
Synthesis of C-Arylnucleoside Analogues


Abstract
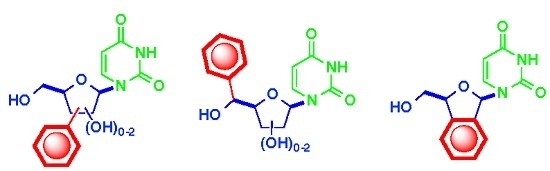
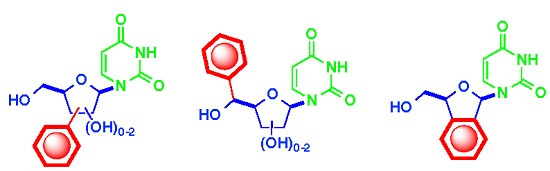
:
1. Introduction
2. Addition of an Aromatic Ring to a Carbonyl Group
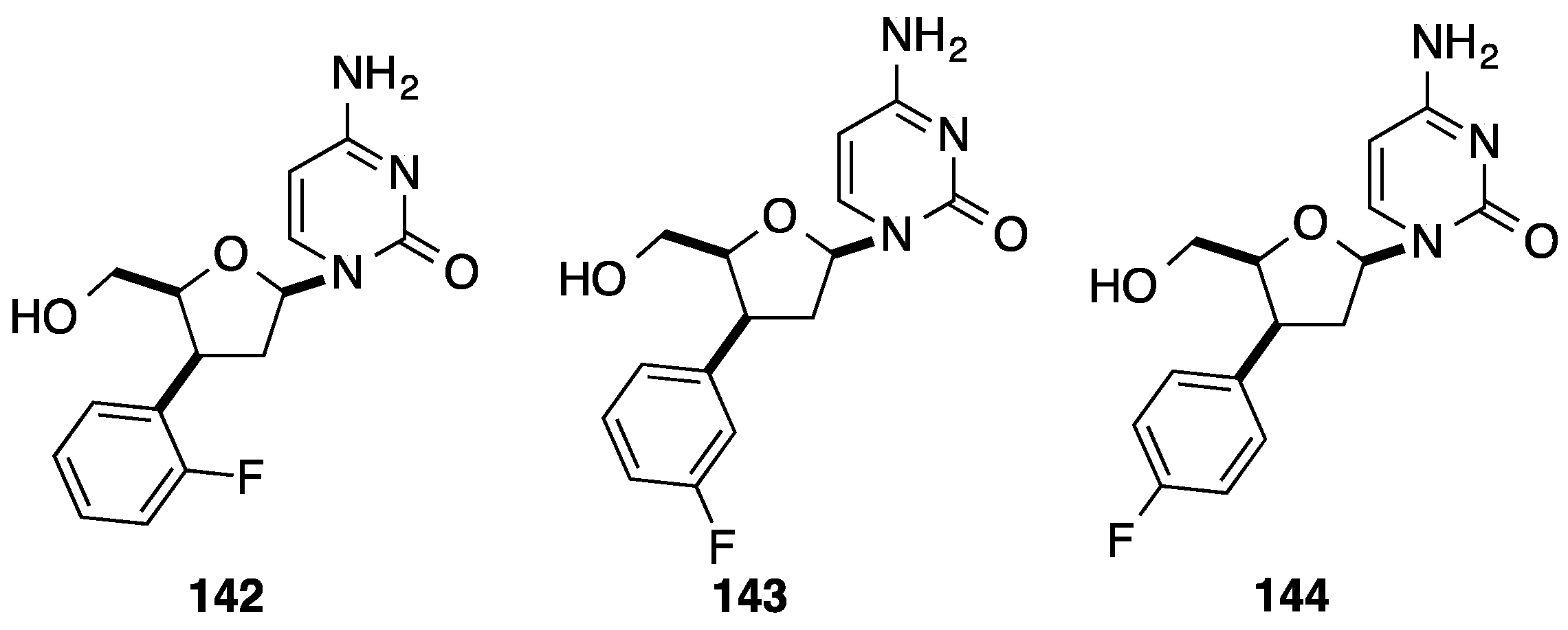
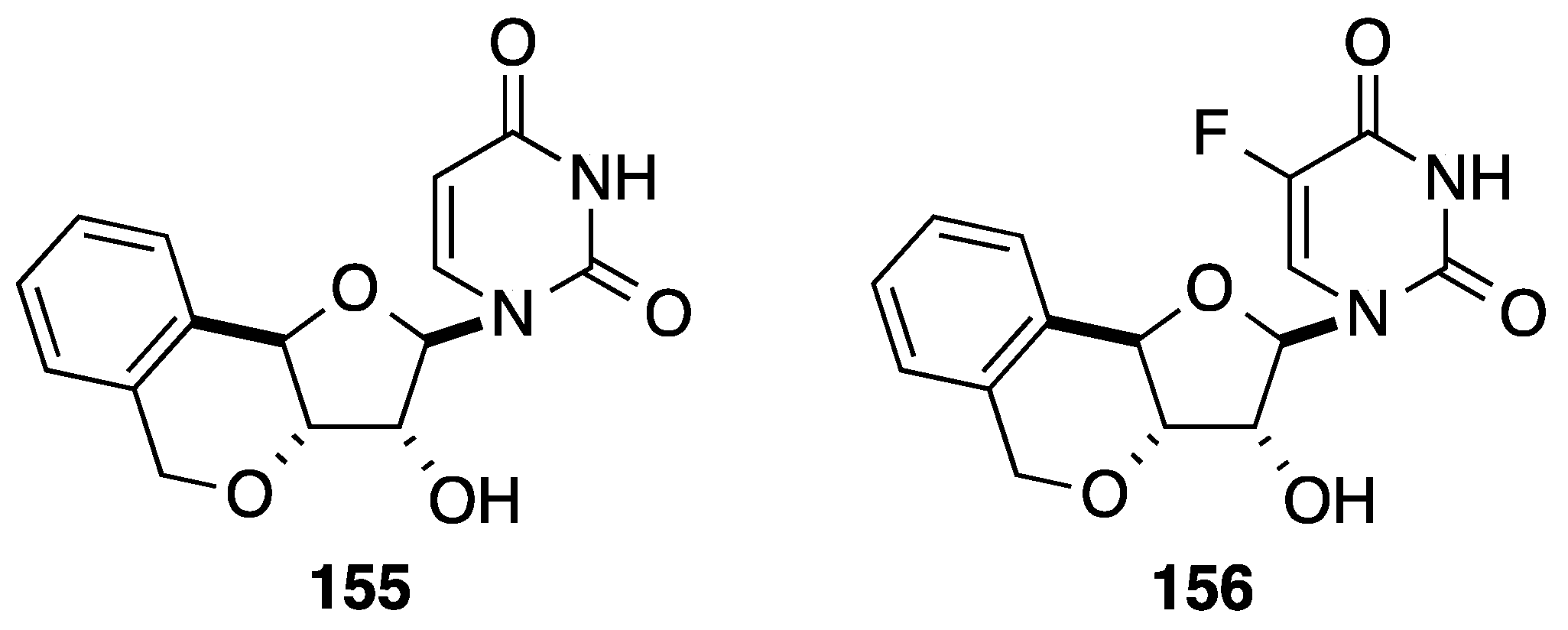
