
σ- versus π-Activation of Alkynyl Benzoates Using B(C6F5)3


Abstract
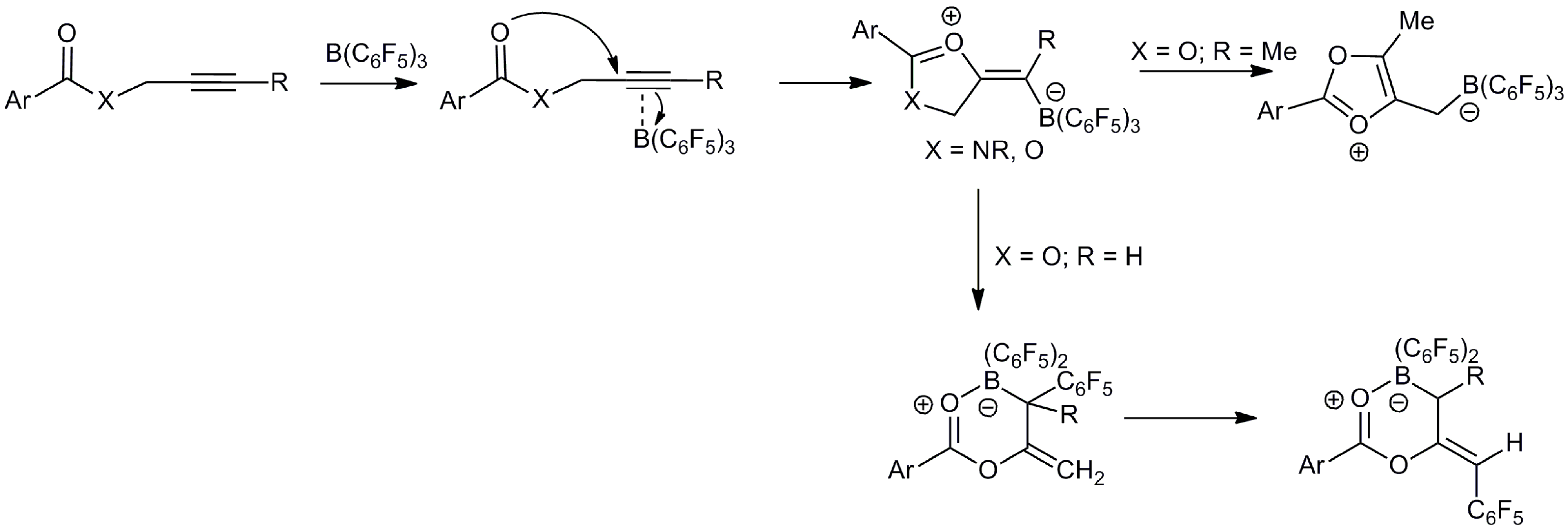
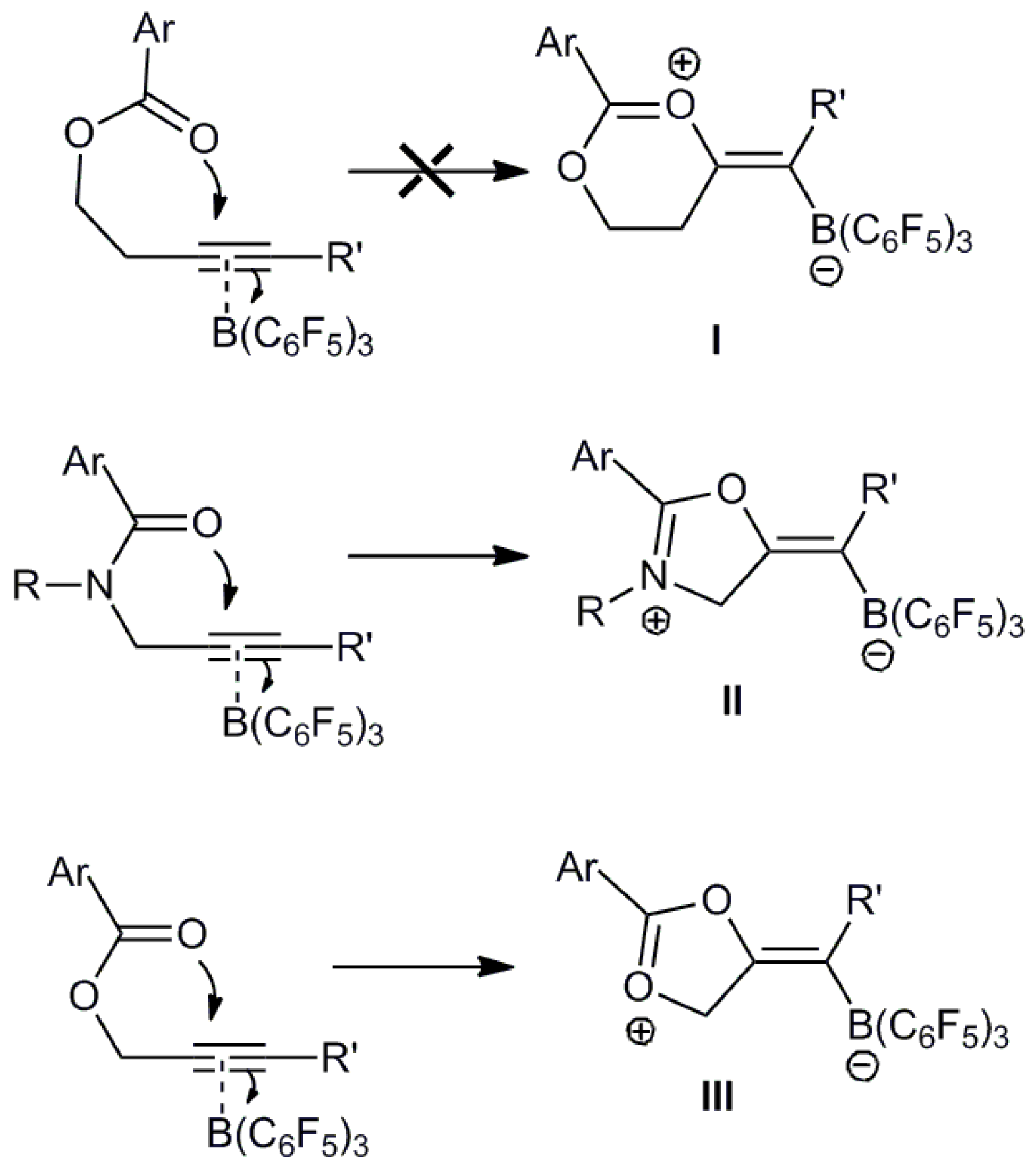
:1. Introduction

2. Results and Discussion





| X | IR Carbonyl Stretching Frequency (cm−1) | ||
|---|---|---|---|
| νCO (1) | νCO (2) | ΔνCO | |
| H (a) | 1717 | 1647 | 70 |
| Me (b) | 1717 | 1647 | 70 |
| OMe (c) | 1713 | 1645 | 68 |
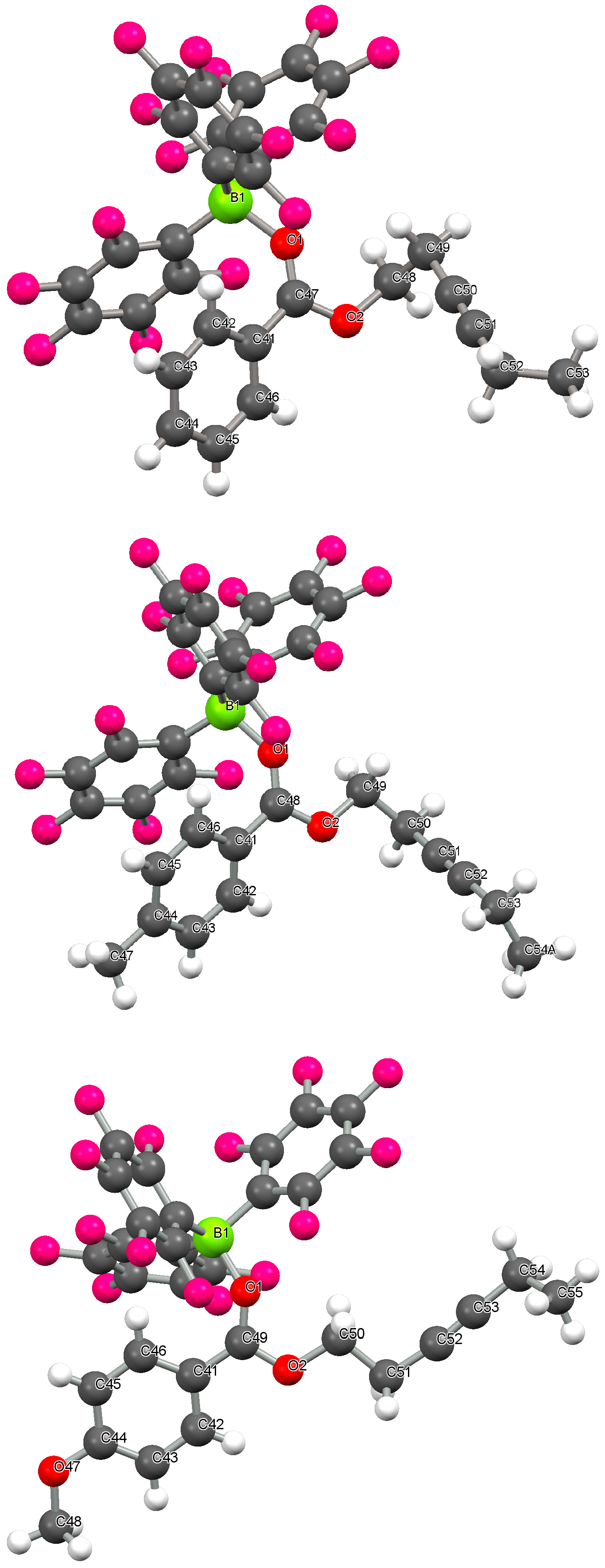
2.1. Crystallographic Studies


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | |||
|---|---|---|---|
| 2a | 2b | 2c | |
| B(1)-O(1)/Å | 1.589(2) | 1.585(2) | 1.565(2) |
| C(1)-O(1)/Å | 1.247(2) | 1.255(2) | 1.255(2) |
| C(2)-C(1) bond length/Å | 1.474(2) | 1.462(2) | 1.451(2) |
| C(1)-O(1)-B(1) angle/° | 135.5(1) | 135.8(1) | 138.4(1) |
| C(2)-C(1)-O(1)-B(1) dihedral angle/° | 23.18 | 29.62 | 33.80 |
| C(3)-C(2)-C(1)-O(1) dihedral angle/° | 32.54 | 31.12 | 13.02 |
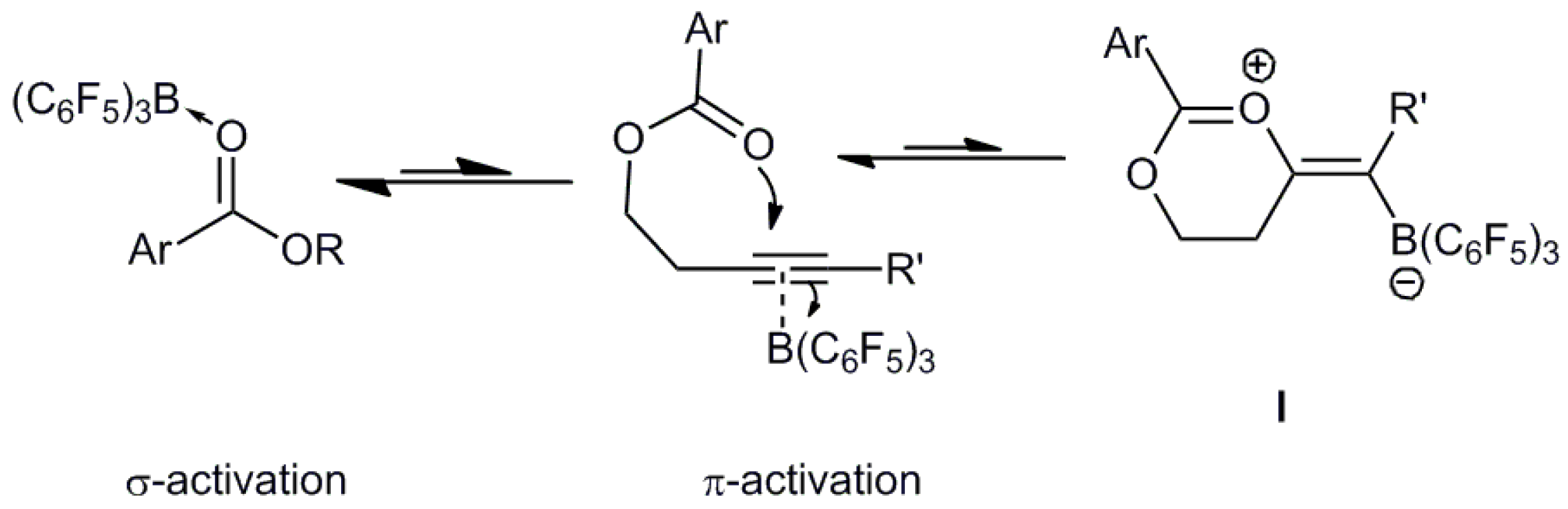
2.2. Computational Studies
| Ester | C=O/Å | Adduct | C-O/Å | B-O/Å | ∆Hadduct/kJ·mol−1 |
|---|---|---|---|---|---|
| 1a | 1.21 | 2a | 1.25 | 1.61 | −8 |
