2.1. Design and Synthesis
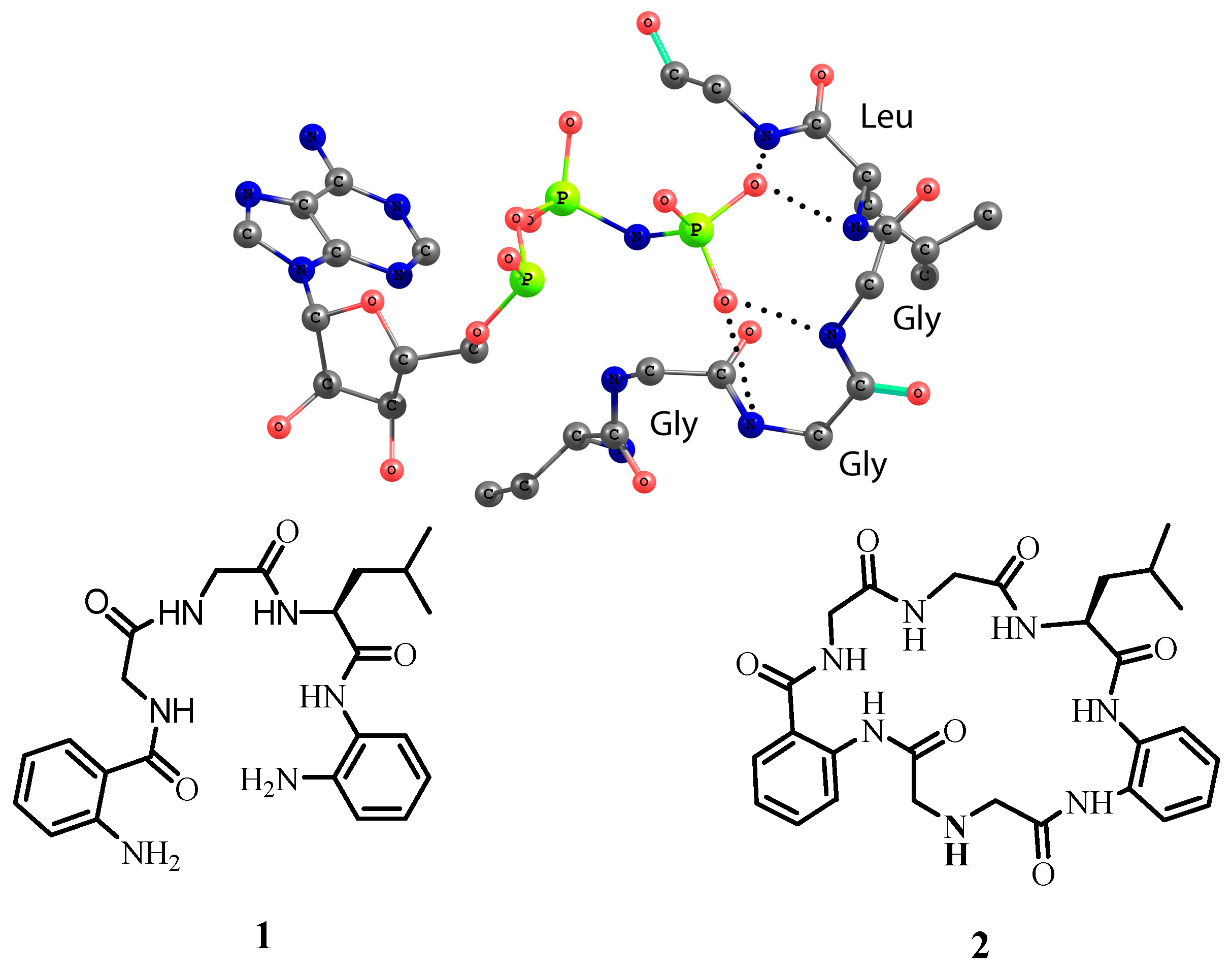
The design for model receptors relies on the structure of the active center of 4-diphosphocytidyl-2C-methyl-
d-erythritol kinase [
19]. The amino acid chain forms a loop around the phosphate with GGLG sequence (
Figure 1). Interestingly, for the coordination of two oxygen atoms only four NH-amide groups of tripeptide GGL are required. To generate a structural model of this binding motif, we functionalized the GGL sequence with 1,2-phenylenediamine units from the one side and with the anthranilic acid from the other side, respectively. The aromatic rings were introduced with the aim to produce amides from both sides of the sequence and rigidify the overall conformation of receptors. There are two reasons why receptor
1 contains two terminal amino groups. Firstly, amino groups can be protonated and generate additional electrostatic interactions between the receptor and the anion. Secondly, receptor
1 can react with diacids bearing either hydrogen-bond-donor or hydrogen-bond-acceptor groups. In this work, we synthesized macrocyclic receptor
2, which contains one secondary amino group. Thus, receptors
1 and
2 differ from each other in structure and in number of amino groups available for protonation. For anion binding studies, we used the following anions as their tetrabutylammonium salts: Cl
−, CH
3COO
−, H
2PO
4−, HSO
4−, and NO
3−. One can expect that interaction of receptors
1 and
2 in organic solvents with hydrogen sulfate and dihydrogen phosphate might be stronger than for other anions because free amino groups can accept the proton from these anions.
Figure 1.
Structure of the active center of 4-diphosphocytidyl-2C-methyl-d-erythritol kinase coordinating AMP-PNP substrate and the structure of model receptors 1 and 2 investigated in this work. Hydrogen bonds are depicted as dotted lines (PDB code 1OJ4).
Figure 1.
Structure of the active center of 4-diphosphocytidyl-2C-methyl-d-erythritol kinase coordinating AMP-PNP substrate and the structure of model receptors 1 and 2 investigated in this work. Hydrogen bonds are depicted as dotted lines (PDB code 1OJ4).