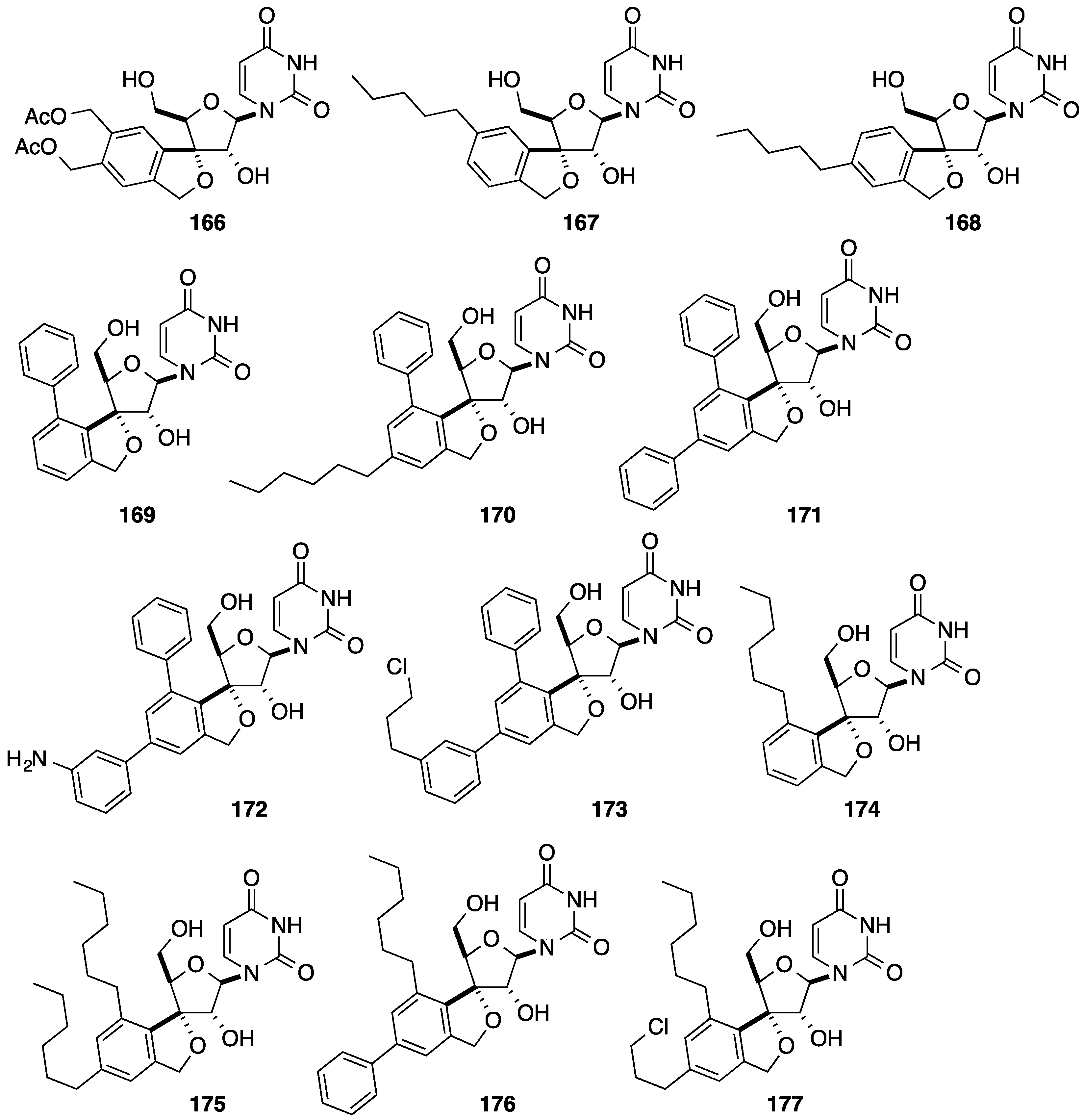
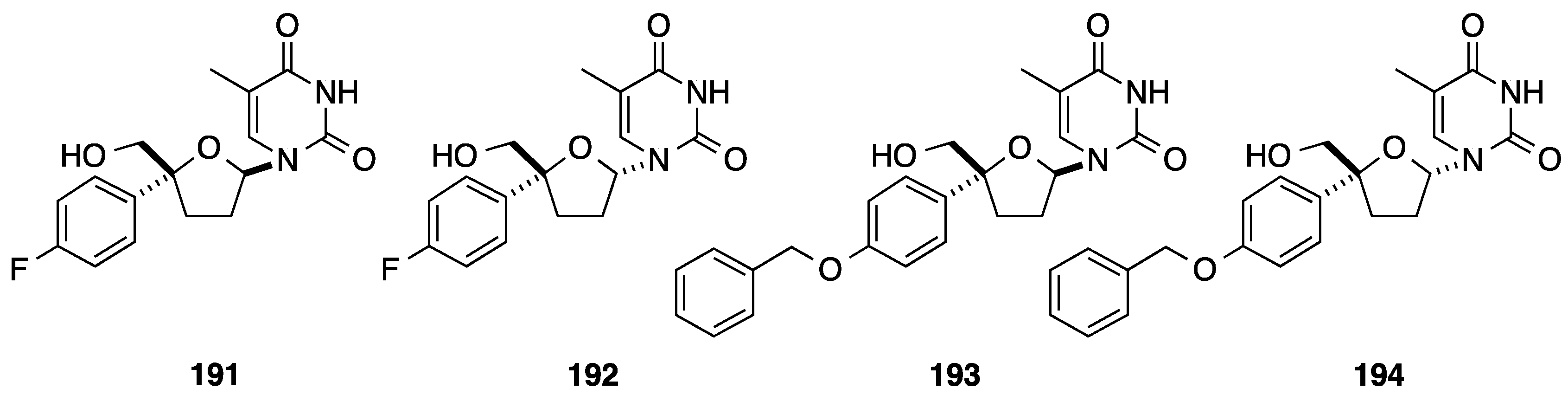
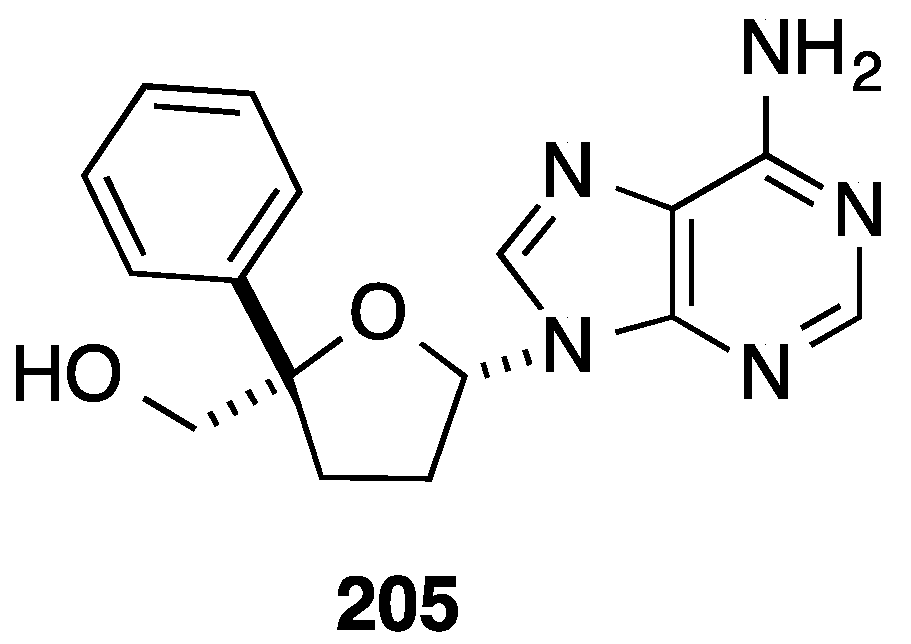