| 1b | 1.21 | 2b | 1.25 | 1.60 | −14 |
| 1c | 1.21 | 2c | 1.25 | 1.60 | −17 |
2.3. Effect of Temperature


3. Experimental Section
3.1. General Information
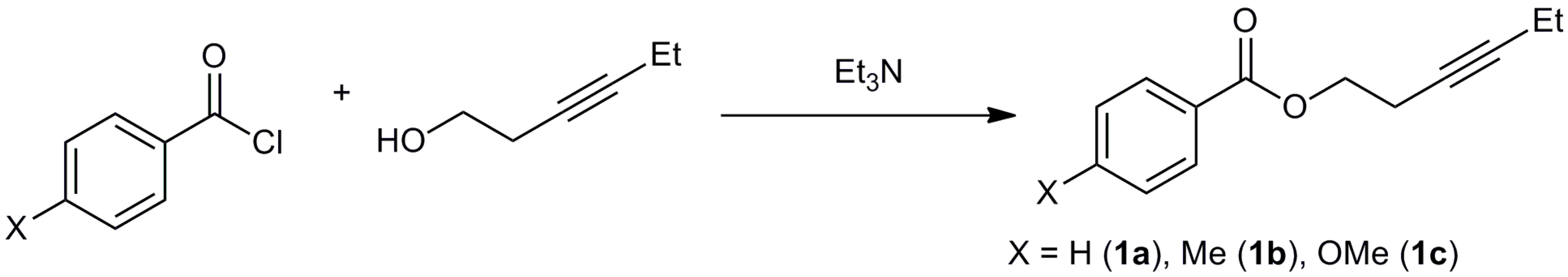
3.2. Synthesis of Starting Materials
3.2.1. Synthesis of Hex-3-yn-1-yl benzoate (1a)
3.2.2. Synthesis of Hex-3-yn-1-yl 4-methylbenzoate (1b)
3.2.3. Synthesis of Hex-3-yn-1-yl 4-methoxybenzoate (1c)
3.2.4. Synthesis of Trispentafluorophenylborane, B(C6F5)3
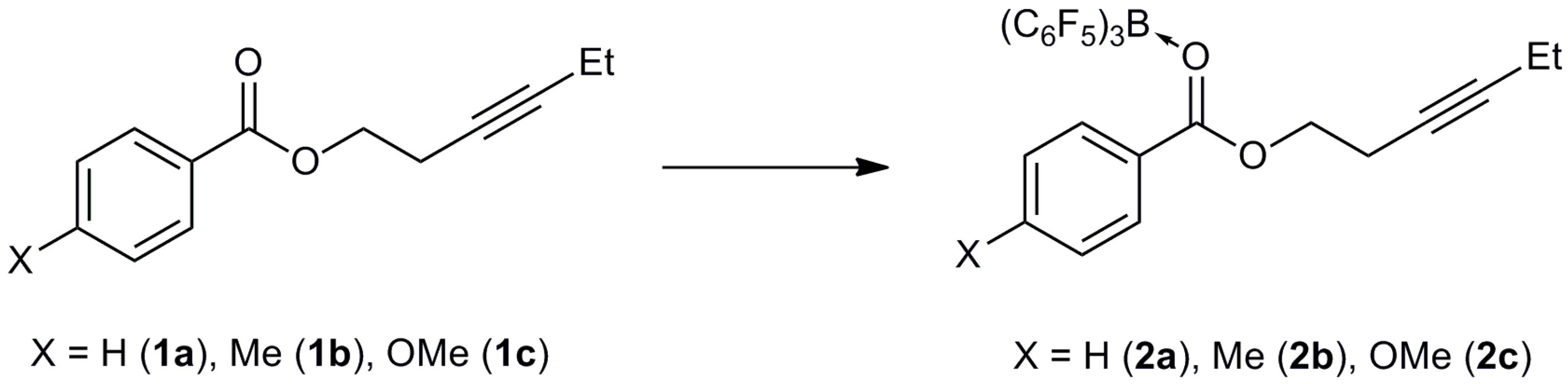
3.3. Synthesis of Adducts
3.3.1. Synthesis of 2a
3.3.2. Synthesis of 2b
3.3.3. Synthesis of 2c
3.4. In Situ NMR Studies of Varying Concentration
3.4.1. NMR Experiments of 2a
3.4.2. NMR Experiments of 2b
3.4.3. NMR Experiments of 2c
3.5. In Situ NMR Studies of Varying Stoichiometry
3.5.1. Excess 2b
3.5.2. Excess B(C6F5)3
3.6. Crystallographic Studies
| Compound | 2a | 2b | 2c |
|---|---|---|---|
| Formula | C31H14BF15O2 * | C32H16BF15O2 | C32H16BF15O3 |
| M | 714.23 | 728.26 | 744.26 |
| Crystal System | Triclinic | Triclinic | Triclinic |
| Space Group | P-1 | P-1 | P-1 |