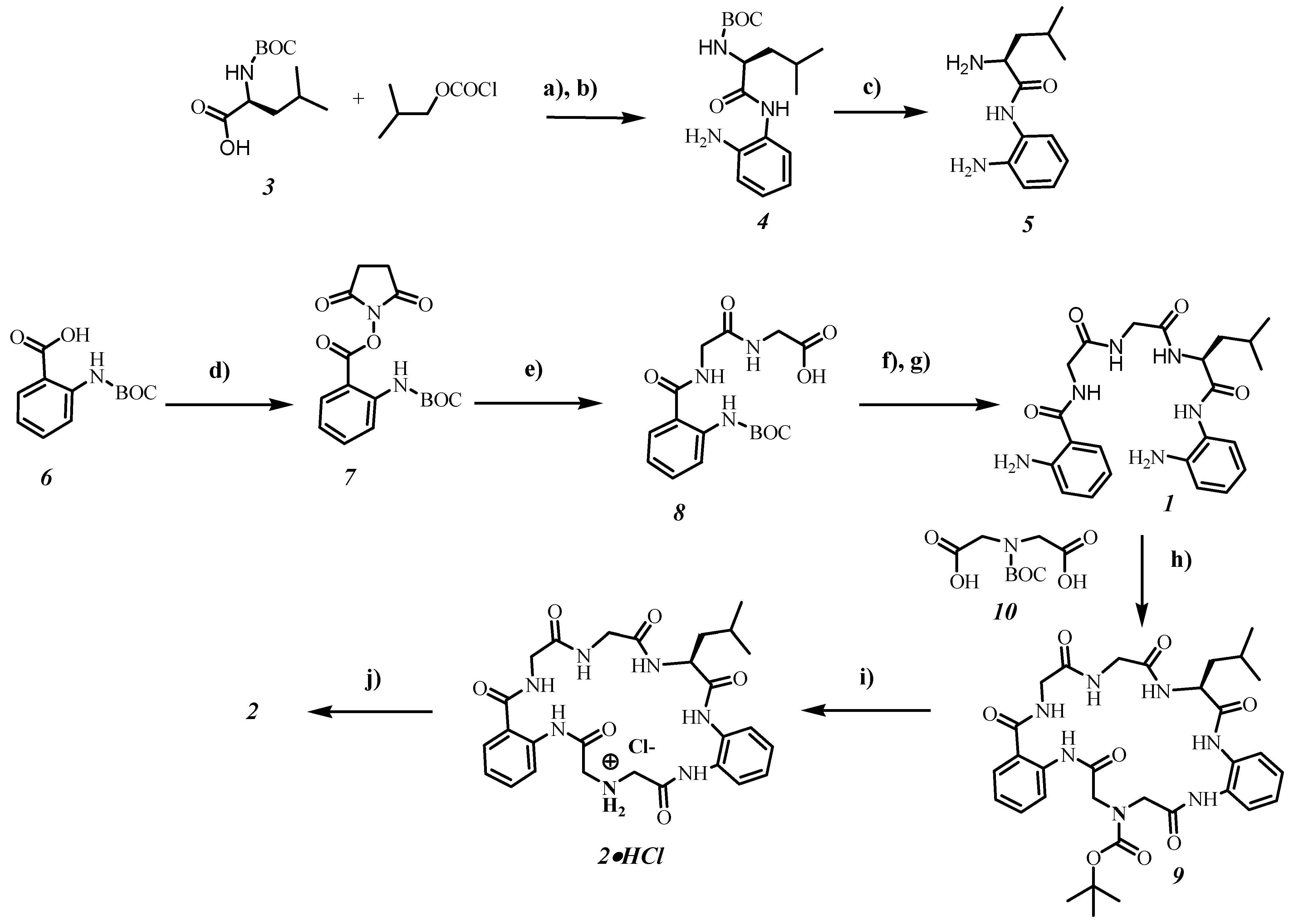
In
Figure 2 the synthesis of receptors
1 and
2 is shown. Target compound
1 was constructed by coupling of protected amine
8 with diamine
5. The deprotection step with trifluoroacetic acid yielded receptor
1 in 37% overall yield based on starting compound
6. The macrocyclization step with the help of Boc-protected iminodiacetic acid
10 towards compound
9 was carried out at high dilution conditions giving the product in 36% yield. Deprotection of macrocycle
9 with trifluoroacetic acid gave only traces of receptor
2 and a mixture of different acyclic side-products, according to the NMR measurements. The best method for the deprotection step was the treatment of
9 with diethyl ether saturated with hydrogen chloride. By using this method, receptor
2 was obtained in 15% overall yield.
Figure 2.
Synthesis of receptors 1 and 2.
Figure 2.
Synthesis of receptors 1 and 2.
Reagents and conditions: (a) diisopropylethylamine (DIPEA); (b) 1,2-phenylenediamine; (c) CHCl3/Et2O saturated with HCl, then NaHCO3; (d) 1-ethyl-3-(3-dimethylaminopropyl)carbo-diimide (EDC), 4-(dimethylamino)-pyridine (DMAP), thriethylamine, N-hydroxysuccinimide (NHS); (e) DIPEA, diglycine; (f) EDC, DIPEA, 1-hydroxybenzotriazole (HOBt) and diamine 5; (g) TFA in dichloromethane; (h) EDC, HOBt, DIPEA; (i) CHCl3/Et2O saturated with HCl; (j) NaHCO3 (5% aq.), extraction with ethyl acetate. BOC—t-butoxycarbonyl.
2.2. Anion Binding Studies
Binding properties of receptors towards different anions were investigated by UV-Vis, fluorescence and NMR titrations in an acetonitrile solution. The receptors bear an anthranilic acid moiety, which exhibits fluorescence emission at 400 nm upon excitation at 250 or 328 nm. Thus, we could use two complementary spectrophotometric methods to determine binding constants for anions. The fitting of binding isotherms was carried out with HypSpec Program [
20]. As inferred from the dilution experiments the host molecules do not form associates at concentrations used in studies.
