract
:
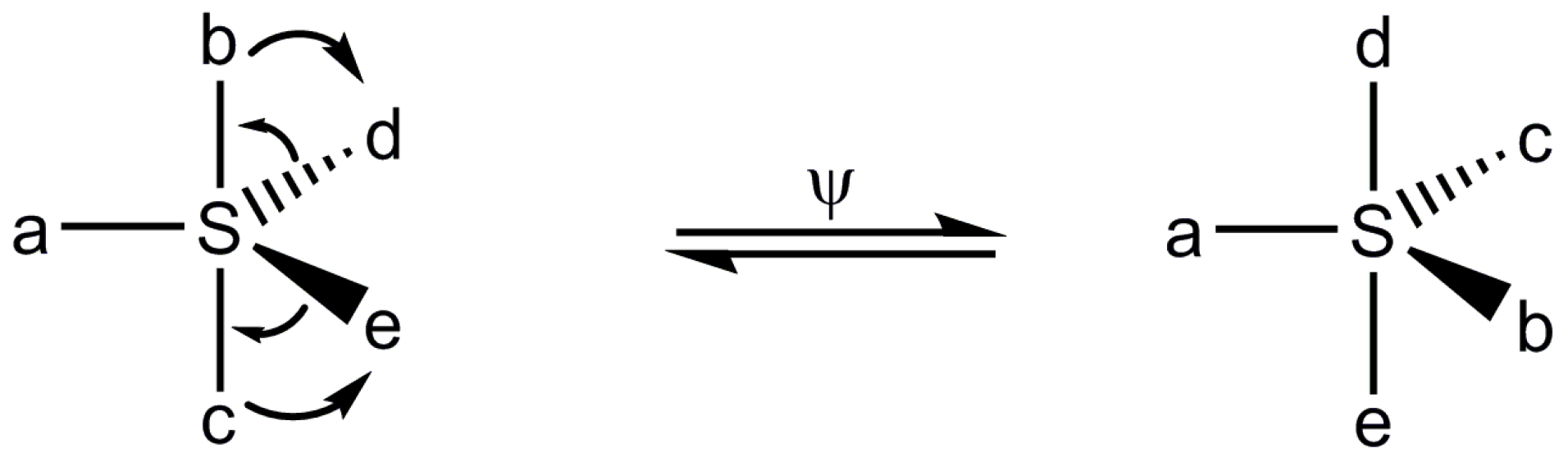
1. Introduction






2. Results and Discussion
2.1. Synthesis of Racemic Sulfinates

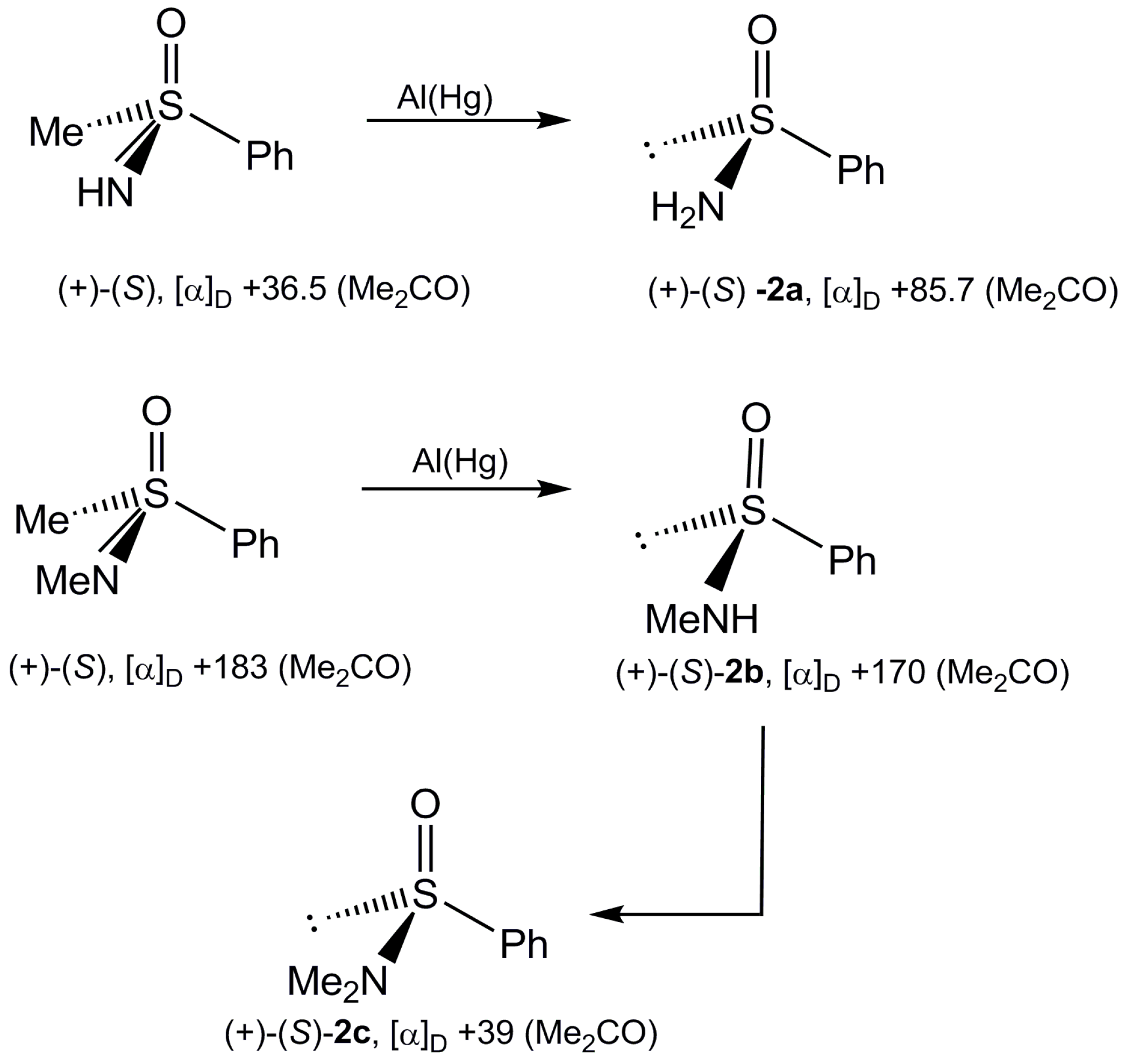
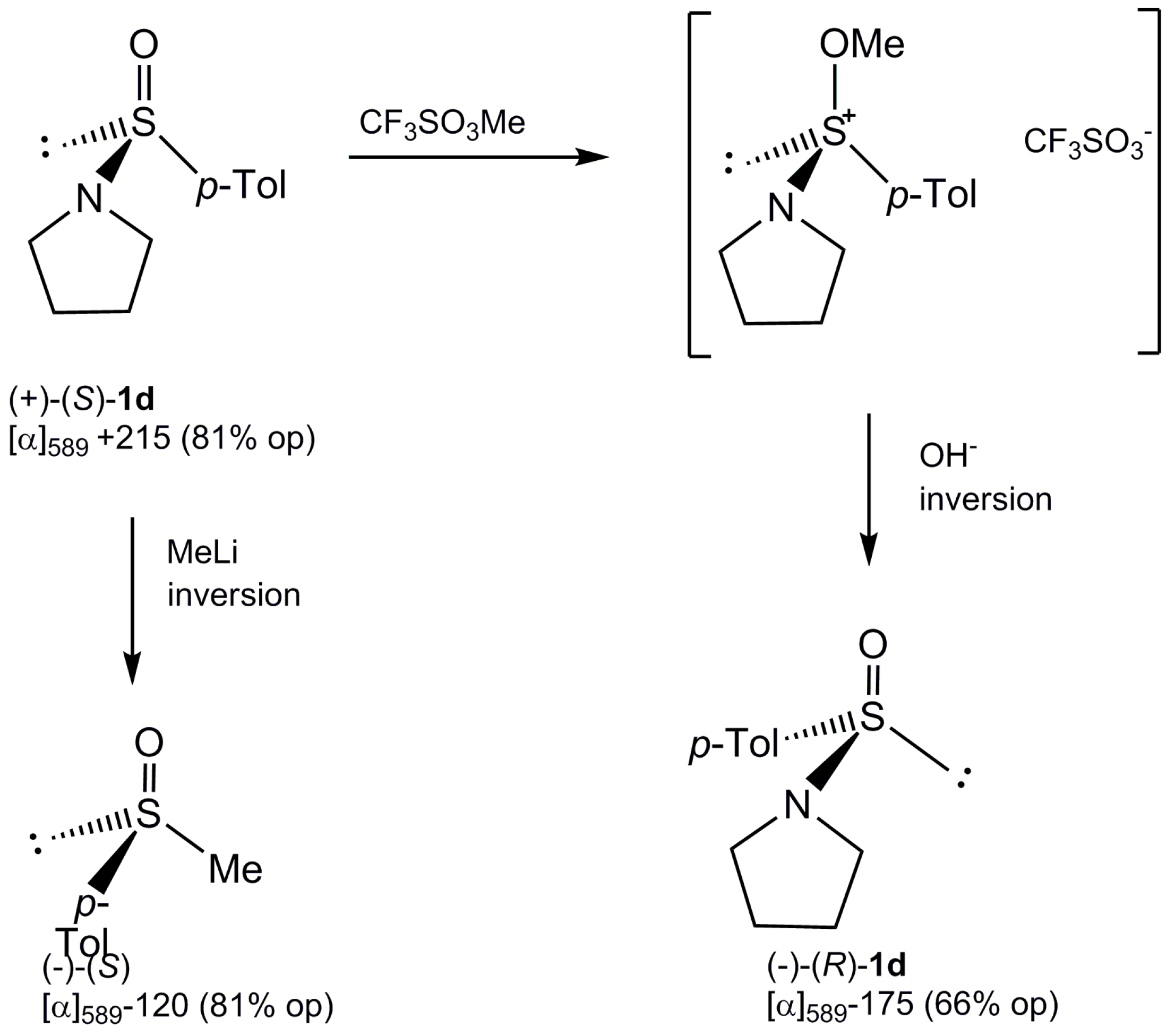
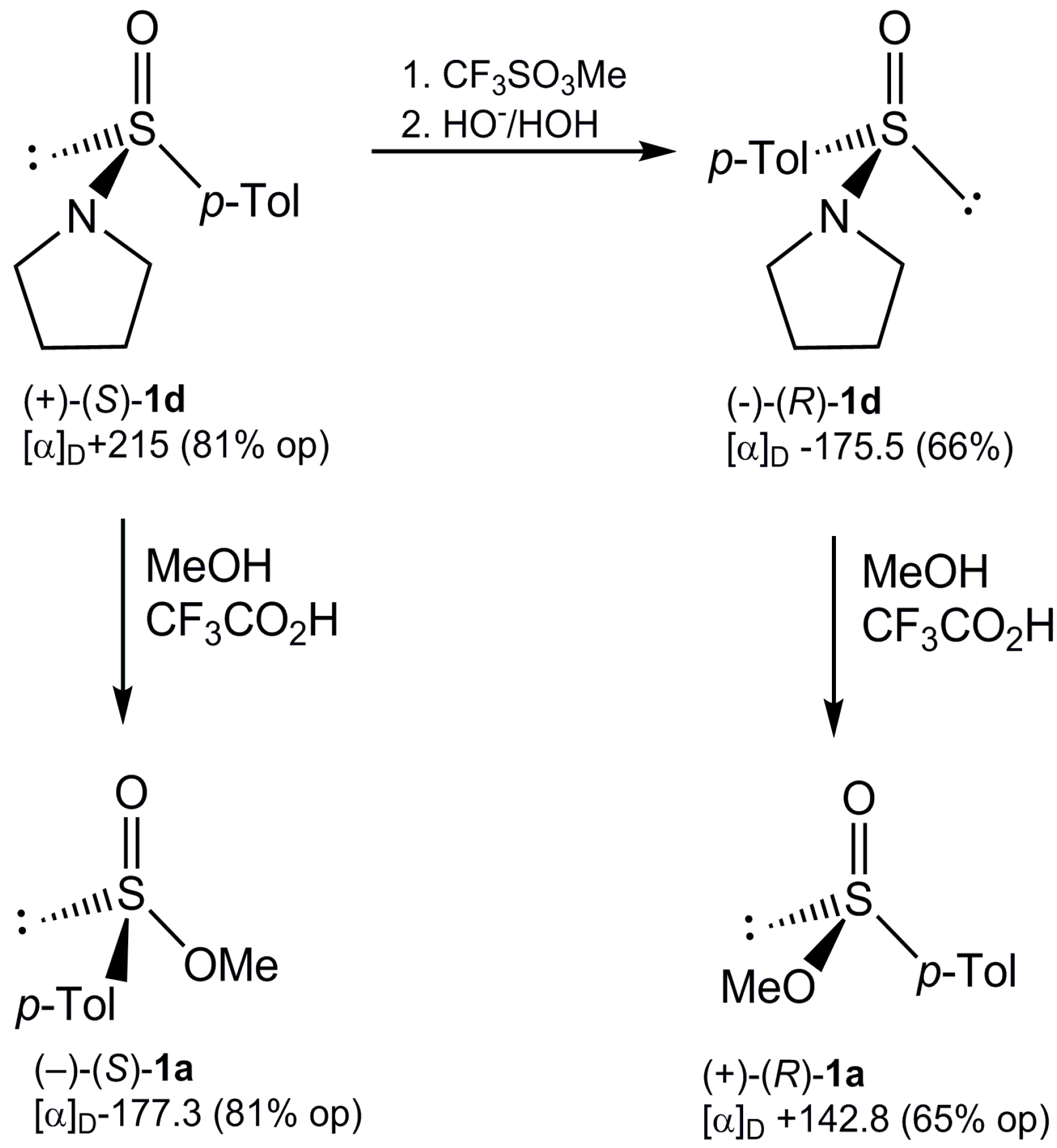
2.2. Synthesis of Optically Active Sulfinamides



{kind=link}
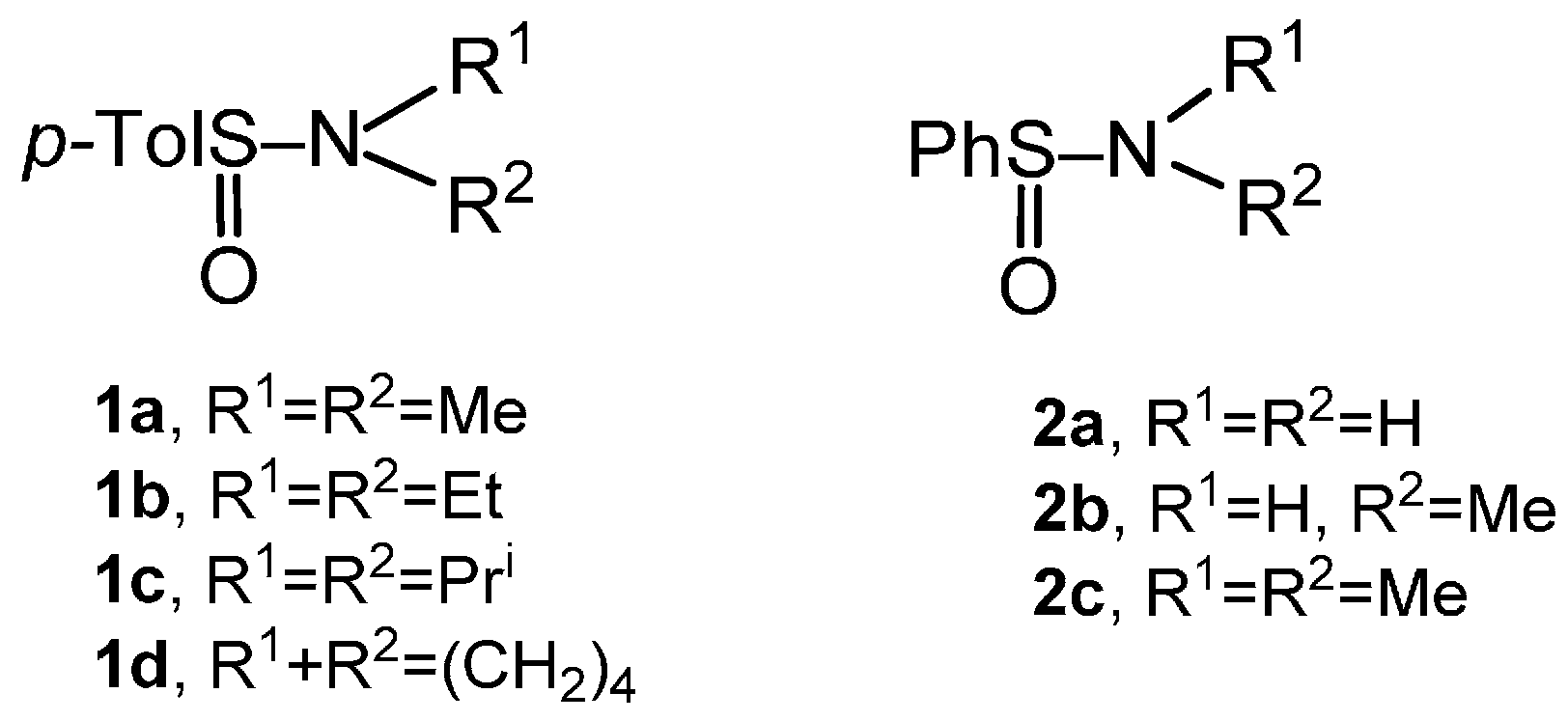{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
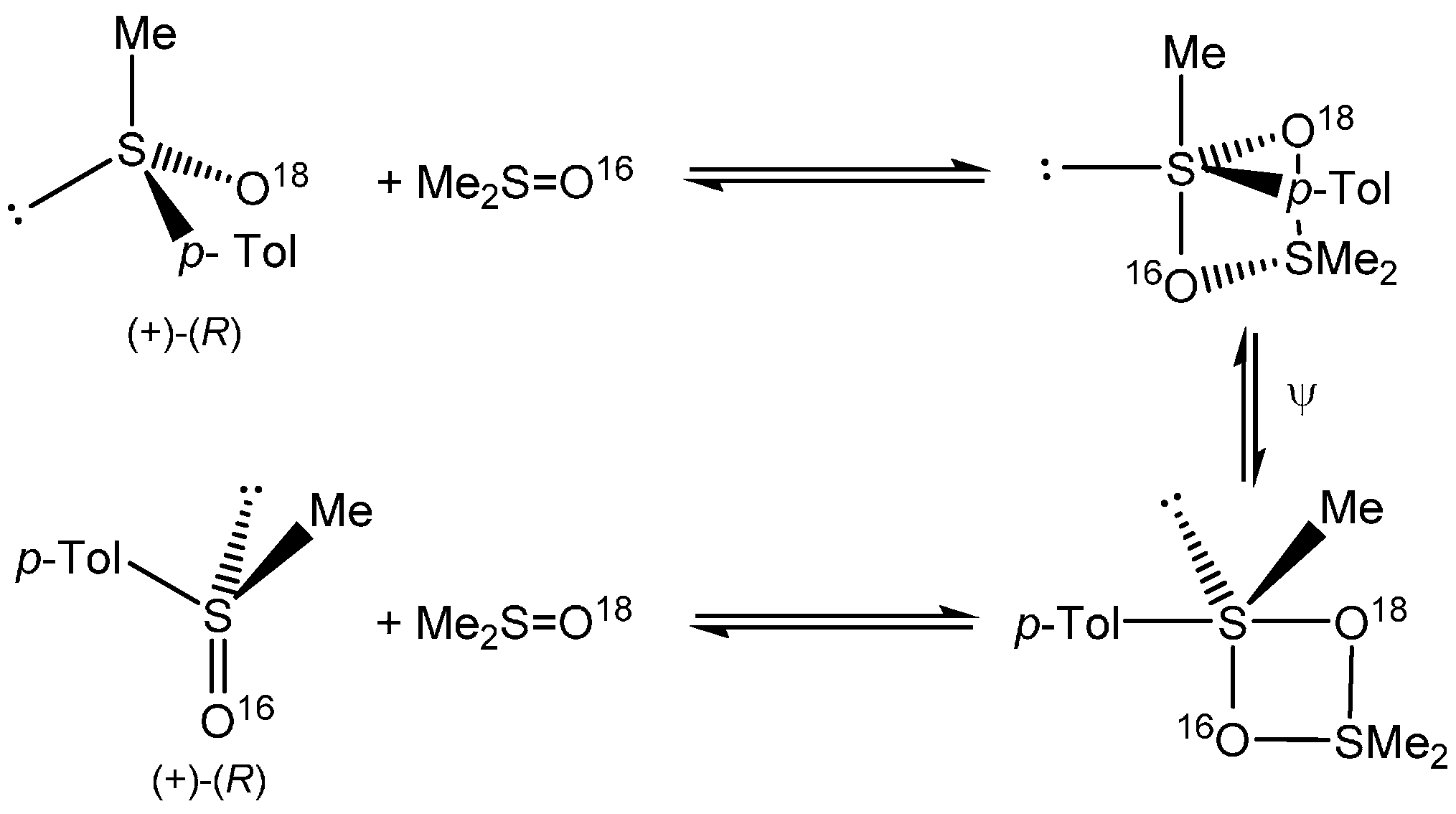{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sulfinate 4 | Reaction Conditions | Sulfinamide 1 | ||||
|---|---|---|---|---|---|---|
| [α]D (Me2CO) | Temp. (°C) | Time (h) | No | Yield (%) a | [α]D (EtOH) | (% op) b |
| −210.0 | 25 | 15 | 1a | 45 | +5.5 | 3.5 |
| −210.0 | 15 | 7 | 1b | 41 | +105.0 | 88 |
| −210.0 | 15 | 20 | 1c | 35 | +104.3 | 54 |
| −198.0 | 25 | 2 | 1c | 65 | +87.0 | 42 |
| −202.0 | 0 | 15 | 1d | 75 | +215.0 | 81 |
| −202.0 | 25 | 15 | 1d | 70 | +135.0 | 51 |

Interconversion of Sulfinamide Enantiomers

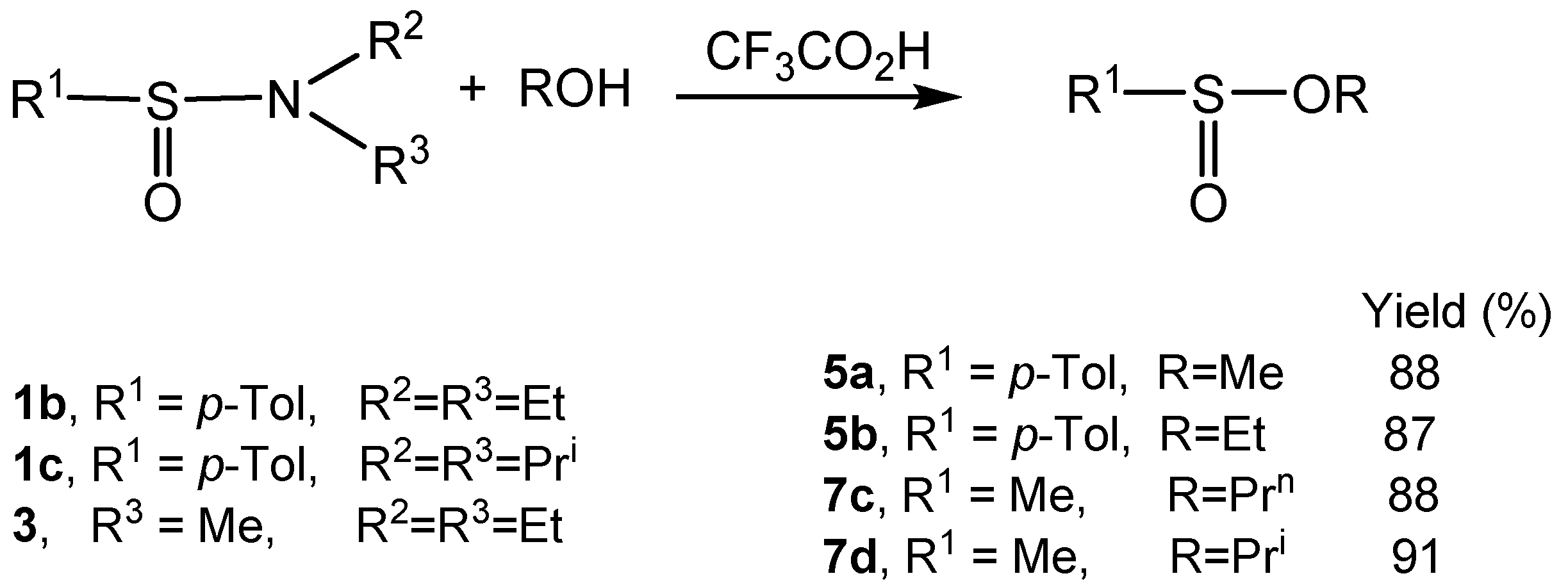
2.3. Stereoselective Synthesis of Optically Active Sulfinates and Stereochemistry of Their Formation

| Sulfinamide 1b | Acid | Sulfinate 5 | Stereochemical Outcome | |||||||
|---|---|---|---|---|---|---|---|---|---|---|
| No | [α]D (Me2CO) | Op (%) | Yield (%) | No | R | [α]D (EtOH) | Op (%) | Selectivity | Inversion (%) | |
| 1b | +107 | 88 | CF3CO2H | 94.0 | 5a | Me | −192.6 | 88 | 100 | 100.0 |
| 1b | +105 | 86 | PhSO3H | 76.5 | 5b | Et | −179.2 | 86 | 100 | 100.0 |
| 1b | +107 | 88 | CF3CO2H | 90.0 | 5b | Et | −137.5 | 66 | 75.5 | 87.7 |
| 1b | +105 | 86 | PhSO3H | 80.0 | 5c | Prn | −161.2 | 84 | 98.2 | 91.1 |
| 1b | +106 | 87 | PhSO3H | 95.0 | 5e | CH2=CHCH2 | −106.5 | 73 | 84.0 | 92.0 |
| 1b | +105 | 86 | CF3CO2H | 84.0 | 5f | HC=CCH2 | −85.9 | 77 | 89.5 | 94.7 |
| 1b | +105 | 86 | PhSO3H | 77.0 | 5g | PhCH2 | −22.6 | 88 | 30 | 65.0 |
| 1b | +96 | 78.5 | CF3CO2H | 87.0 | 5d | Pri | −109.3 | 54 | 69.5 | 84.7 |
| 1b | +105 | 86 | PhSO3H | 53.0 | 5d | Pri | −100.7 | 50 | 58 | 79.0 |
| 1b | +104.7 | 86 | CF3CO2H | 86.0 | 5d | Pri | −107.9 | 54 | 62.7 | 81.5 |