| a | 10.9048(8) | 10.5395(4) | 11.1660(5) |
| b | 11.4463(6) | 11.3370(4) | 12.5550(5) |
| c | 13.7289(6) | 13.2062(4) | 12.6939(4) |
| α | 84.827(4) | 102.149(3) | 78.409(3) |
| β | 74.262(5) | 97.888(3) | 68.678(4) |
| γ | 63.545(6) | 94.962(3) | 65.281(4) |
| V | 1475.75(16) | 1517.15(9) | 1503.32(13) |
| T/K | 150(2) | 200(2) | 150(2) |
| Z | 2 | 2 | 2 |
| Dc | 1.607 | 1.594 | 1.644 |
| θmin, θmax | 4.264–73.720 | 3.468–74.078 | 3.744–73.995 |
| Crystal size | 0.26 × 0.08 × 0.07 | 0.33 × 0.28 × 0.23 | 0.46 × 0.34 × 0.12 |
| μ/mm−1 | 1.479 | 1.451 | 1.505 |
| F(000) | 712 | 728 | 744 |
| Total Reflections | 9959 | 10254 | 10549 |
| Independent Reflections | 5788 | 5926 | 5876 |
| Rint | 0.0211 | 0.0164 | 0.0164 |
| R1 (I > 2s(I)) | 0.0349 | 0.0439 | 0.0323 |
| wR2 (all data) | 0.1043 | 0.1377 | 0.0945 |
| S | 1.020 | 1.045 | 1.013 |
| Min/max e−/Å3 | +0.28/−0.27 | +0.78/−0.31 | +0.30/−0.24 |
3.7. Computational Studies
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Massey, A.G.; Park, A.J.; Stone, F.G.A. Tris(pentafluorophenyl)boron. Proc. Chem. Soc. 1963, 212. [Google Scholar]