Receptor
1 has an acyclic structure and it was expected from the beginning that it could bind all anions in question. However, chloride and nitrate induced very small changes in UV-Vis spectra so that fitting of the data was not possible. In spite of this fact, the chloride anion induced considerable changes in fluorescence titrations and the determined affinity constant was 520 M
−1. Receptor
1 showed high affinity for dihydrogen phosphate, hydrogen sulfate and acetate. For the latter two anions we observed a stepwise binding mode—1:1 and 1:2 (receptor:anion). The Job’s plot analysis confirmed this stoichiometry (see Supporting information). Interestingly, receptor
1 binds two dihydrogen phosphate anions almost simultaneously, as inferred from the fitting model. The first binding event with a 1:1 stoichiometry is likely too weak to assess it by the curve-fitting analysis. Additional proof of a 1:2 binding mode was obtained from the NMR titration experiment. The affinity constant for the binding of two dihydrogen phosphate anions to receptor
1 was logβ
12 = 8.98(10) (
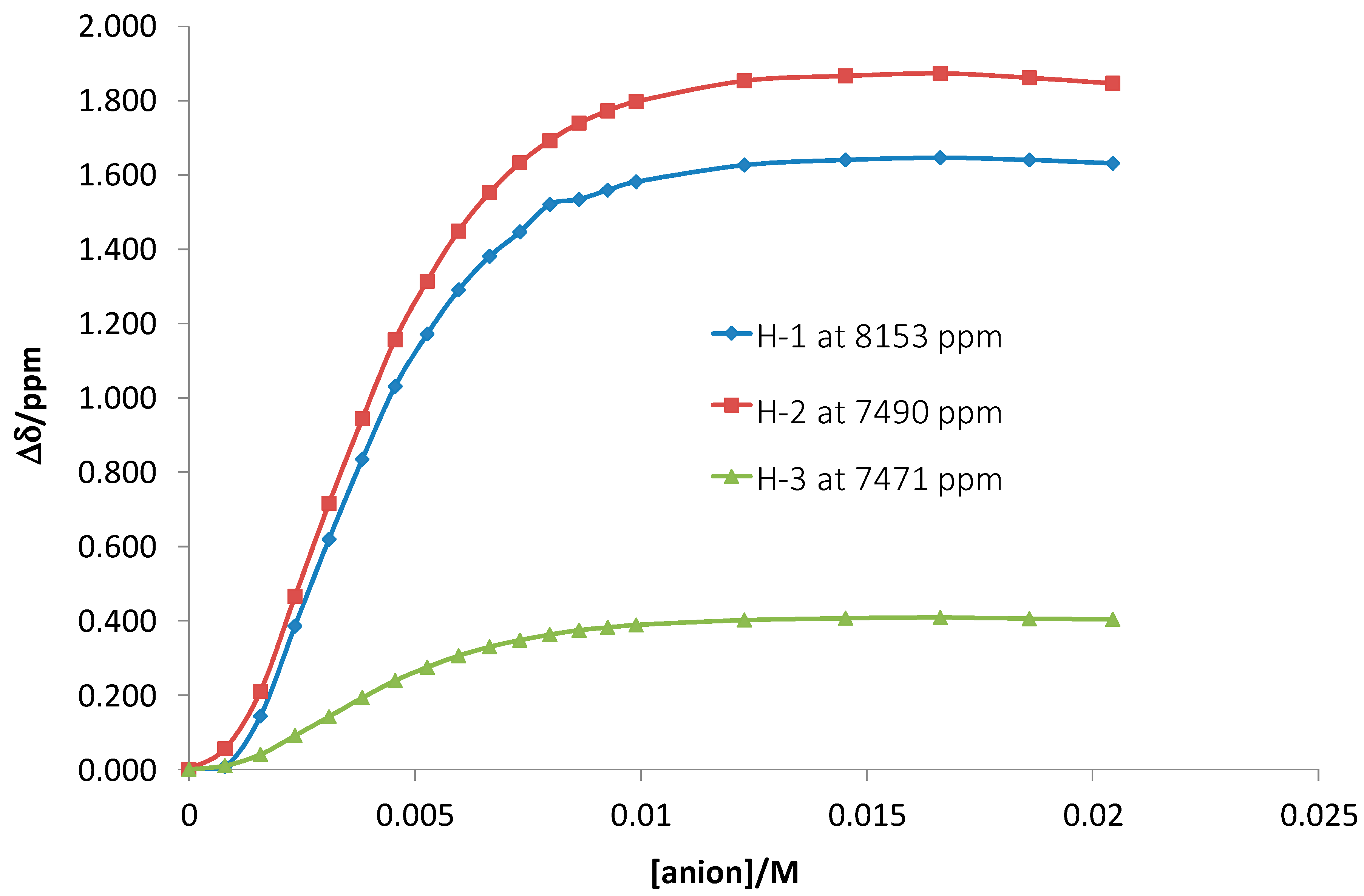
Figure 3). This value is in agreement with the value obtained by using spectrophotometric measurements. Interestingly, the strongest shifts were observed for amide protons H1 (the anthranilic acid fragment) and H2 (the phenylenediamine fragment) belonging to aromatic systems. Smaller shifts were detected for amide protons of the amino acids (e.g., H3).
Figure 3.
1H-NMR chemically induced resonance shifts upon addition of tetrabutylammonium dihydrogen phosphate to receptor 1. H1 is the amide-NH of the anthranilic acid, H2 is the amide-NH connected to the phenylenediamine. H3 is the amide-NH of one of the amino acid residues.
Figure 3.
1H-NMR chemically induced resonance shifts upon addition of tetrabutylammonium dihydrogen phosphate to receptor 1. H1 is the amide-NH of the anthranilic acid, H2 is the amide-NH connected to the phenylenediamine. H3 is the amide-NH of one of the amino acid residues.