| 1b | +105 | 86 | HSbF6 | 61.0 | 5d | Pri | −134.2 | 67 | 77 | 88.5 |
| 1b | +104.7 | 86 | CF3CO2H | 55.0 | 5i | Bui | −29.85 | 23 | 27.4 | 63.7 |
| 1b | +104.7 | 86 | CF3SO3+CF3CO2H | 55.5 | 5i | Bui | −10.6 | 8.3 | 9.7 | 54.8 |
| Sulfinamide 1c | Sulfinate 5 | Inversion (%) or Retention (%) | ||||
|---|---|---|---|---|---|---|
| [α]D (Me2CO) | Op (%) | No | R | [α]D (EtOH) | Op. (%) | |
| +94.4 | 45.3 | 5a | Me | −35.0 | 16.0 | 68.75, Inv |
| +94.4 | 45.3 | 5b | Et | −35.0 | 3.4 | 53.75, Inv |
| +94.4 | 45.3 | 5c | Prn | −13.9 | 7.3 | 58.0, Inv |
| +94.4 | 45.3 | 5d | Pri | +15.8 | 7.9 | 58.7, Ret |
| +86.9 | 42.3 | 5d' | (CD3)2CH | +10.1 | 4.6 | 55.5, Ret |
| +86.9 | 42.3 | 5h | (CF3)2CH | +3.4 | 1.7 | 52.0, Ret |
| +95.0 | 45.3 | 5j | Hexc | +41.0 | 22.4 | 74.5, Ret |
| +95.0 | 45.3 | 5k | Penc | +3.3 | 1.8 | 52.0, Ret |
| +95.0 | 45.3 | 5l | Et2CH | −4.4 | 2.3 | 52.5, Inv |
| +95.0 | 45.3 | 5m | Bui | −2.7 | 1.4 | 51.5, Inv |
| Sulfinamide 2 | Sulfinate 6 | Inversion (%) | |||||
|---|---|---|---|---|---|---|---|
| No | [α]D (Me2CO) | Op (%) | No | R | [α]D (EtOH) | Op. (%) | |
| 2a | +85.7 | 100 | 6a | Me | −64.0z | 24.0 | 62.0 |
| 2a | +85.7 | 100 | 6b | Et | −23.0 | 10.8 | 55.4 |
| 2b | +170.0 | 100 | 6a | Me | −204.2 | 76.5 | 87.2 |
| 2b | +170.0 | 100 | 6b | Et | −167.0 | 78.0 | 89.0 |
| 2c | +39.0 | 15.6 | 6a | Me | −45.5 | 15.6 | 100.0 |
| 2c | +39.0 | 15.6 | 6b | Et | −33.4 | 15.6 | 100.0 |


| Sulfinate 5 | Inversion/Retention Ratio with AgClO4 | Inversion/Retention Ratio without AgClO4 |
|---|---|---|
| 5a, Me | 100/0 | 68.7/31.3 |
| 5b, Et | 91/9 | 53.7/46.3 |
| 5c, Prn | 100/0 | 58.0/42.0 |
| 5d, Pri | 82/18 | 41.3/58.7 |
| 5j, Hexc | 65.5/34.5 | 25.5/74.5 |

| KA | Prevailing Stereochemistry | KA | Prevailing Stereochemistry |
|---|---|---|---|
| CoCl2 | 55% Retention | Co(NO3)3 | 73.0% Inversion |
| NiC2O4 | 71% Retention | Ni(NO3)2 | 66.0% Inversion |
| Ag2CO3 | 65% Retention | AgClO4 | 82.0% Inversion |
| Ag2Cr2O7 | 67% Retention | AgNO3 | 53.0% Inversion |
| Ag2SO4 | 63% Retention | Ce(NO3)3 | 71.0% Inversion |
| HgBr2 | 69% Retention | CrCl3 | 50.5% Inversion |
| Cd(OAc)2 | 68% Retention |