- Massey, A.G.; Park, A.J. Tris(pentafluorophenyl)boron. J. Organomet. Chem. 1964, 2, 245–250. [Google Scholar] [CrossRef]
- Yang, X.; Stern, C.L.; Marks, T.J. Cationic Zirconocene Olefin Polymerization Catalysts Based on the Organo-Lewis Acid Tris(pentafluorophenyl)borane. A Synthetic, Structural, Solution Dynamic, and Polymerization Catalytic Study. J. Am. Chem. Soc. 1994, 116, 10015–10031. [Google Scholar] [CrossRef]
- Parks, D.J.; Piers, W.E. Tris(pentafluorophenyl)boron-Catalyzed Hydrosilation of Aromatic Aldehydes, Ketones, and Esters. J. Am. Chem. Soc. 1996, 118, 9440–9441. [Google Scholar] [CrossRef]
- Piers, W.E.; Chivers, T. Pentafluorophenylboranes: From obscurity to applications. Chem. Soc. Rev. 1997, 26, 345–354. [Google Scholar] [CrossRef]
- Blackwell, J.M.; Foster, K.L.; Beck, V.H.; Piers, W.E. B(C6F5)3-Catalyzed Silation of Alcohols: A Mild, General Method for Synthesis of Silyl Ethers. J. Org. Chem. 1999, 64, 4887–4892. [Google Scholar] [CrossRef] [PubMed]
- Blackwell, J.M.; Sonmor, E.R.; Scoccitti, T.; Piers, W.E. B(C6F5)3-Catalyzed Hydrosilation of Imines via Silyliminium Intermediates. Org. Lett. 2000, 2, 3921–3923. [Google Scholar]