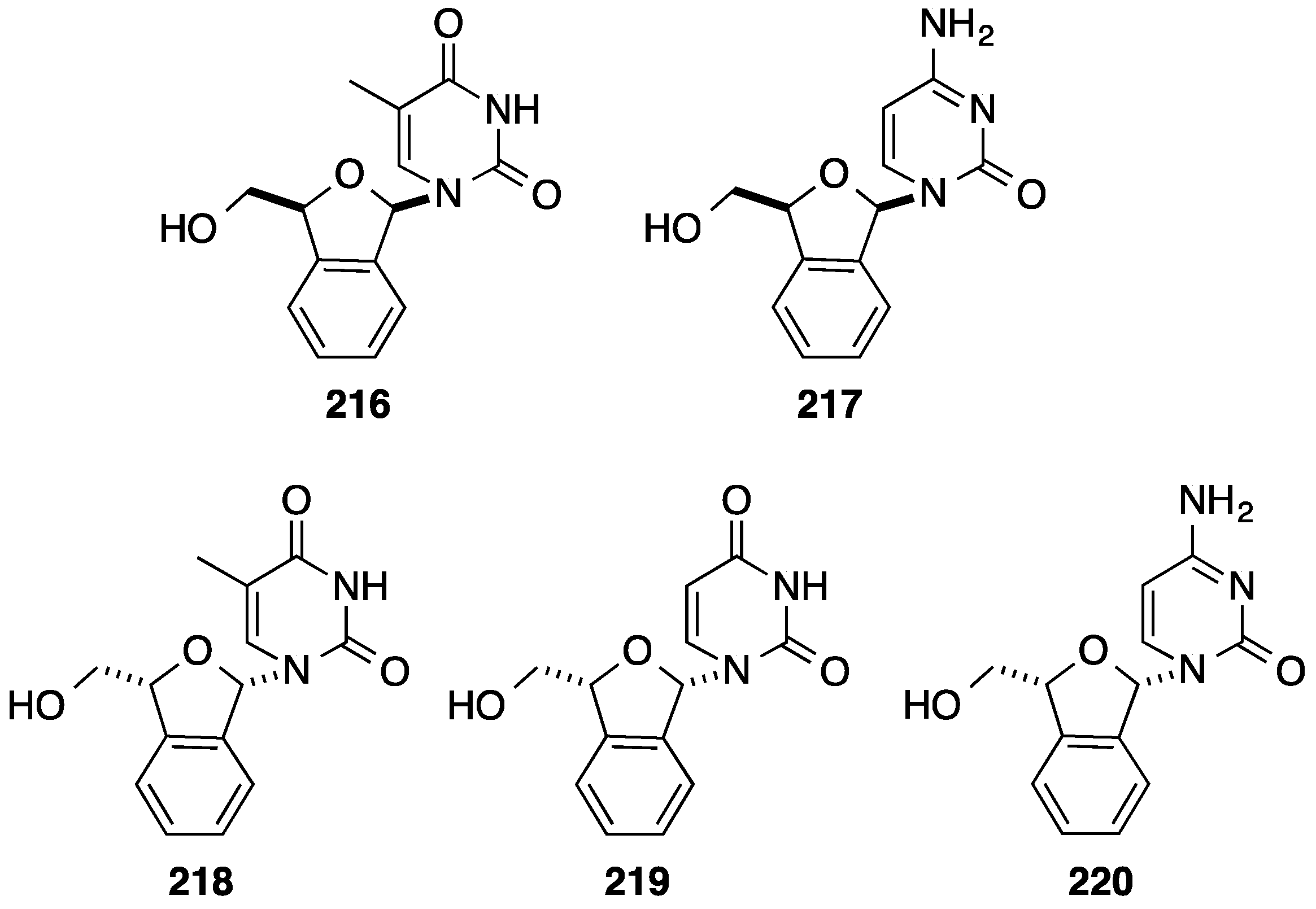
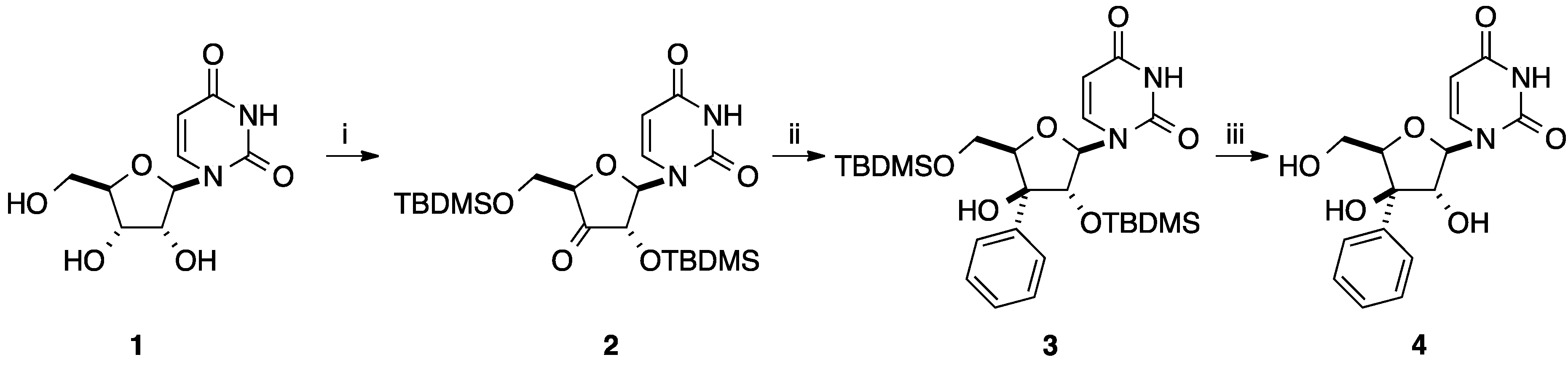
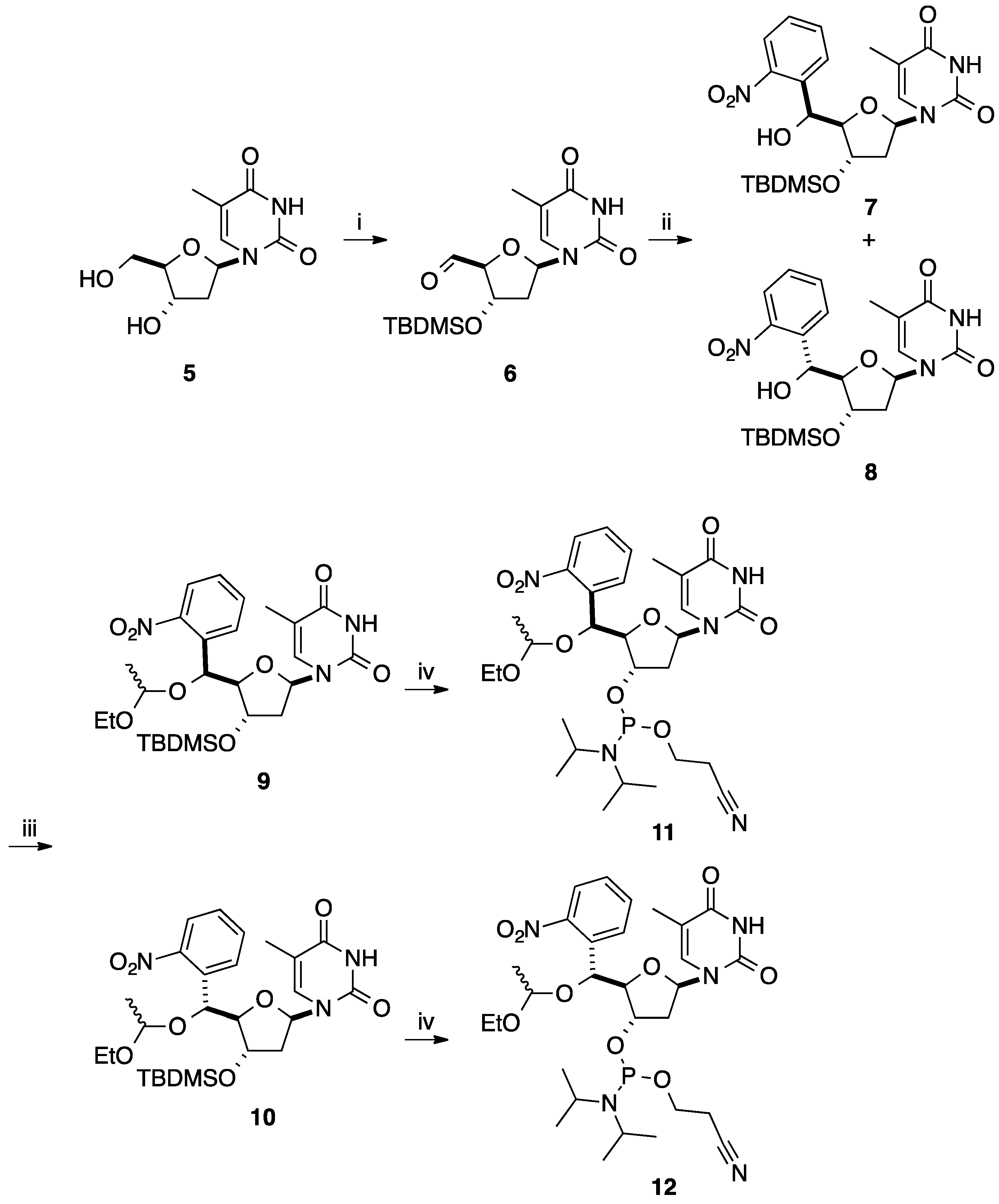