| Solvent | Inv/Ret Ratio |
|---|---|
| CHCl3 | 55/45 |
| C6H6 | 56/46 |
| C6H14n | 58/42 |
| CH3CN | 49/51 |
2.4. Reaction Kinetics
| Run | Sulfinamide | Alcohol | Temp. (K) | k1(10−4s−1) | |
|---|---|---|---|---|---|
| 1 | 1a, | p-TolS(O)NMe2 | PriOH | 310.0 | 16.0 ± 0.35 |
| 2 | 1b, | p-TolS(O)NEt2 | PriOH | 298.0 | 2.32 ± 0.35 |
| 3 | 1b, | 307.1 | 3.75 ± 0.09 | ||
| 4 | 1b, | 310.1 | 5.22 ± 0.10 | ||
| 5 | 1b, | 318.0 | 8.64 ± 0.15 | ||
| 6 | 1b, | 328.0 | 19.5 ± 0.30 | ||
| 7 | 1b, | p-TolS(O)NEt2 | MeOH | 298.0 | 33.0 ± 1.2 |
| 8 | 1b, | p-TolS(O)NEt2 | CH3OD | 298.0 | 47.9 ± 1.5 |
| 9 | 1c, | p-TolS(O)NPr2i | PriOH | 303.7 | 0.521 ± 0.015 |
| 10 | 1c, | 310.0 | 0.935 ± 0.04 | ||
| 11 | 1c, | 316.7 | 1.57 ± 0.04 | ||
| 12 | 2, | p-TolS(O)NMe2 | PriOH | 310.0 | 5.60 ± 0.10 |
| Reaction | Ea (kJ mol−1/kJ mol−1) | ΔS≠ (J mol−1 k−1/e.u.) |
|---|---|---|
| 1b | 58.3/14 | −145.5/−34.8 |
| 1c | 68.1/16.3 | −110.4/−26.4 |



| Concentration of CF3CO2Na | Rate Contant | Prevailing Stereochemistry |
|---|---|---|
| 0 mol | k = (2.73 ± 0.26) 10−4 s−1 | 58% Ret |
| 16 mol | k = (3.29 ± 0.06) 10−4 s−1 | 58% Inv |
| 32 mol | k = (3.56 ± 0.18) 10−4 s−1 | 59% Inv |
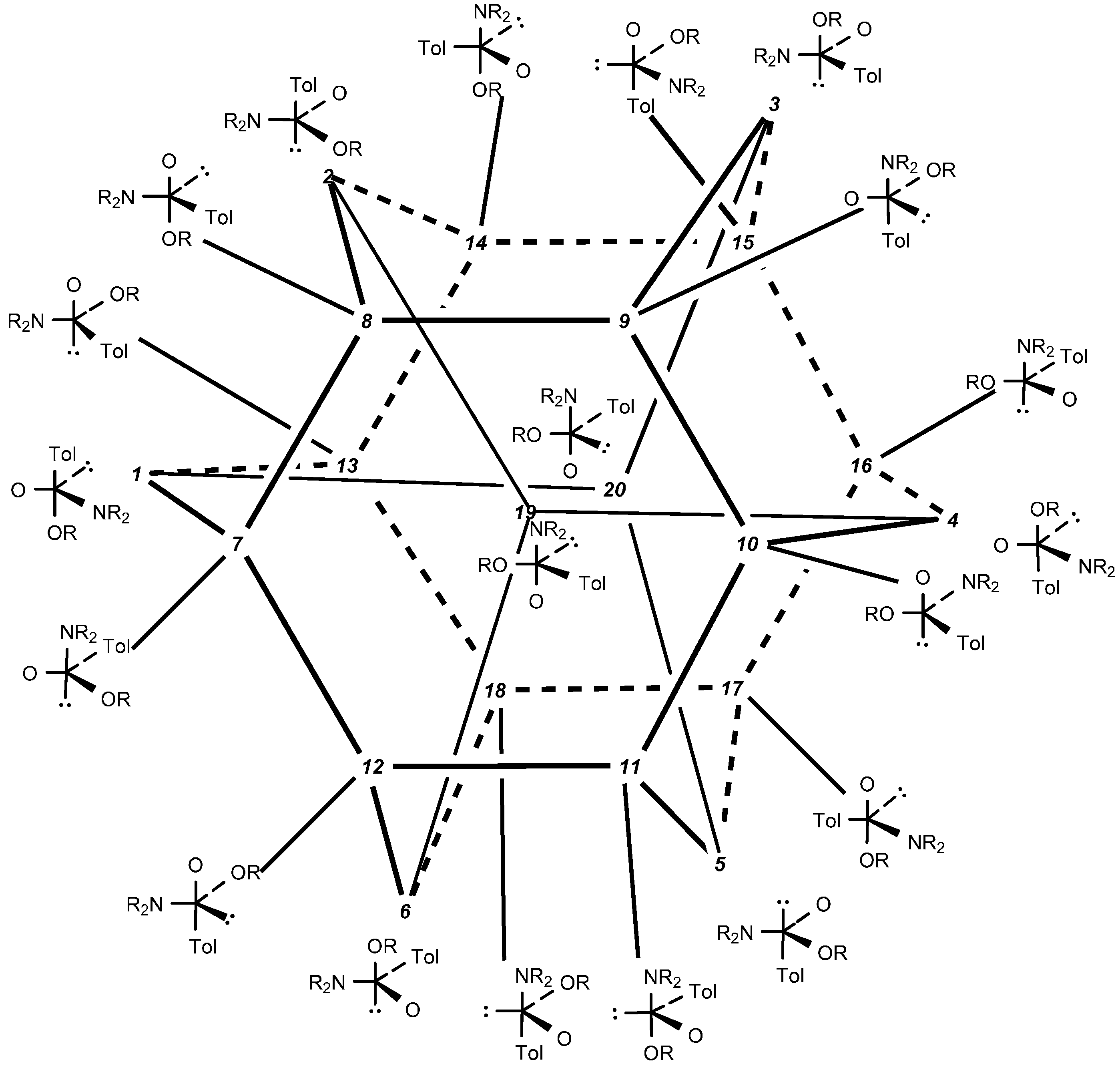
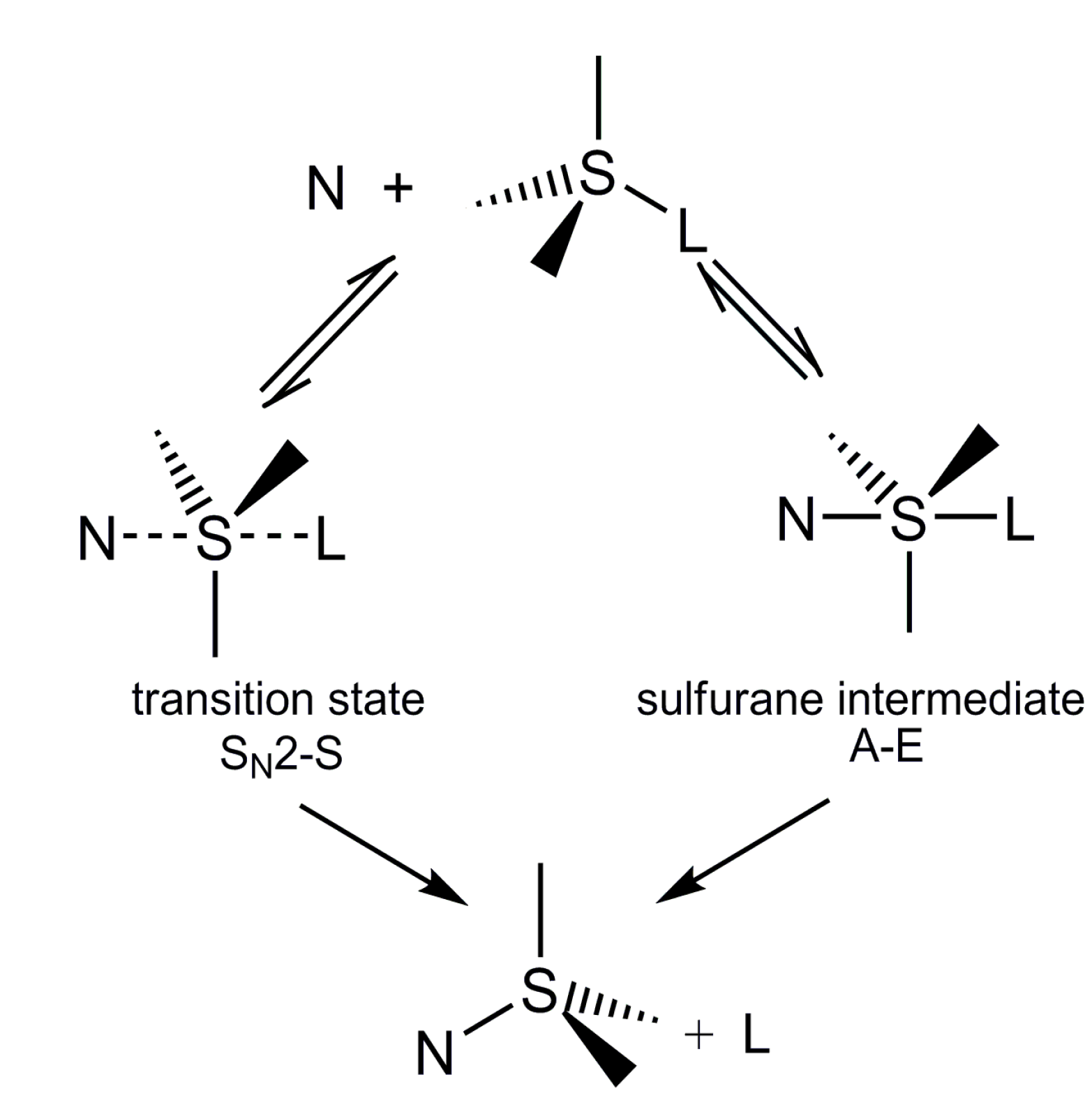
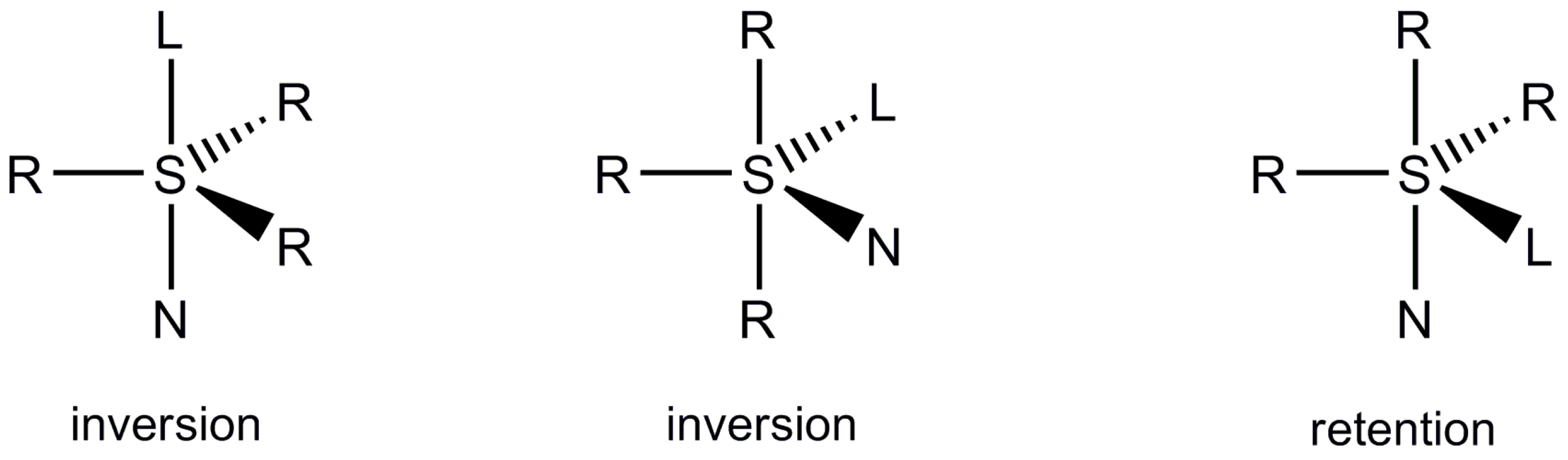
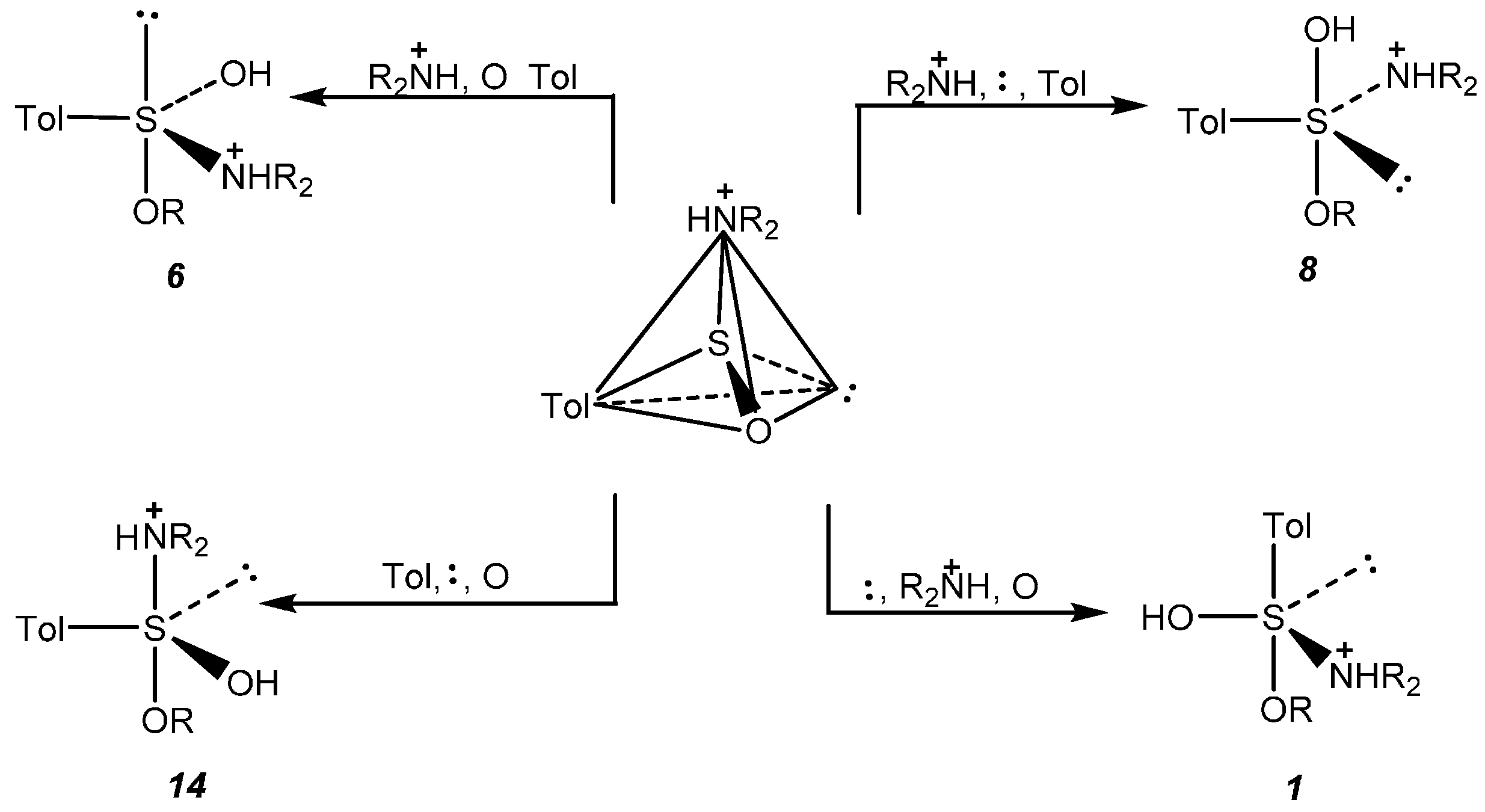
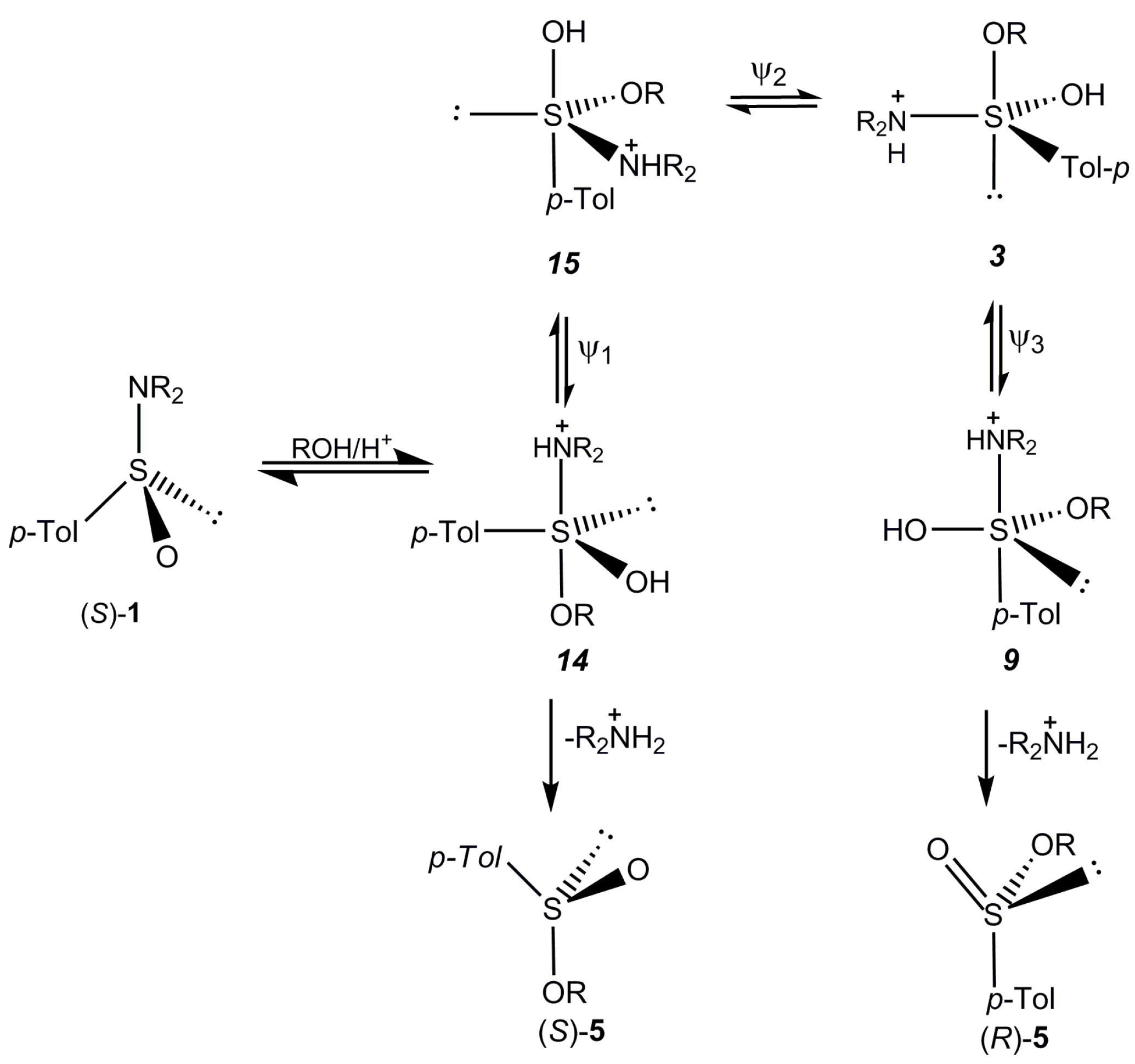
2.5. Discussion




3. Experimental Section
3.1. General
3.2. Synthesis of Optically Active p-toluenesulfinamides 1
General Procedure
3.3. Optically Active Alkyl p-Toluenesulfinates 5 and Alkyl Benzenesulfinates 6 from Acid Catalyzed Alcoholysis of Optically Active Sulfinamides 1 and 2
General Procedure
| No | Ar | R | IR(S=O) cm−1 | 1H-NMR (60 MHz, CDCl3 δ (ppm) |
|---|---|---|---|---|
| 5a | p-Tol | Me | 1126 | 7.45 and 7.25 (AB-system, 4H, H-Ar, JAB = 7.5 Hz); 3.30 (s, 3H, CH3O); 2.35 (s, 3H, CH3-Ar). |
| 5b | p-Tol | Et | 1120 | 7.45 and 7.25 (AB-system, 4H, H-Ar, JAB = 8 Hz); 3.90 and 3.50 (ABX3-system, 2H, CH3-O, JAB = 10.5 Hz, JAX = JBX = 7 Hz); 2.40 (s, 3H, CH3-Ar); 1.20 (t, 3H, CH3CH2, J = 7 Hz) |