Binding constants obtained by fluorescence measurements were in a good agreement with those obtained by UV-Vis titrations (
Table 1). As a rule, all anions enhance the fluorescence of
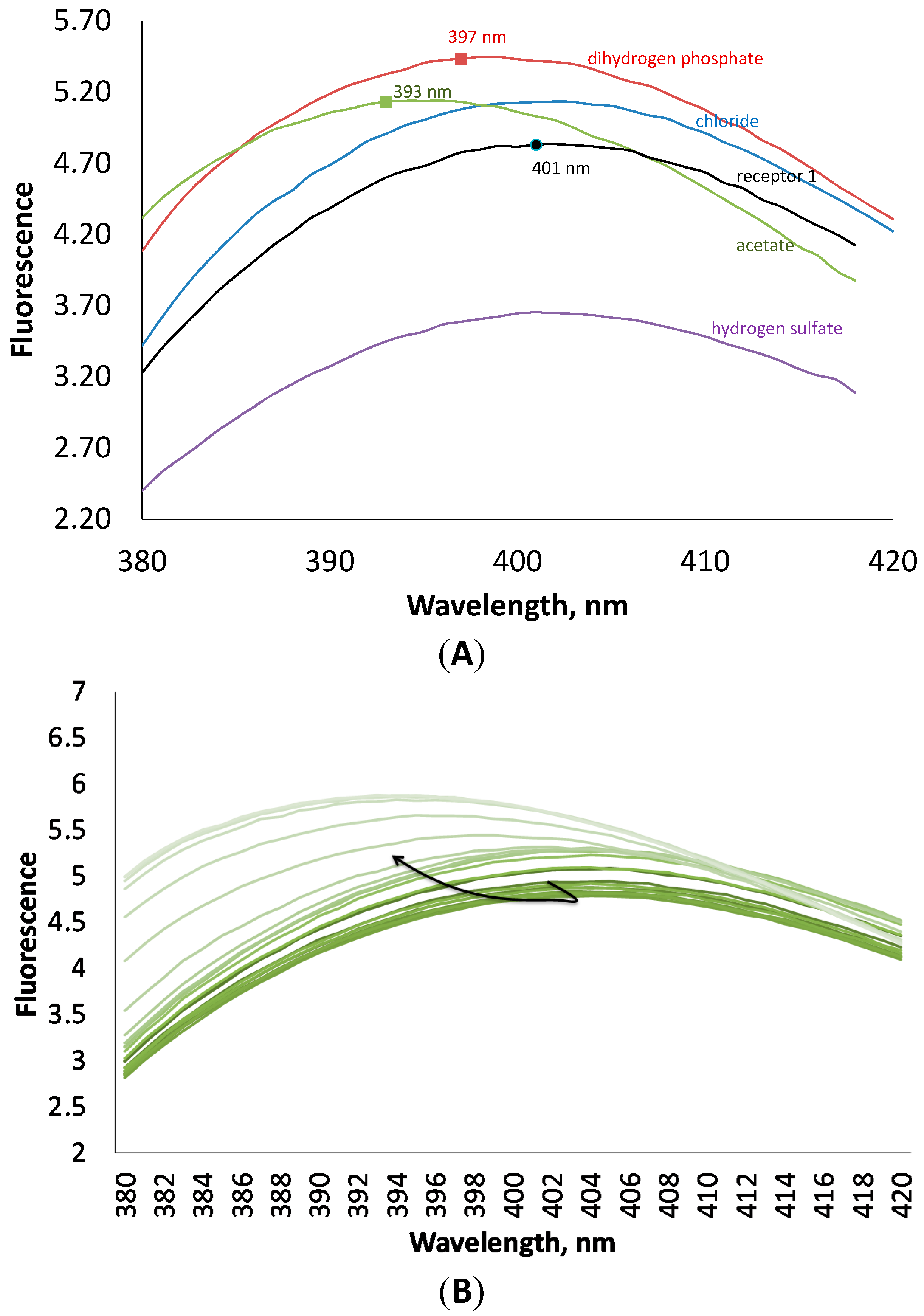
1, except hydrogen sulfate. The latter anion quenches the fluorescence of the receptor. The addition of anions induced also a small hypsochromic shift (
Figure 4). These interesting changes in fluorescence can be explained in terms of strong interactions through hydrogen bonds between the anions and the anthranilic acid moiety, which is responsible for fluorescence.
Figure 4.
(A) Fluorescence spectra of receptor 1 at 10−4 M concentration in the presence of 4 equivalents of different anions in an acetonitrile solution. (B) Changes in fluorescence of 1 upon addition of tetrabutylammonium dihydrogen phosphate.
Figure 4.
(A) Fluorescence spectra of receptor 1 at 10−4 M concentration in the presence of 4 equivalents of different anions in an acetonitrile solution. (B) Changes in fluorescence of 1 upon addition of tetrabutylammonium dihydrogen phosphate.
Table 1.
Association constants logK of receptor 1 with different anions in an acetonitrile solution measured at 25 °C. a Small changes were observed upon the addition of the anion, so that it was not possible to fit the data; b Calculation of the first binding event was not possible due to the high fitting error.
Table 1.
Association constants logK of receptor 1 with different anions in an acetonitrile solution measured at 25 °C. a Small changes were observed upon the addition of the anion, so that it was not possible to fit the data; b Calculation of the first binding event was not possible due to the high fitting error.
| Anion | UV-Vis | Fluorescence |
|---|
| Cl− | - a | logβ11 = 2.72 ± 0.01 a |
| CH3COO− | logK11 = 5.51 ± 0.08
logK12 = 3.99 ± 0.06 | logK11 = 5.00 ± 0.06
logK12 = 3.93 ± 0.06 |
| H2PO4− | logβ12 = 7.85 ± 0.01
- b | logβ12 = 7.95 ± 0.03
- b |
| HSO4− | logK11 = 6.40 ± 0.08
logK12 = 2.88 ± 0.06 | logK11 = 6.00 ± 0.04
logK12 = 4.87 ± 0.05 |
According to the titration experiments, receptor 2 binds anions stepwise in 1:1 and 1:2 modes, except for chloride. This behavior is similar to that observed for receptor 1. While for receptor 1 we observed almost no changes in UV-Vis titrations after addition of chloride, in case of the macrocyclic receptor chloride induced considerable changes in spectra so that we could determine the affinity constant. Interestingly the affinities of 1 and 2 for chloride are the same. All these facts indicate that the coordination of chloride induces considerable changes in the conformation of the macrocyclic receptor, likely orienting NH-donor groups towards the chloride anion. Because the affinity constant for chloride is much less, than that for phosphate, one can suggest that the orientation of NH-donors fits better the geometry of phosphate.