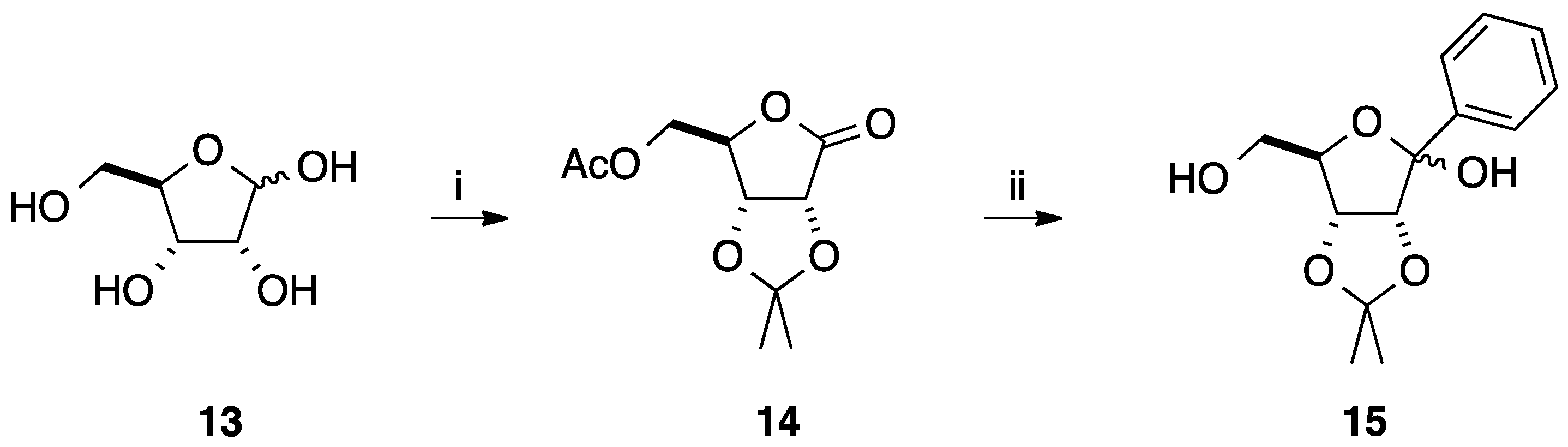
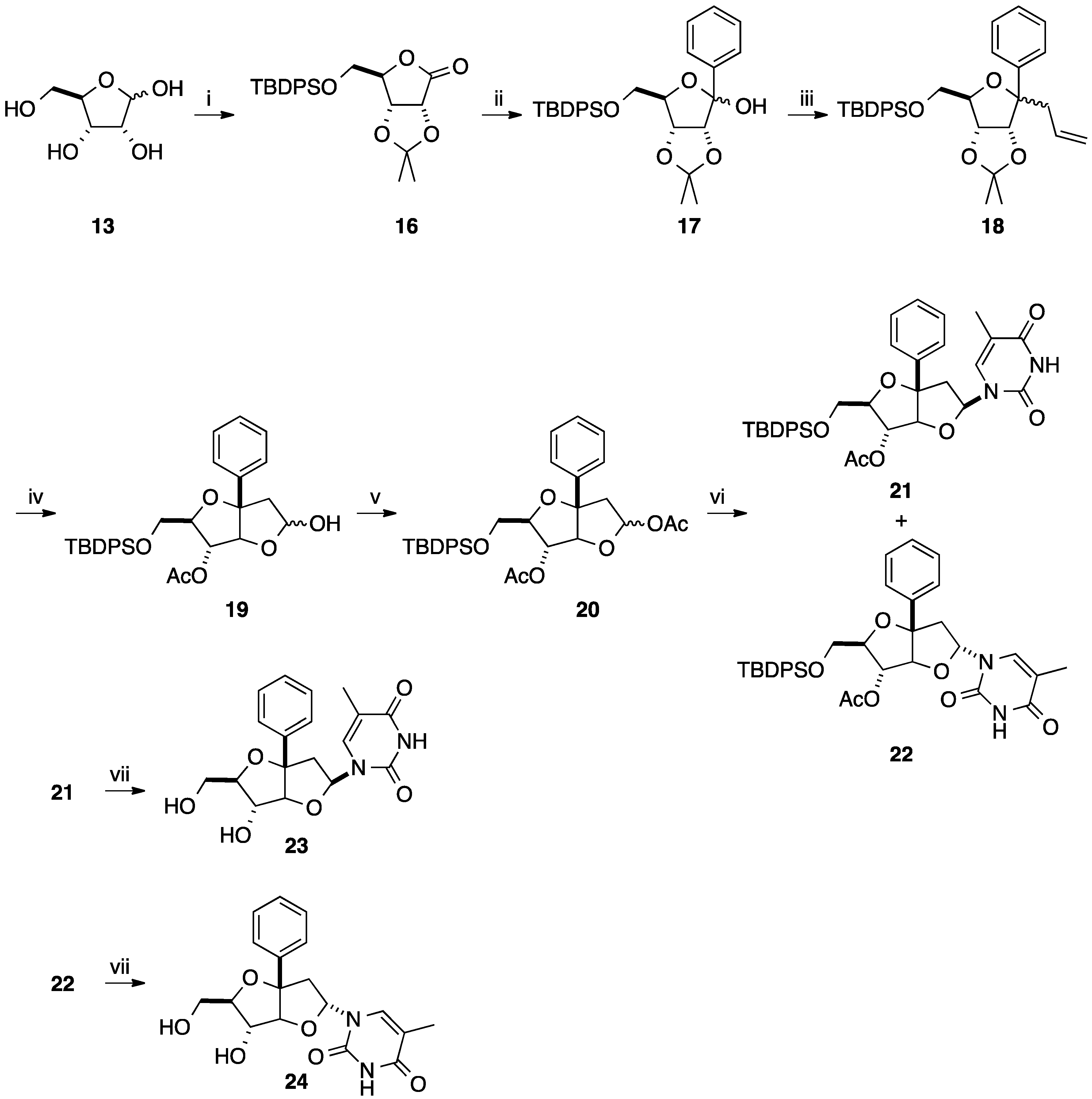
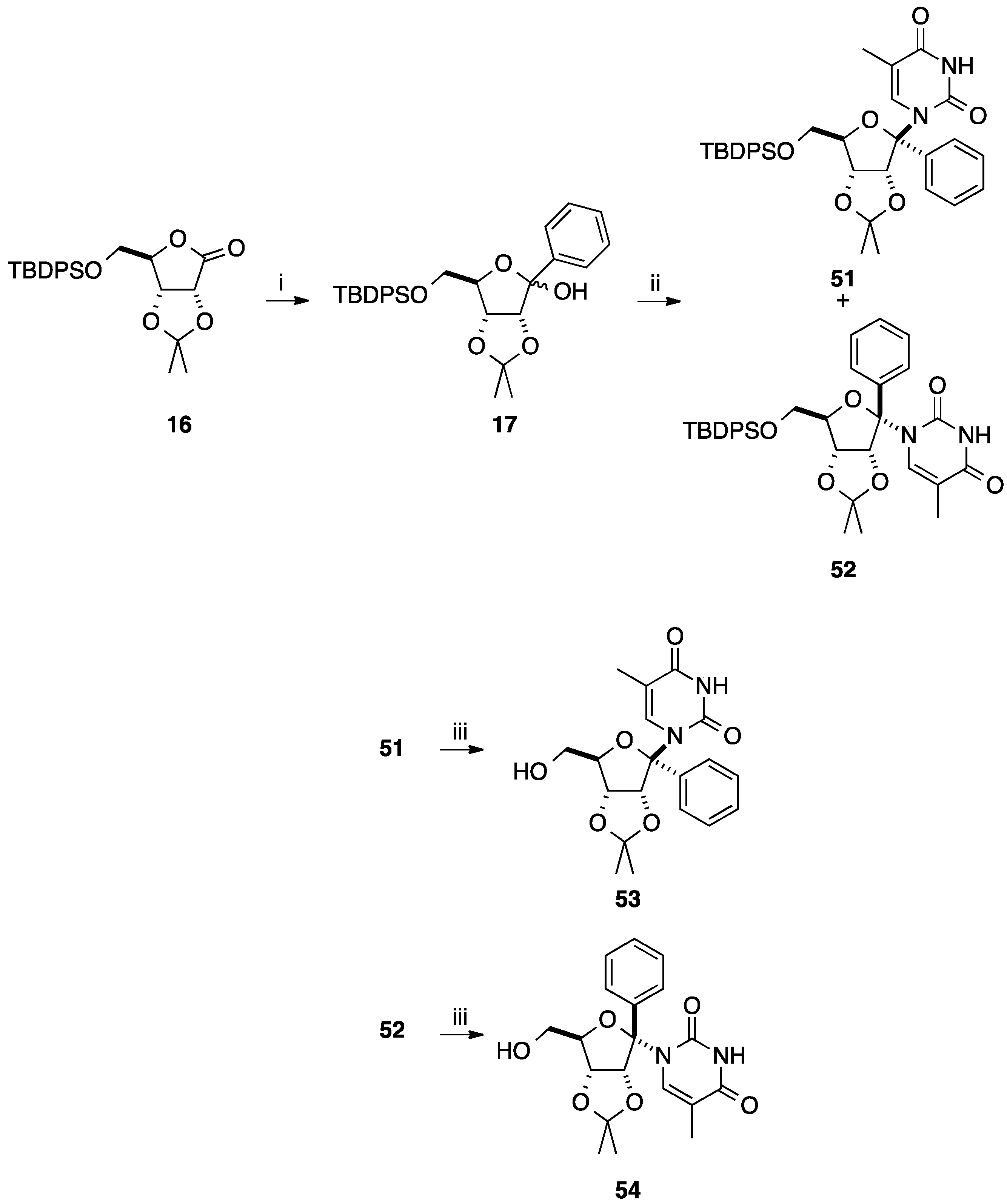