- Roesler, R.; Har, B.J.N.; Piers, W.E. Synthesis and Characterization of (Perfluoroaryl)borane-Functionalized Carbosilane Dendrimers and Their Use as Lewis Acid Catalysts for the Hydrosilation of Acetophenone. Organometallics 2002, 21, 4300–4302. [Google Scholar] [CrossRef]
- Piers, W.E. The chemistry of perfluoroaryl boranes. Adv. Organomet. Chem. 2004, 52, 1–76. [Google Scholar]
- Erker, G. Tris(pentafluorophenyl)borane: A special boron Lewis acid for special reactions. Dalton Trans. 2005, 11, 1883–1890. [Google Scholar] [CrossRef] [PubMed]
- Rendler, S.; Oestreich, M. Conclusive Evidence for an SN2-Si Mechanism in the B(C6F5)3-Catalyzed Hydrosilylation of Carbonyl Compounds: Implications for the Related Hydrogenation. Angew. Chem. Int. Ed. 2008, 47, 5997–6000. [Google Scholar] [CrossRef]
- Piers, W.E.; Marwitz, A.J.V.; Mercier, L.G. Mechanistic Aspects of Bond Activation with Perfluoroarylboranes. Inorg. Chem. 2011, 50, 12252–12262. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. “Frustrated Lewis pairs”: A concept for new reactivity and catalysis. Org. Biomol. Chem. 2008, 6, 1535–1539. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. Frustrated Lewis pairs: A new strategy to small molecule activation and hydrogenation catalysis. Dalton Trans. 2009, 3129–3136. [Google Scholar] [CrossRef]
- Stephan, D.W. Activation of dihydrogen by non-metal systems. Chem. Commun. 2010, 46, 8526–8533. [Google Scholar] [CrossRef]
- Stephan, D.W.; Erker, G. Frustrated Lewis Pairs: Metal-free Hydrogen Activation and More. Angew. Chem. Int. Ed. 2010, 49, 46–76. [Google Scholar] [CrossRef]
- Stephan, D.W. “Frustrated Lewis pair” hydrogenations. Org. Biomol. Chem. 2012, 10, 5740–5746. [Google Scholar] [CrossRef] [PubMed]
- Stephan, D.W. Frustrated Lewis Pairs: From Concept to Catalysis. Acc. Chem. Res. 2015, 48, 306–316. [Google Scholar] [CrossRef] [PubMed]
- Topics in Current Chemistry: Frustrated Lewis Pairs I; Stephan, D.W.; Erker, G. (Eds.) Springer Press: New York, NY, USA, 2013.
- Topics in Current Chemistry: Frustrated Lewis Pairs II; Stephan, D.W.; Erker, G. (Eds.) Springer Press: New York, NY, USA, 2013.