| 5c | p-Tol | Prn | 1125 | 7.45 and 7.23 (AB-system, 4H, H-Ar, JAB = 8.0 Hz); 3.82 and 3.44 (ABX2-system, 2H, CH2O, JAB = 8.5 Hz, JAX = JBX = 7 Hz); 1.60 (sx, 2H, CH3CH2, J = 7 Hz); 0.9 (t, 3H, CH3CH2, J = 7Hz) |
| 5d | p-Tol | Pri | 1130 | 7.50 and 7.30 (AB-syst, 4H, H-Ar, JAB = 8 Hz); 4.50 (sp, 1H, (CH3)2CHO, J = 6 Hz); 2,35 (s, 3H, CH3-Ar); 1.31 and 1.18 (d,d, 6H, (CH3)2CH, J = 6 Hz) |
| 5d' | p-Tol | (CD3)2CH | 1130 | 7.45 and 7.20 (AB-system, 4H, H-Ar, JAB = 8.5 Hz); 4.44 (m, 1H, CH(CD3)2); 2.35 (s,3H, CH3-Ar) |
| 5d'' | p-Tol | (CF3)2CH | – | 7.50 and 7.30 (AB-system, 4H, H-Ar. JAB = 8.5 Hz) 4.35 (sp, 1H, (CF3)2CH, JF-H = 6 Hz); 2.40 (s, 3H, CH3-Ar).19F-NMR (CDCl3); δ = +95.2, (d, 6F, (CF3)CH, JF-H = 6Hz) (C6F6 as internal standard) |
| 5j | p-Tol | Hexc | 1130 | 7.40 and 7.25 (AB-system, 4H, H-Ar, JAB = 8 Hz); 4.10–4.35 (m, 1H, CHO); 2.35 (s, 3H, CH3-Ar); 2.10–1.05 (m, 10H, Cy-H) |
| 5k | p-Tol | Penc | 1125 | 7.45 and 7.20 (AB-system, 4H, H-Ar, JAB = 8 Hz); 4.60(m, 1H, CHO); 2.40 (s, 3H, CH3-Ar);1.95–1.30 (m, 8H, Cp-H) |
| 5m | p-Tol | Bui | 1128 | 7.60 and 7.35 (AB-system, 4H, H-Ar, JAB = 8 Hz); 4.00–3.20 (m, 2H, OCH2CH); 2.40 (s, 3H, CH3-Ar); 1.85 (m, 1H, (CH3)2CH); 0.90 (d, 6H, (CH3)2CH, J = 6 Hz) |
| 5n | p-Tol | But | 1125 | 7.45 and 7.20 (AB-system, 4H, H-Ar, JAB = 8.5 Hz); 2.35 (s, 3H, CH3Ar); 1.45 (s, 9H, (CH3)3CO) |
| 5l | p-Tol | Pentyl-3 | – | 7.70 and 7.35 (AB-system, 4H, H-Ar, JAB = 8 Hz); 4.20 (mc, 1H, CHO); 2.35 (s, 3H, CH3-Ar); 1.90–1.20 (m, 4H, CH3CH2-O); 0.95 and 0.90 (t,t, 6H, CH3CH2CHO, J = 6 Hz) |
| 6a | Ph | Me | 1125 | 7.60–7.35 (m, 5H, H-Ar), 3.32 (s, 3H, CH3O) |
| 6b | P | Et | – | 7.70–7.40 (m, 5H, H-Ar), 4.00 (q, 2H, CH3CH2O, J = 7 Hz), 1.30 (t, 3H, CH3CH2, J = 7 Hz) |
3.4. Conversion of Selected Sulfinates 5 into Methyl p-tolyl Sulfoxide
General Procedure
| p-Tol S(O)OR | MeS(O)-Tol-p | |||||
|---|---|---|---|---|---|---|
| No | R | [α]D | Conf. | [α]D | Op (%) | Conf. |
| 5e | CH2=CH–CH2 | −106.5 | S | +108.0 | 73 | R |
| 5f | CH2=CHCH2 | −86.0 | S | +111.3 | 77 | R |
| 5d' | (CD3)2CH | −19.8 | S | +13.6 | 9.2 | R |
| 5d'' | (CF3)2CH | −10.3 | S | +7.5 | 5.1 | R |