While the acyclic receptor did not show selectivity for a particular oxyanion, the macrocyclic receptor has a remarkable preference to bind dihydrogen phosphate selectively. This can be better seen from the comparison of the cumulative affinity constants for anions, e.g., for acetate logβ
12 is 8.46, for dihydrogen phosphate logβ
12 is 10.51 (
Table 2). Thus, the macrocyclic structure allowed us to achieve selectivity for dihydrogen phosphate in an acetonitrile solution. Interestingly, hydrogen sulfate induced no changes in fluorescence but small changes in UV-Vis. By using the latter method, we were able to determine the affinity constant for hydrogen sulfate, which is two orders of magnitude lower than that for dihydrogen phosphate.
Table 2.
Association constants logK of receptor 2 with different anions in an acetonitrile solution measured at 25 °C. a Small changes were observed upon the addition of the anion, so that it was not possible to fit the data.
Table 2.
Association constants logK of receptor 2 with different anions in an acetonitrile solution measured at 25 °C. a Small changes were observed upon the addition of the anion, so that it was not possible to fit the data.
| Anion | UV-Vis | Fluorescence |
|---|
| Cl− | logK11 = 2.70 ± 0.003 | logK11 = 2.72 ± 0.01 |
| CH3COO− | logK11 = 4.63 ± 0.006
logK12 = 3.83 ± 0.09 | logK11 = 4.60 ± 0.10
logK12 = 4.18 ± 0.03 |
| H2PO4− | logK11 = 6.42 ± 0.07
logK12 = 4.09 ± 0.07 | logK11 = 6.00 ± 0.05
logK12 =4.16 ± 0.05 |
| HSO4− | logK11 = 3.94 ± 0.02
logK12 = 2.84 ± 0.05 | -
a |
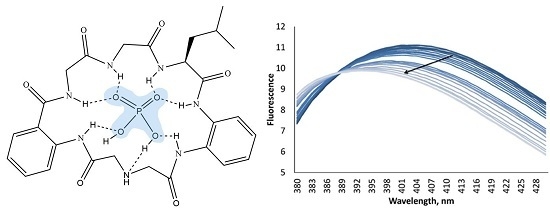
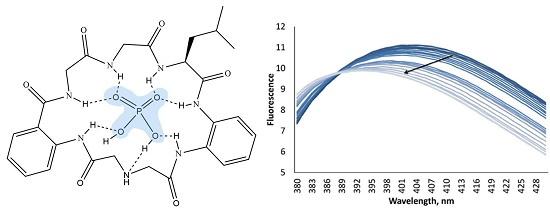
According to fluorescence measurements, receptor
2 demonstrated a different fluorometric answer towards anions, as compared to receptor
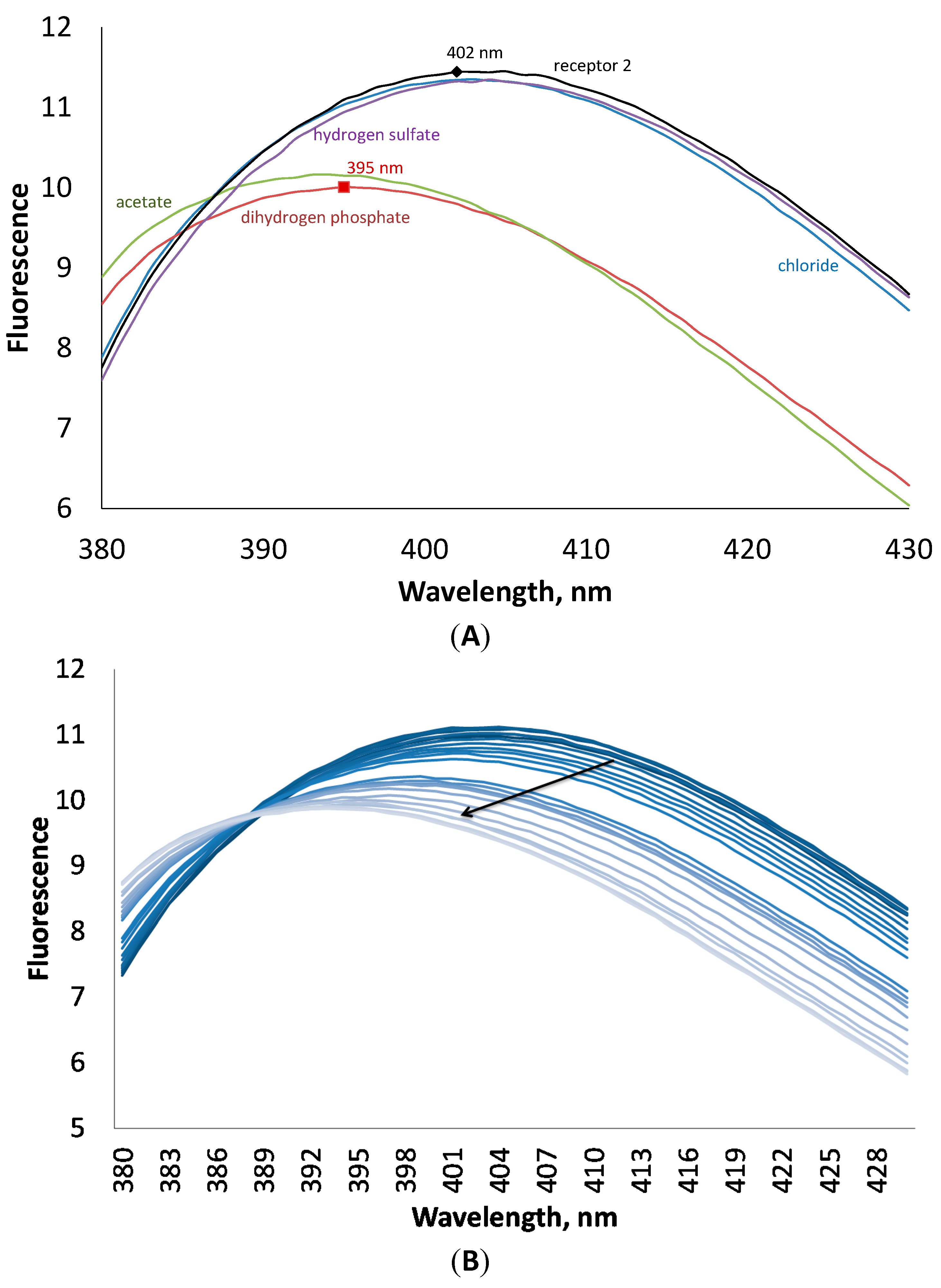
1. The addition of anions induced quenching of fluorescence. As can be seen in
Figure 5, the addition of four equivalents of chloride and hydrogen sulfate has almost no influence on the fluorescence of the host, while acetate and dihydrogen phosphate decrease the fluorescence intensity and shift the maximum towards lower wavelength numbers on 7 nm. This interesting behavior of the receptor to shift the fluorescence maximum upon addition of anions can be investigated in future studies in details for the design of ratiometric probes for anions [
21].