- Voss, T.; Chen, C.; Kehr, G.; Nauha, E.; Erker, G.; Stephan, D.W. Cyclizations via Frustrated Lewis Pairs: Lewis Acid Induced Intramolecular Additions of Amines to Olefins and Alkynes. Chem. Eur. J. 2010, 16, 3005–3008. [Google Scholar] [CrossRef] [PubMed]
- Voss, T.; Mahdi, T.; Otten, E.; Fröhlich, R.; Kehr, G.; Stephan, D.W.; Erker, G. Frustrated Lewis Pair Behavior of Intermolecular Amine/B(C6F5)3 Pairs. Organometallics 2012, 31, 2367–2378. [Google Scholar] [CrossRef]
- McCahill, J.S.J.; Welch, G.C.; Stephan, D.W. Reactivity of “Frustrated Lewis Pairs”: Three-Component Reactions of Phosphines, a Borane, and Olefins. Angew. Chem. Int. Ed. 2007, 46, 4968–4971. [Google Scholar] [CrossRef]
- Chen, C.; Fröhlich, R.; Kehr, G.; Erker, G. Remarkably variable reaction modes of frustrated Lewis pairs with non-conjugated terminal diacetylenes. Chem. Commun. 2010, 46, 3580–3582. [Google Scholar] [CrossRef]
- Liedtke, R.; Fröhlich, R.; Kehr, G.; Erker, G. Frustrated Lewis Pair Reactions with Bis-Acetylenic Substrates: Exploring the Narrow Gap Separating Very Different Competing Reaction Pathways. Organometallics 2011, 30, 5222–5232. [Google Scholar] [CrossRef]
- Dureen, M.A.; Stephan, D.W. Terminal Alkyne Activation by Frustrated and Classical Lewis Acid/Phosphine Pairs. J. Am. Chem. Soc. 2009, 131, 8396–8397. [Google Scholar] [CrossRef] [PubMed]
- Dureen, M.A.; Brown, C.C.; Stephan, D.W. Addition of Enamines or Pyrroles and B(C6F5)3 “Frustrated Lewis Pairs” to Alkynes. Organometallics 2010, 29, 6422–6432. [Google Scholar] [CrossRef]
- Jiang, C.; Blacque, O.; Berke, H. Activation of Terminal Alkynes by Frustrated Lewis Pairs. Organometallics 2010, 29, 125–133. [Google Scholar] [CrossRef]
- Mömming, C.M.; Kehr, G.; Wibbeling, B.; Fröhlich, R.; Schirmer, B.; Grimme, S.; Erker, G. Formation of Cyclic Allenes and Cumulenes by Cooperative Addition of Frustrated Lewis Pairs to Conjugated Enynes and Diynes. Angew. Chem. Int. Ed. 2010, 49, 2414–2427. [Google Scholar] [CrossRef]
- Chen, C.; Eweiner, F.; Wibbeling, B.; Fröhlich, R.; Senda, S.; Ohki, Y.; Tatsumi, K.; Grimme, S.; Kehr, G.; Erker, G. Exploring the Limits of Frustrated Lewis Pair Chemistry with Alkynes: Detection of a System that Favors 1,1-Carboboration over Cooperative 1,2-P/B-Addition. Chem. Asian J. 2010, 5, 2199–2208. [Google Scholar] [CrossRef] [PubMed]
- Dureen, M.A.; Brown, C.C.; Stephan, D.W. Deprotonation and Addition Reactions of Frustrated Lewis Pairs with Alkynes. Organometallics 2010, 29, 6594–6607. [Google Scholar] [CrossRef]
- Winkelhaus, D.; Neumann, B.; Stammler, H.G.; Mitzel, N.W. Intramolecular Lewis acid–base pairs based on 4-ethynyl-2,6-lutidine. Dalton Trans. 2012, 41, 9143–9150. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Kehr, G.; Fröhlich, R.; Erker, G. Carbon−Carbon Bond Activation by 1,1-Carboboration of Internal Alkynes. J. Am. Chem. Soc. 2010, 132, 13594–13595. [Google Scholar] [CrossRef] [PubMed]
- Ekkert, O.; Kehr, G.; Fröhlich, R.; Erker, G. P−C Bond Activation Chemistry: Evidence for 1,1-Carboboration Reactions Proceeding with Phosphorus−Carbon Bond Cleavage. J. Am. Chem. Soc. 2011, 133, 4610–4616. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Voss, T.; Fröhlich, R.; Kehr, G.; Erker, G. 1,1-Carboboration of 1-Alkynes: A Conceptual Alternative to the Hydroboration Reaction. Org. Lett. 2011, 13, 62–65. [Google Scholar] [CrossRef] [PubMed]
- Möbus, J.; Bonnin, Q.; Ueda, K.; Fröhlich, R.; Itami, K.; Kehr, G.; Erker, G. The 1,1-Carboboration of Bis(alkynyl)phosphanes as a Route to Phosphole Compounds. Angew. Chem. Int. Ed. 2012, 51, 1954–1957. [Google Scholar] [CrossRef]