| 5j | Hexc | −23.9 | S | +20.6 | 13.8 | R |
| 5k | Penc | −6.5 | S | +5.2 | 3.5 | R |
| 5l | Et2CH | −11.7 | S | +9.2 | 6.2 | R |
| 5m | Bui | −8.2 | S | +6.2 | 4.2 | R |
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Mikołajczyk, M.; Drabowicz, J. Chiral Organosulfur Compounds. In Topics in Stereochemistry; Allinger, N.L., Eliel, E.L., Wilen, S.H., Eds.; John Wiley and Sons, Inc.: New York, NY, USA, 1982; Volume 13, pp. 333–468. [Google Scholar]
- Drabowicz, J.; Łyżwa, P.; Mikołajczyk, M. High-coordinated Sulfur Compounds. In Supplement S: The Chemistry of Sulfur-Containing Functional Groups; Patai, S., Rappoport, Z., Eds.; John Wiley and Sons: Chichester, UK, 1993; pp. 799–956. [Google Scholar]
- Drabowicz, J. Hypervalent Sulfuranes as Transient and Isolable Structures: Occurence, Synthesis and Reactivity. In Chemistry of Hypervalent Compounds; Akiba, K., Ed.; John Wiley and Sons: New York, NY, USA, 1999; pp. 211–240. [Google Scholar]
- Drabowicz, J.; Halaba, G. Stereochemical aspect of the chemistry of hypervalent chalcogen compounds. Rev. Heteroatom Chem. 2000, 22, 1–31. [Google Scholar]
- Allenmark, S. Recent advances in spirochalcogenurane stereochemistry—A mini review. Chirality 2008, 20, 544–551. [Google Scholar] [CrossRef] [PubMed]
- Mikołajczyk, M.; Drabowicz, J.; Ślebocka-Tilk, H. Nucleophilic substitution at sulfur. Kinetic evidence for inversion of configuration at sulfinyl sulfur in acid-catalyzed transesterification of sulfonates. J. Am. Chem. Soc. 1979, 101, 1302–1303. [Google Scholar] [CrossRef]
- Mikołajczyk, M. Nucleophilic substitution at sulfinyl and sulfonyl centres: Stereochemical and kinetic studies. Phosphorus Sulfur Relat. Elem. 1986, 27, 31–42. [Google Scholar] [CrossRef]
- Oae, S.; Yokoyama, M.; Kise, M.; Furukawa, N. Oxygen exchange reaction of sulfoxides with dimethyl sulfoxide. Tetrahedron Lett. 1968, 9, 4131–4134. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Drabowicz, J.; Bujnicki, B. Acid-catalysed conversion of sulfinamides into sulfinates. A new synthesis of optically active sulfinates. J. Chem. Soc. Chem. Commun. 1976, 568–569. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Drabowicz, J.; Bujnicki, B. Nucleophilic substitution at sulfinyl sulfur. Factors affecting the inversion to retention ratio in acid-catalyzed alcoholysis of chiral N,N-diizopropyl p-toluenesulfinamide. Tetrahedron Lett. 1985, 26, 5699–5702. [Google Scholar] [CrossRef]