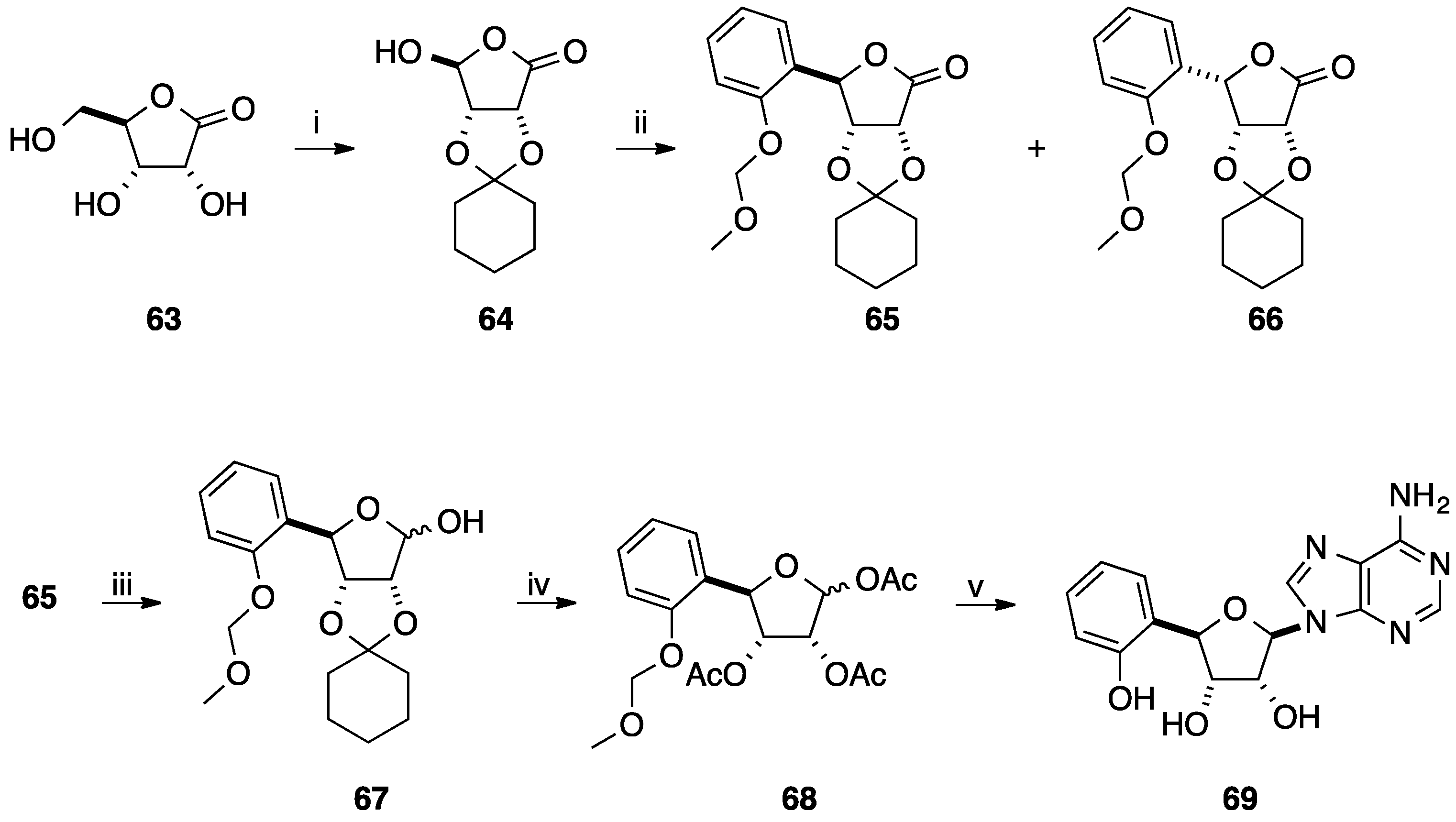
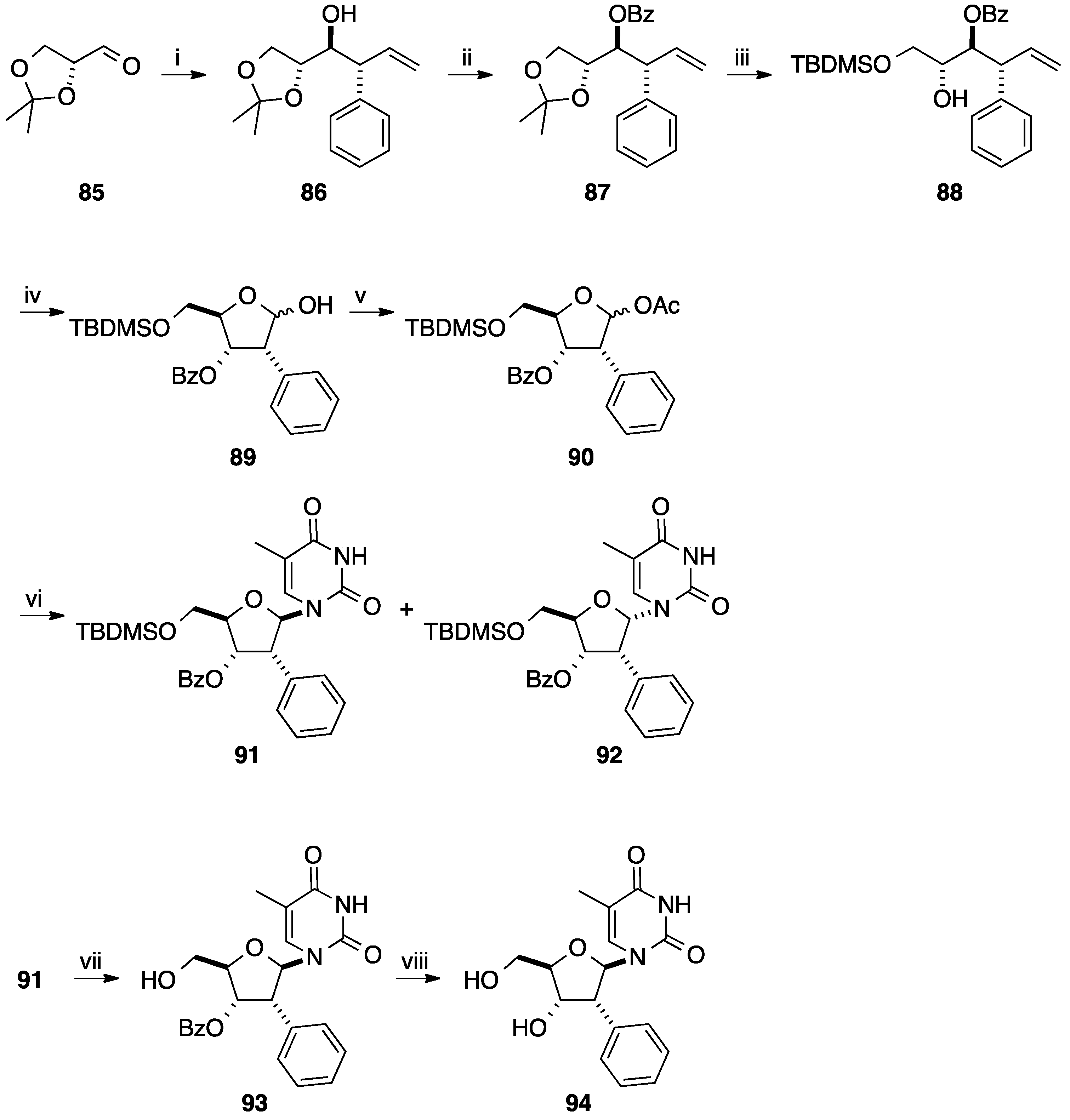
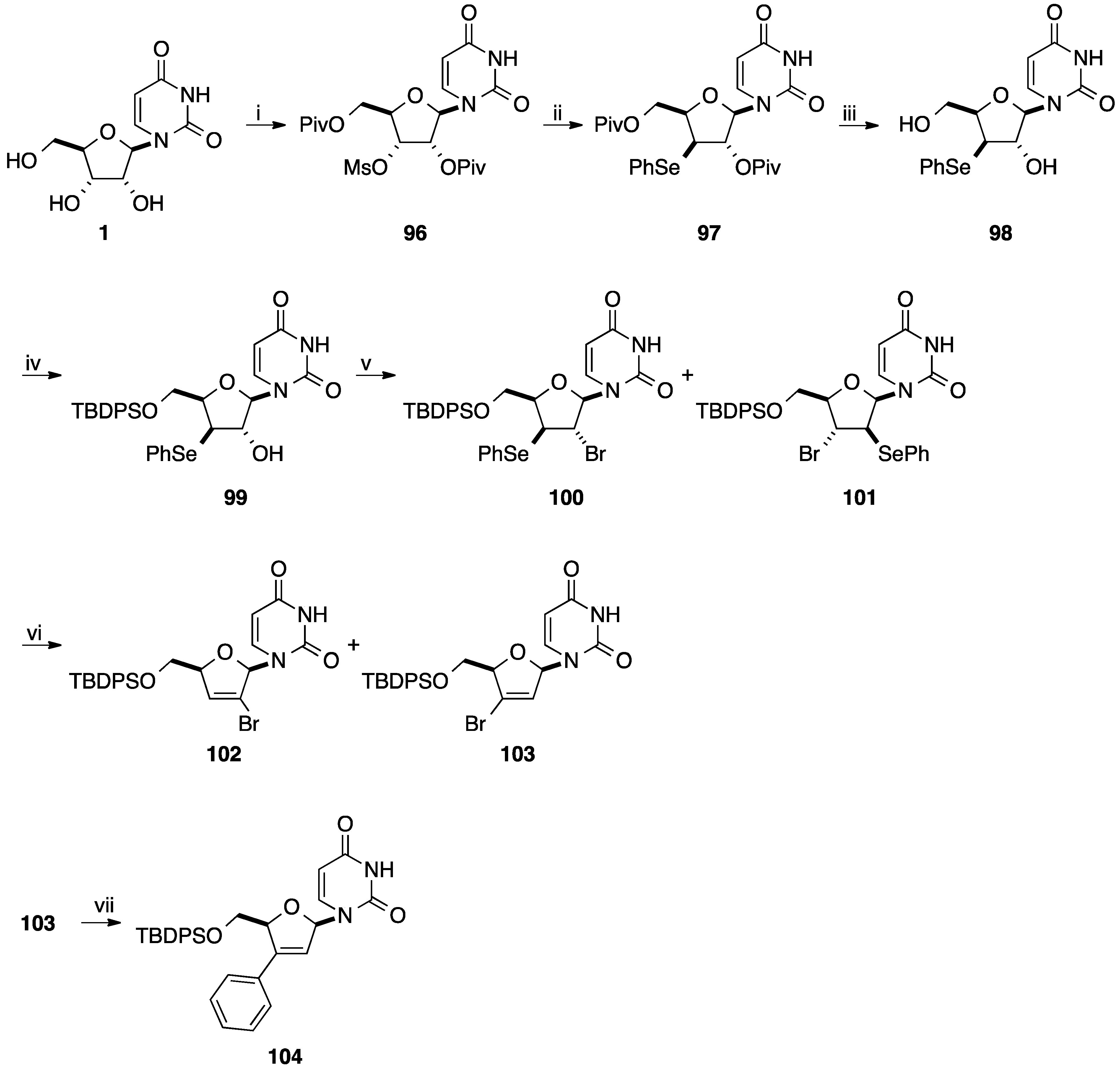
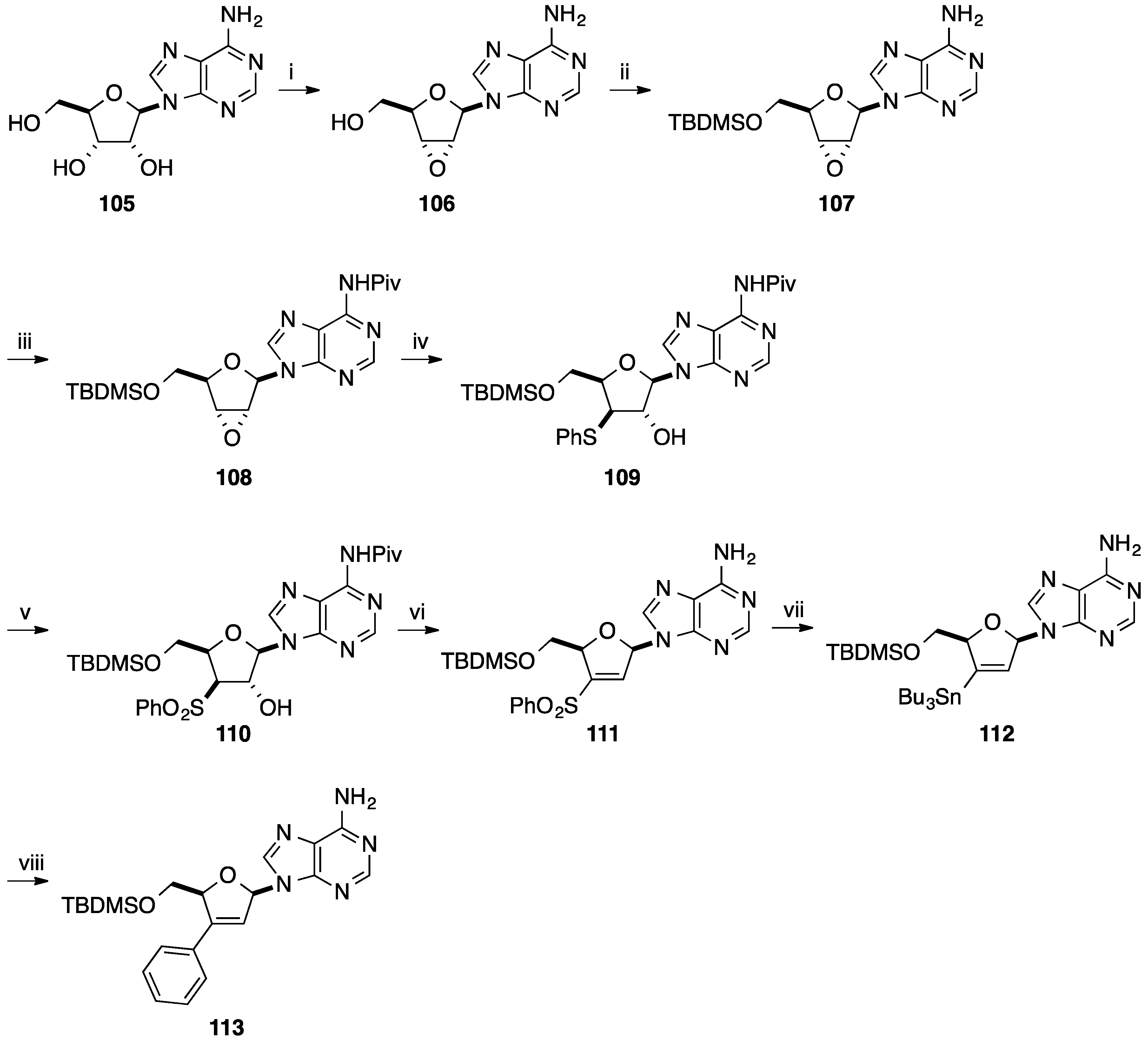