- Kehr, G.; Erker, G. 1,1-Carboboration. Chem. Commun. 2012, 48, 1839–1850, and references therein. [Google Scholar] [CrossRef]
- Eller, C.; Kehr, G.; Daniliuc, C.G.; Fröhlich, R.; Erker, G. Facile 1,1-Carboboration Reactions of Acetylenic Thioethers. Organometallics 2013, 32, 384–386. [Google Scholar] [CrossRef]
- Melen, R.L. Applications of pentafluorophenyl boron reagents in the synthesis of heterocyclic and aromatic compounds. Chem. Commun. 2014, 50, 1161–1174, and references therein. [Google Scholar] [CrossRef]
- Melen, R.L.; Hansmann, M.M.; Lough, A.J.; Hashmi, A.S.K.; Stephan, D.W. Cyclisation versus 1,1-Carboboration: Reactions of B(C6F5)3 with Propargyl Amides. Chem. Eur. J. 2013, 19, 11928–11938. [Google Scholar] [CrossRef]
- Hansmann, M.M.; Melen, R.L.; Rominger, F.; Hashmi, A.S.K.; Stephan, D.W. Activation of Alkynes with B(C6F5)3–Boron Allylation Reagents Derived from Propargyl Esters. J. Am. Chem. Soc. 2014, 136, 777–782. [Google Scholar] [CrossRef] [PubMed]
- Hansmann, M.M.; Melen, R.L.; Rominger, F.; Hashmi, A.S.K.; Stephan, D.W. B(C6F5)3 promoted cyclisation of internal propargyl esters: Structural characterisation of 1,3-dioxolium compounds. Chem. Commun. 2014, 50, 7243–7245. [Google Scholar] [CrossRef]
- Parks, D.J.; Piers, W.E.; Parvez, M.; Atencio, R.; Zaworotko, M.J. Synthesis and Solution and Solid-State Structures of Tris(pentafluorophenyl)borane Adducts of PhC(O)X (X = H, Me, OEt, NPri2). Organometallics 1998, 17, 1369–1377. [Google Scholar] [CrossRef]
- Hammett, L.P. The Effect of Structure upon the Reactions of Organic Compounds. Benzene Derivatives. J. Am. Chem. Soc. 1937, 59, 96–103. [Google Scholar] [CrossRef]
- Sana, M.; Leroy, G.; Wilante, C. Enthalpies of formation and bond energies in lithium, beryllium, and boron derivatives. 2. Dative, single, and triple bonds. Organometallics 1992, 11, 781–787, and references therein. [Google Scholar] [CrossRef]
- Lancaster, S. Alkylation of boron trifluoride with pentafluorophenyl Grignard reagent; Tris(pentafluorophenyl)boron; borane. ChemSpider SyntheticPages 2003. [Google Scholar] [CrossRef]
- CrysAlisPro; Version 1.171.37.33; Agilent Technologies: Santa Clara, CA, USA, 2014.
- SHELXTL; Bruker AXS: Madison, WI, USA, 2008.
- Spek, A.L. PLATON. Acta Crystallogr. 2009, D65, 148–155. [Google Scholar]
- Jaguar v.8.5; Schrödinger LLC: New York, NY, USA, 2014.
- Lee, C.; Yang, W.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar] [CrossRef]
- Raghavachari, K.; Binkley, J.S.; Seeger, R.; Pople, J.A. Self-consistent molecular orbital methods. XX. A basis set for correlated wave functions. J. Chem. Phys. 1980, 72, 650–654. [Google Scholar] [CrossRef]
- Sample Availability: Samples of alkynyl benzoates 1 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bähr, A.; Wilkins, L.C.; Ollegott, K.; Kariuki, B.M.; Melen, R.L. σ- versus π-Activation of Alkynyl Benzoates Using B(C6F5)3. Molecules 2015, 20, 4530-4547. https://doi.org/10.3390/molecules20034530
Bähr A, Wilkins LC, Ollegott K, Kariuki BM, Melen RL. σ- versus π-Activation of Alkynyl Benzoates Using B(C6F5)3. Molecules. 2015; 20(3):4530-4547. https://doi.org/10.3390/molecules20034530
Chicago/Turabian StyleBähr, Alexander, Lewis C. Wilkins, Kevin Ollegott, Benson M. Kariuki, and Rebecca L. Melen. 2015. "σ- versus π-Activation of Alkynyl Benzoates Using B(C6F5)3" Molecules 20, no. 3: 4530-4547. https://doi.org/10.3390/molecules20034530
APA StyleBähr, A., Wilkins, L. C., Ollegott, K., Kariuki, B. M., & Melen, R. L. (2015). σ- versus π-Activation of Alkynyl Benzoates Using B(C6F5)3. Molecules, 20(3), 4530-4547. https://doi.org/10.3390/molecules20034530