- Montanari, F.; Giovini, R.; Colonna, S. Stereochemistry of nucleophilic substitution at the sulphur atom. The absolute configuration of sulphinamides. Chemical Commun. 1968, 865–866. [Google Scholar] [CrossRef]
- Schroeck, C.W.; Johnson, C.R. Chemistry of sulfoxides and related compounds. XXXI. Aluminum amalgam reduction of aryl sulfoximines and related compounds. J. Am. Chem. Soc. 1971, 93, 5305–5306. [Google Scholar] [CrossRef]
- Schroeck, C.W.; Johnson, C.R. Chemistry of sulfoxides and related compounds. XLV. Asymmetric syntheses using optically active oxosulfonium alkylides. J. Am. Chem. Soc. 1973, 95, 7418–7423. [Google Scholar] [CrossRef]
- Johnson, C.R.; McCants, D., Jr. Nucleophilic displacement on sulfur. The inversion of sulfoxide configurations. J. Am. Chem. Soc. 1965, 87, 5404–5409. [Google Scholar] [CrossRef]
- Minato, H.; Yamaguchi, K.; Kobayashi, M. Formation of alkoxyaminosulfonium ions. Chem. Lett. 1975, 4, 991–994. [Google Scholar] [CrossRef]
- Minato, H.; Yamaguchi, K.; Okuma, K.; Kobayashi, M. Syntheses of some alkoxy-, dialkoxy-, and alkoxyamino-sulfonium ions and their O-methylene and N-methylene PMR chemical shifts. Bull. Chem. Soc. Jpn. 1976, 49, 2590–2593. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Drabowicz, J. Optically active O-alkyl alkylsulphinates. Tetrahedron Lett. 1972, 13, 2379–2382. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Drabowicz, J. Asymmetric synthesis of sulphinic esters with the sulphur atom as a sole chirality centre. J. Chem. Soc. D, Chem. Commun. 1974, 547–548. [Google Scholar] [CrossRef]
- Mikołajczyk, M.; Drabowicz, J. One-step synthesis of alkyl t-alkanesulfinates. Synthesis 1974, 124–126. [Google Scholar] [CrossRef]
- Asefi, H.; Tillett, J.G. Nucleophilic substitution at sulphur. Part 2. The acid-catalysed hydrolysis of arenesulphinamides. J. Chem. Soc. Perkin Trans. 2 1979, 1579–1582. [Google Scholar] [CrossRef]