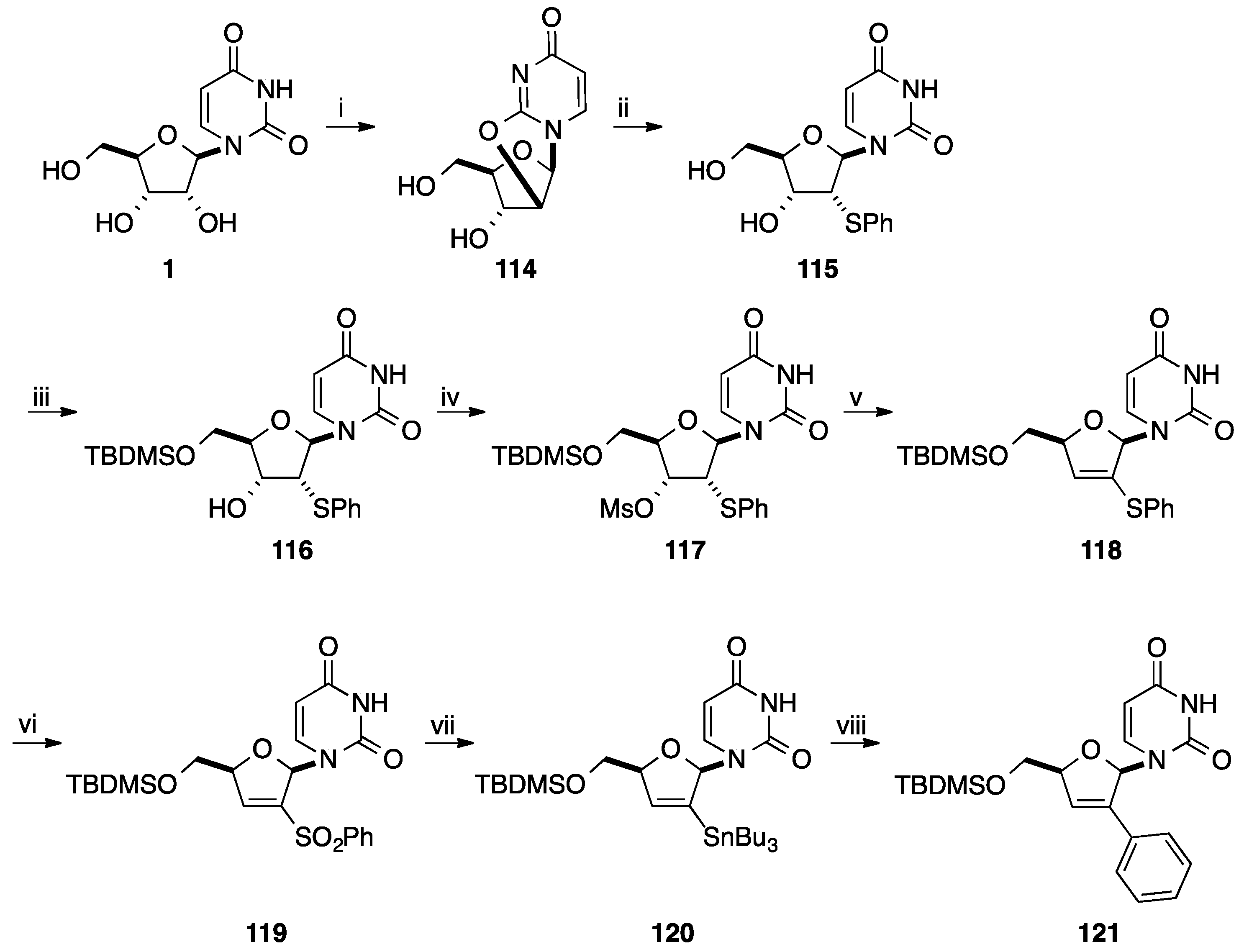
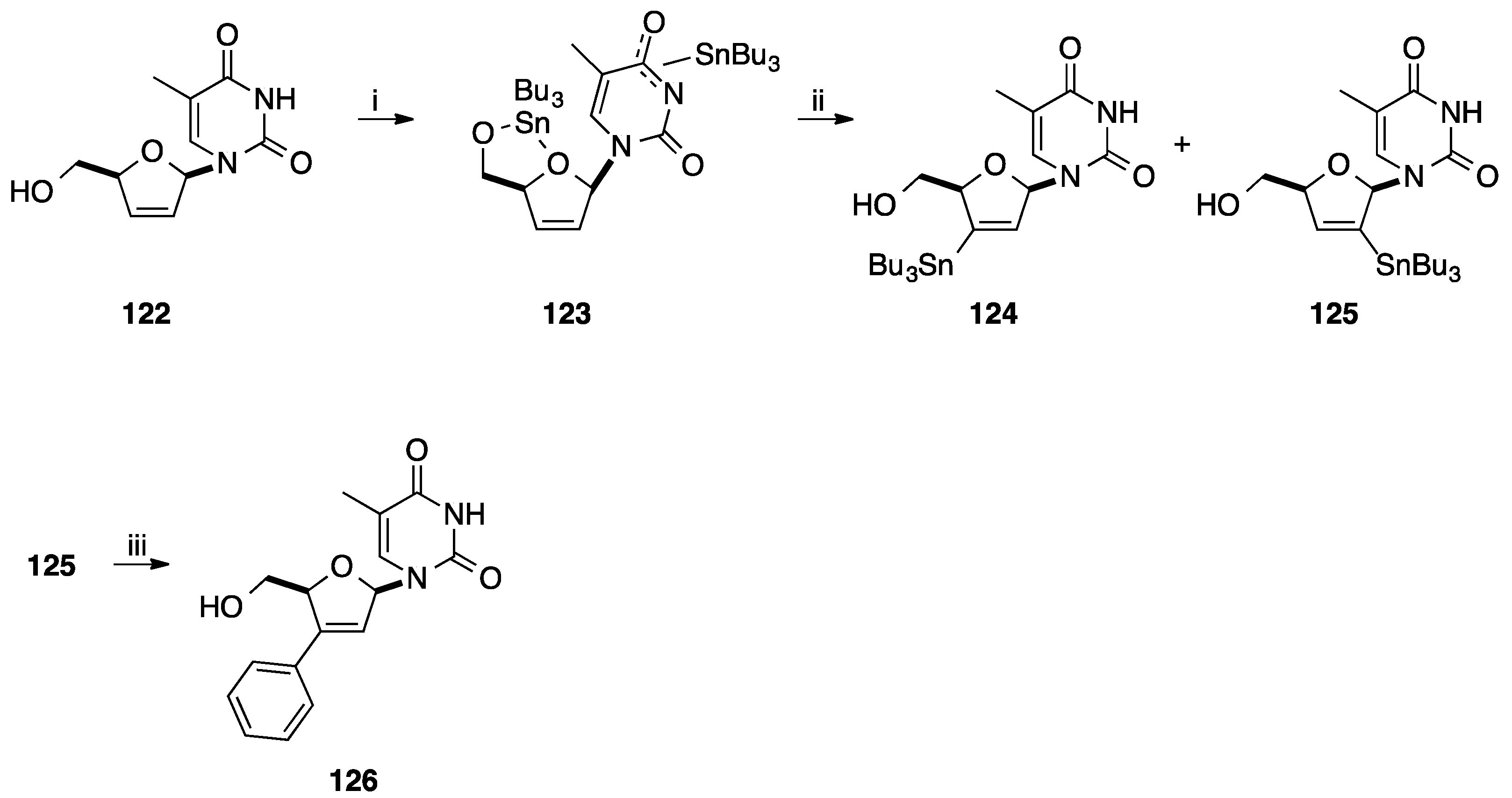
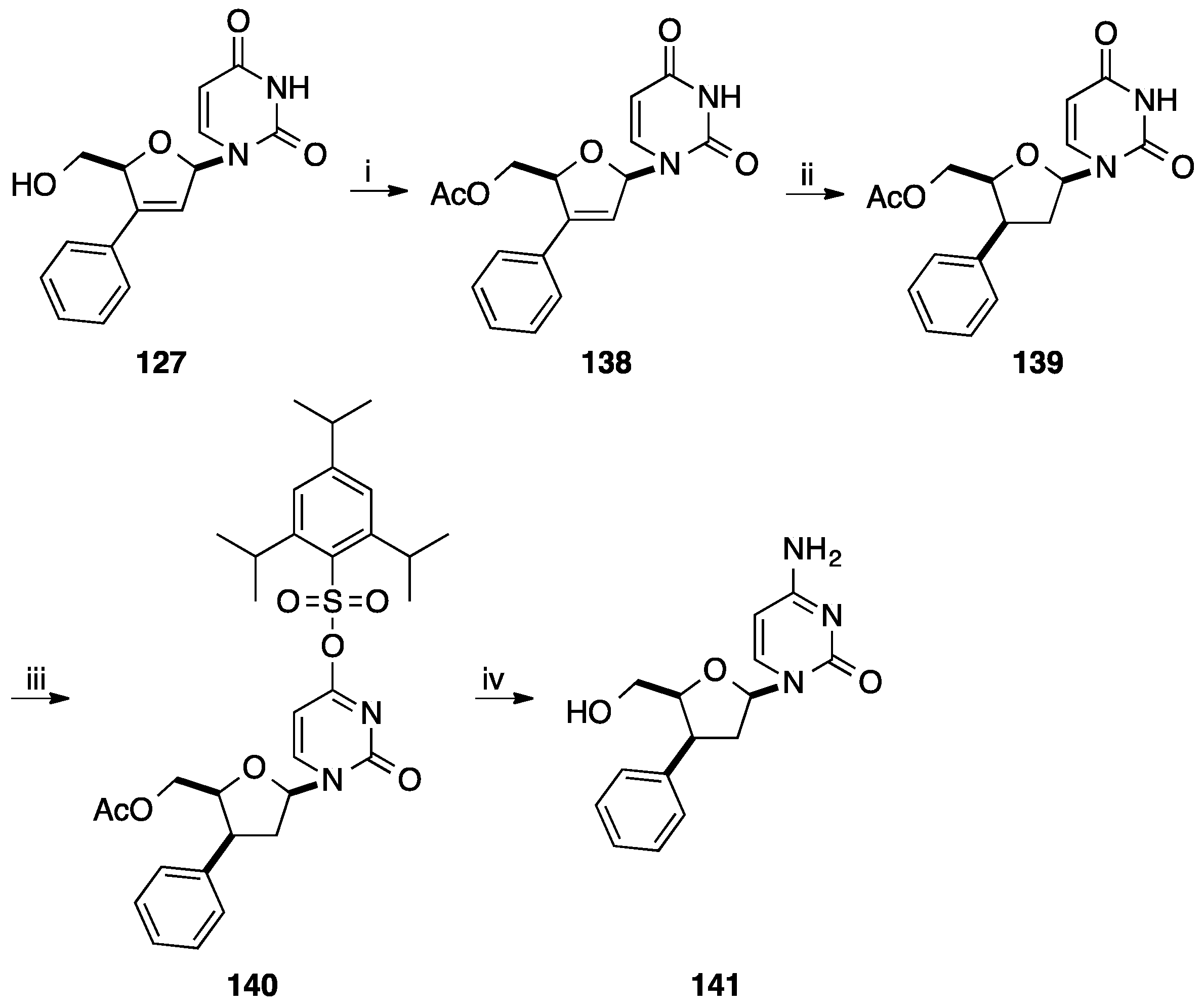