Figure 5.
(A) Fluorescence spectra of receptor 2 at 10−4 M concentration in the presence of 4 equivalents of different anions in an acetonitrile solution. (B) Changes in fluorescence of 2 upon addition of tetrabutylammonium dihydrogen phosphate.
Figure 5.
(A) Fluorescence spectra of receptor 2 at 10−4 M concentration in the presence of 4 equivalents of different anions in an acetonitrile solution. (B) Changes in fluorescence of 2 upon addition of tetrabutylammonium dihydrogen phosphate.
The property of receptors
1 and
2 to bind two anions, though the cavity does not allow this, is rather unusual. However, this behavior can have three different reasons. First, the coordination of two dihydrogen phosphate anions can be explained by dimerization of the phosphate anion in the presence of a synthetic receptor through the P-O…H-O-P hydrogen bond. Such a precedent has been already reported in the literature [
22]; Second, HSO
4− and H
2PO
4− anions have protons, which can be transferred to free amino groups during the anion coordination enabling the binding of the second anion to the positively charged receptor. This will lead to the formation of very stable complexes, in which the receptor and an anion have both electrostatic and hydrogen bonding interactions; Third, the receptor has a conformation, in which two pairs of donor NH-groups are oriented in different directions so that two anions could be bound from different sides. Such a situation has been observed for the urea-based receptors, which bind two acetate anions stepwise [
23] and for the phosphate receptor reported by us earlier [
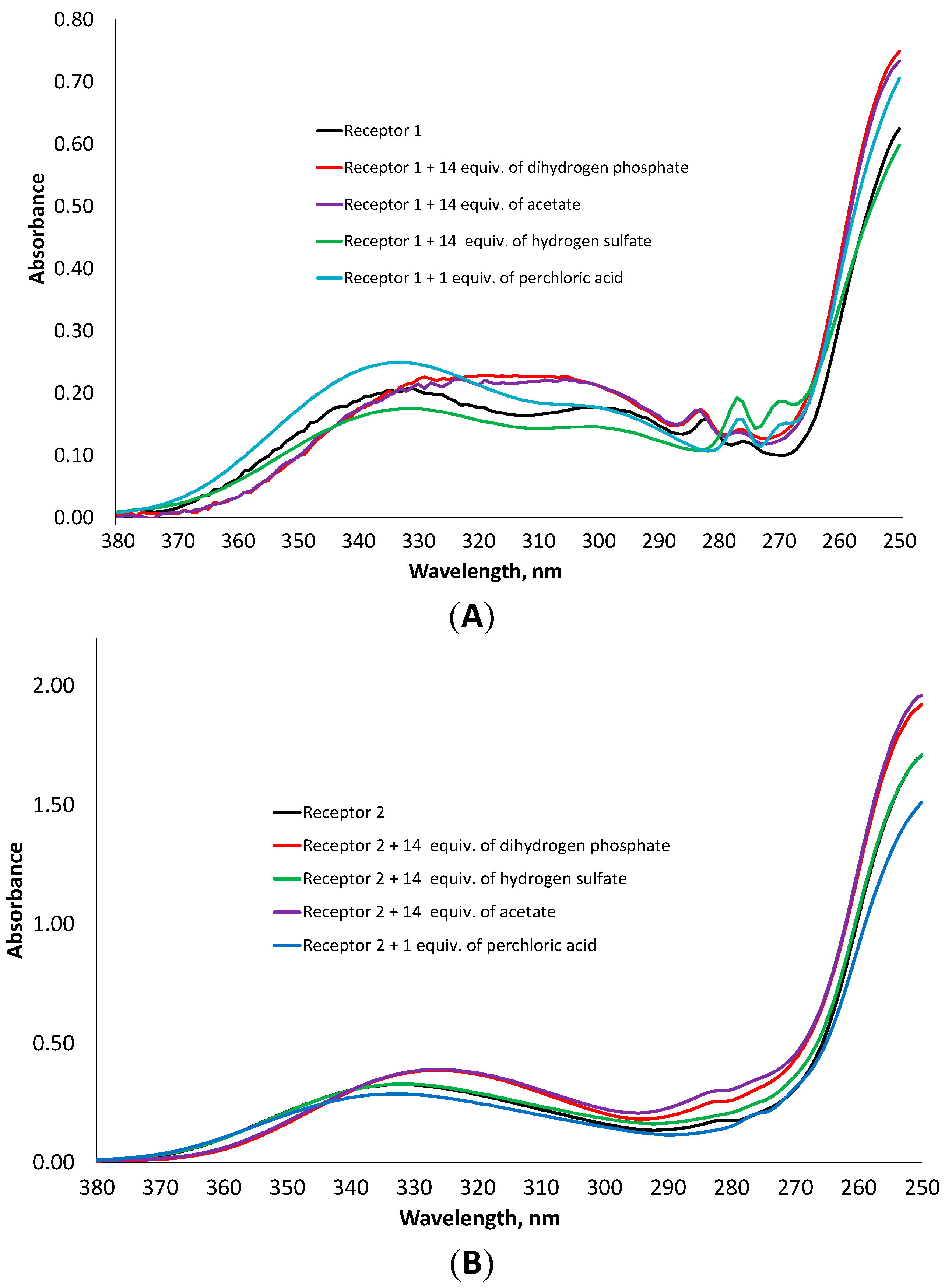
24]. In order to understand the binding of anions by the synthesized receptors, we added to a solution of the receptor one equivalent of perchloric acid in acetonitrile, measured UV-Vis spectrum and compared this spectrum with those obtained after addition of different oxyanions. According to UV-Vis and fluorescence measurements, the receptors do not coordinate the perchlorate anion, therefore the addition of perchloric acid can be considered as a pure protonation effect. As can be seen in
Figure 6, the spectrum of the protonated receptor
1 has the same structure as that obtained by the addition of hydrogen sulfate. An increase in 277 nm band and a decrease in 285 nm band were observed. In case of receptor
2, the addition of the acid or hydrogen sulfate leads to the disappearance of a small shoulder at 282 nm, while the addition of dihydrogen phosphate or acetate induces a considerable hypsochromic shift.