- Piggott, A.M.; Karuso, P. Hydrolysis rates of alkyl and aryl sulfinamides: Evidence of general acid catalysis. Tetrahedron Lett. 2007, 48, 7452–7455. [Google Scholar]
- Bujnicki, A.; Drabowicz, J.; Mikołajczyk, M.; Kolbe, A.; Stefaniak, L. Protonation of sulfinamides. Does it occur at oxygen or nitrogen? J. Org. Chem. 1996, 61, 7593–7596. [Google Scholar] [CrossRef] [PubMed]
- DeBruin, K.E.; Naumann, K.; Zon, G.; Mislow, K. Topological representation of the stereochemistry of displacement reactions at phosphorus in phosphonium salts and cognate systems. J. Am. Chem. Soc. 1969, 91, 7031–7040. [Google Scholar] [CrossRef]
- Westheimer, F.H. Pseudorotation in the hydrolysis of phosphate esters. Acc. Chem. Res. 1968, 1, 70–78. [Google Scholar] [CrossRef]
- Nicoud, J.-F.; Cherkaoui, M.Z. A new synthesis of enantiomerically pure n-alkyl 4-bromobenzenesulfinates and their configurational assignment. Tetrahedron Asym. 1995, 6, 1941–1946. [Google Scholar] [CrossRef]
- Fernandez, I.; Khiar, N. Recent developments in the synthesis and utilization of chiral sulfoxides. Chem. Rev. 2003, 103, 3651–3705. [Google Scholar] [CrossRef] [PubMed]
- Nojima, T.; Hirano, Y.; Ishiguro, K.; Sawaki, Y. Dramatic change of carbonyl oxidereactivity by the potent electron-withdrawing trifluoromethyl group. J. Org. Chem. 1997, 62, 2387–2395. [Google Scholar] [CrossRef] [PubMed]
- Sato, S.; Zhang, S.-Z.; Furukawa, N. The Pummerer-like reaction of 2,5-bis(trimethylsilyl)thiophene S-oxide with trifluoroacetic anhydride: intermediary formation of sulfurane [10-S-4(C2O2)] (λ4-sulfane). Heteroatom. Chem. 2001, 12, 444–450. [Google Scholar] [CrossRef]
- Volonterio, A.; Bravo, P.; Pesenti, C.; Zanda, M. The “non-oxidative” chloro-Pummerer reaction: A highly stereoselective entry to β-chloro amines and aziridines. Tetrahedron Lett. 2001, 42, 3985–3988. [Google Scholar] [CrossRef]
- Clennan, E.L.; Pan, G. Isotope effects as mechanistic probes in solution and in intrazeolite photooxygenations. The formation of a hydroperoxysulfonium ylide. J. Org. Chem. 2003, 68, 5174–5179. [Google Scholar] [CrossRef] [PubMed]
- González-Núñez, M.E.; Mello, R.; Royo, J.; Asensio, G.; Monzó, I.; Tomás, F.; Rios, J.V.; López, J.G.; Ortiz, F.L. Mechanism of the oxidation of sulfides by dioxiranes: Conformational mobility and transannular interaction in the oxidation of thianthrene 5-oxide. J. Org. Chem. 2004, 69, 9090–9099. [Google Scholar] [CrossRef] [PubMed]
- Nagao, Y.; Miyamoto, S.; Miyamoto, M.; Takeshige, H.; Hayashi, K.; Sano, S.; Shiro, M.; Yamaguchi, Y.; Sei, Y. Highly stereoselective asymmetric Pummerer reactions that Incorporate intermolecular and intramolecular nonbonded S···O interactions. J. Am. Chem. Soc. 2006, 128, 9722–9729. [Google Scholar] [CrossRef] [PubMed]
- Boschi-Muller, S.; Gand, A.; Branlant, G. The methionine sulfoxide reductases: Catalysis and substrate specificities. Arch. Biochim. Biophys. 2008, 474, 266–273. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bujnicki, B.; Drabowicz, J.; Mikołajczyk, M. Acid Catalyzed Alcoholysis of Sulfinamides: Unusual Stereochemistry, Kinetics and a Question of Mechanism Involving Sulfurane Intermediates and Their Pseudorotation. Molecules 2015, 20, 2949-2972. https://doi.org/10.3390/molecules20022949
Bujnicki B, Drabowicz J, Mikołajczyk M. Acid Catalyzed Alcoholysis of Sulfinamides: Unusual Stereochemistry, Kinetics and a Question of Mechanism Involving Sulfurane Intermediates and Their Pseudorotation. Molecules. 2015; 20(2):2949-2972. https://doi.org/10.3390/molecules20022949
Chicago/Turabian StyleBujnicki, Bogdan, Józef Drabowicz, and Marian Mikołajczyk. 2015. "Acid Catalyzed Alcoholysis of Sulfinamides: Unusual Stereochemistry, Kinetics and a Question of Mechanism Involving Sulfurane Intermediates and Their Pseudorotation" Molecules 20, no. 2: 2949-2972. https://doi.org/10.3390/molecules20022949
APA StyleBujnicki, B., Drabowicz, J., & Mikołajczyk, M. (2015). Acid Catalyzed Alcoholysis of Sulfinamides: Unusual Stereochemistry, Kinetics and a Question of Mechanism Involving Sulfurane Intermediates and Their Pseudorotation. Molecules, 20(2), 2949-2972. https://doi.org/10.3390/molecules20022949