Figure 6.
Comparison of UV-Vis spectra of receptor 1 (A) and 2 (B) in the presence of one equivalent of HClO4 and 14 equivalents of dihydrogen phosphate, hydrogen sulfate and acetate, respectively.
Figure 6.
Comparison of UV-Vis spectra of receptor 1 (A) and 2 (B) in the presence of one equivalent of HClO4 and 14 equivalents of dihydrogen phosphate, hydrogen sulfate and acetate, respectively.
These facts indicate that both receptors are protonated after addition of HSO
4−, but not after addition of dihydrogen phosphate, acetate or chloride. The interaction of hydrogen sulfate with the receptors leads to the protonation so that the resulting SO
42− anion can be also located outside the cavity. Such a situation was often observed in the solid state for positively-charged cryptand receptors [
25]. Analysis of NMR spectra of receptor
1 and
2 in the presence of anions in a CD
3CN solution supports this hypothesis. The binding of dihydrogen phosphate or acetate leads to sharpening of the proton signals, while binding of hydrogen sulfate induces broadening of both NH and CH signals (see Supporting Information).
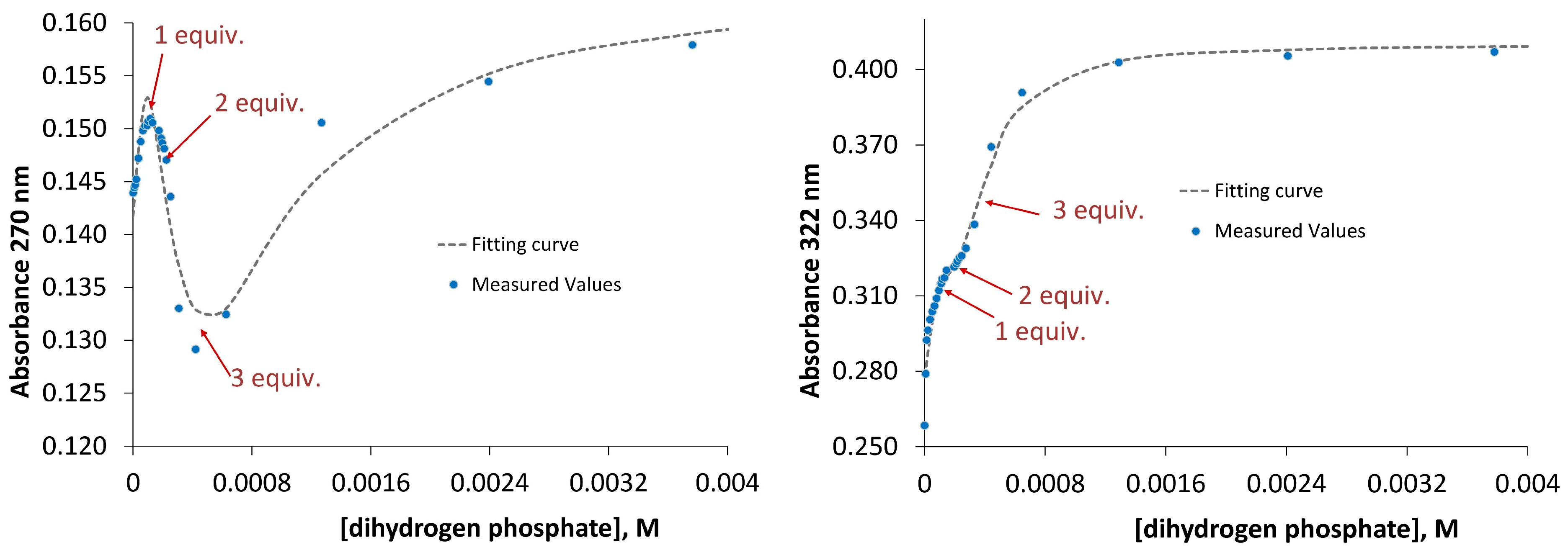
In order to explain a 1:2 stoichiometry of oxyanion binding we proposed that both receptors possess a specific conformation that enables binding of two anions. To prove our suggestion we protonated the receptors with one equivalent of perchloric acid and titrated the resulting salts with dihydrogen phosphate anion. If a receptor is able to accommodate only one anion then the corresponding positively charged receptor should bind two dihydrogen phosphate anions because it is added in excess and one anion will be located in the binding cavity and the other—outside the cavity stabilized by electrostatic interactions. If a receptor is able to accommodate two anions then the corresponding positively charged receptors should bind three dihydrogen phosphate anions. Exactly the latter situation we observed in UV-Vis titrations of salts
1•HClO
4 and
2•HClO
4 with tetrabutylammonium dihydrogen phosphate. As can be seen in
Figure 7 the titration curve changes its direction almost exactly at points 1, 2 and 3 equivalents of dihydrogen phosphate, respectively. Fitting of the experimental data with HypSpec Program resulted in cumulative association constants β
13(for
1) = 12.82 ± 0.01 and β
13(for
2) = 13.69 ± 0.01 (
Figure 7).
Figure 7.
UV-Vis titration data and fitting curves obtained for the addition of dihydrogen phosphate to the mixture of receptor 1 (left) and receptor 2 (right) with one equivalent of perchloric acid.
Figure 7.
UV-Vis titration data and fitting curves obtained for the addition of dihydrogen phosphate to the mixture of receptor 1 (left) and receptor 2 (right) with one equivalent of perchloric acid.
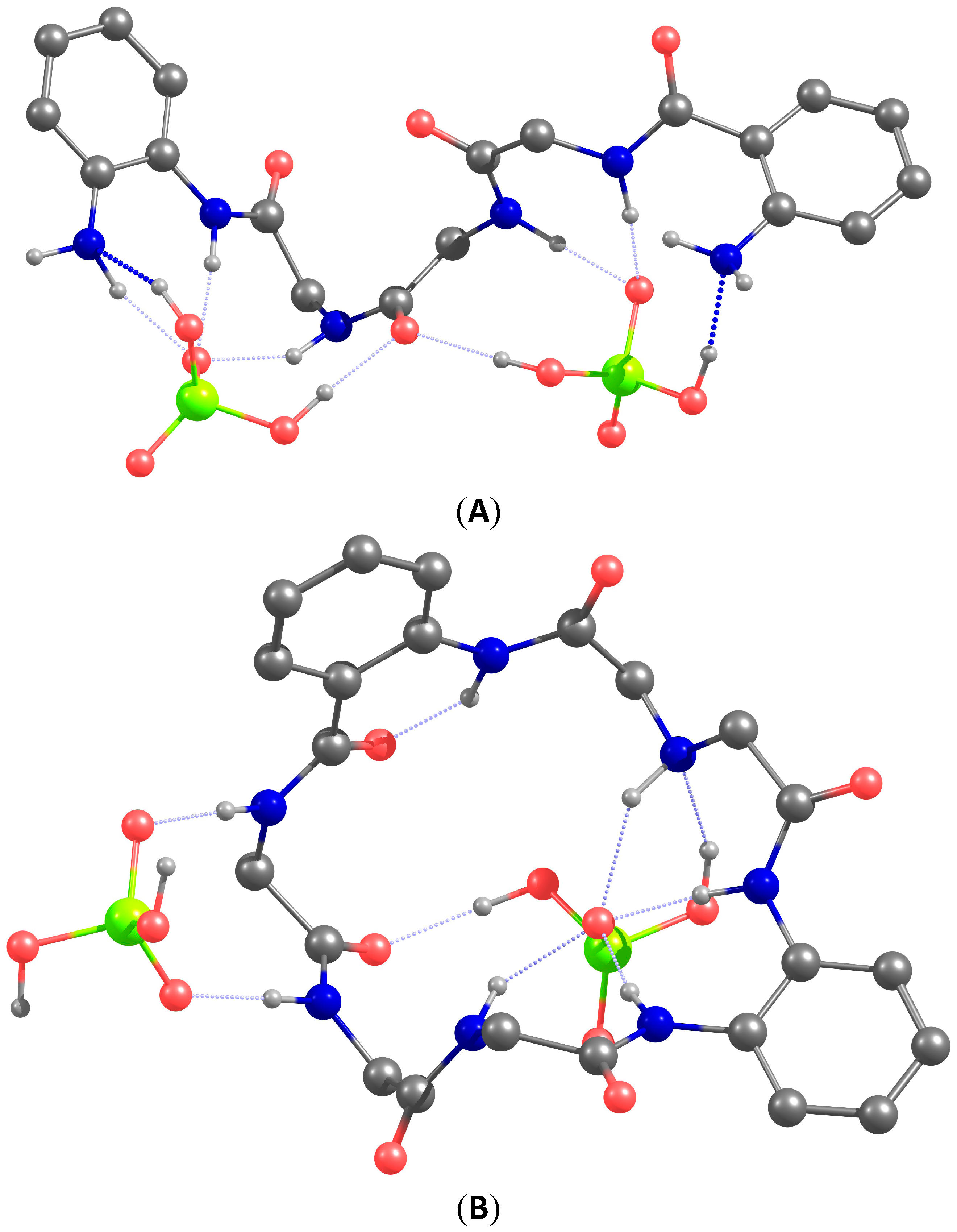
To get an understanding how two dihydrogen phosphate anions can be coordinated by our receptors we carried our DFT modeling of the complexes. The optimized structures of complexes
1•(H
2PO
4−)
2 and
2•(H
2PO
4−)
2 are depicted in
Figure 8. The following conclusions can be drawn from the analysis of the structures of the optimized host-guest complexes. In the acyclic receptor one amino acid subunit bearing two NH-sites coordinates one oxygen atom of phosphate. This binding mode is similar to that of phosphate coordination by the kinase (
Figure 1). Interestingly, in both complexes the amide oxygen atoms play a role of hydrogen bond acceptor, as well as amino groups, which facilitates the coordination of the protonated phosphate anion. Macrocyclic receptor has a rigid structure and coordinates two dihydrogen phosphate anions from different sides. One of the anions is stabilized only with two NH-sites. This coordination mode hints how two acetate or two sulfate anions can be bound to the receptor. Unexpected for us was the fact that the phenylenediamine subunit forms an almost planar arrangement of NH-sites, which bind one oxygen atom of the phosphate anions. This arrangement explains the affinity of the receptor for the chloride anion.
Figure 8.
Calculated structures of complexes 1•(H2PO4−)2 (A) and 2•(H2PO4−)2 (B). The isopropyl substituent and most of the hydrogen atoms were omitted for clarity. The atoms are shown in different color Red: oxygen; green: phosphorus; blue: nitrogen; gray: carbon.
Figure 8.
Calculated structures of complexes 1•(H2PO4−)2 (A) and 2•(H2PO4−)2 (B). The isopropyl substituent and most of the hydrogen atoms were omitted for clarity. The atoms are shown in different color Red: oxygen; green: phosphorus; blue: nitrogen; gray: carbon.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}