2.1. Addition of Aromatic Organolithiums to Carbonyl Groups





{kind=link}
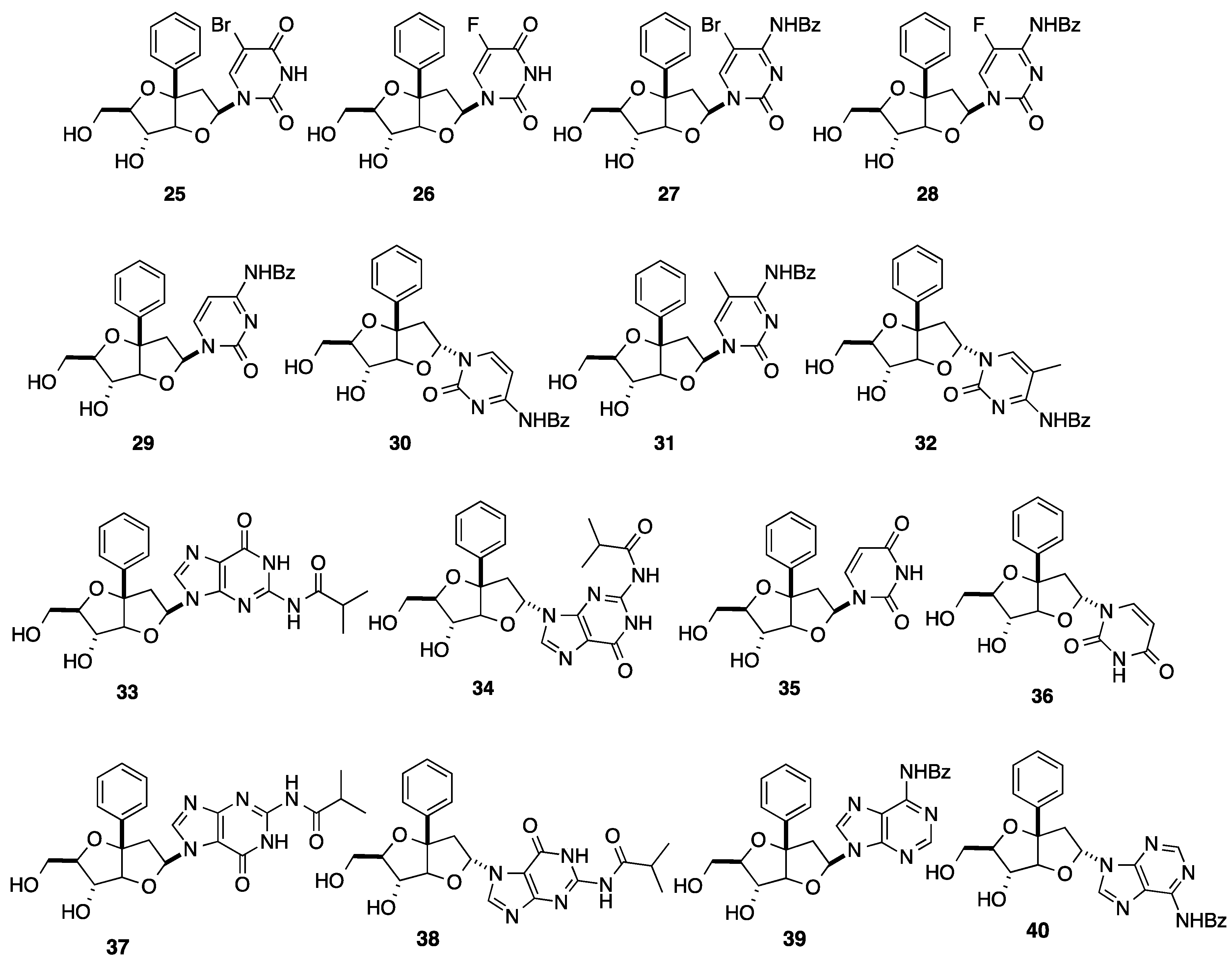{kind=link}
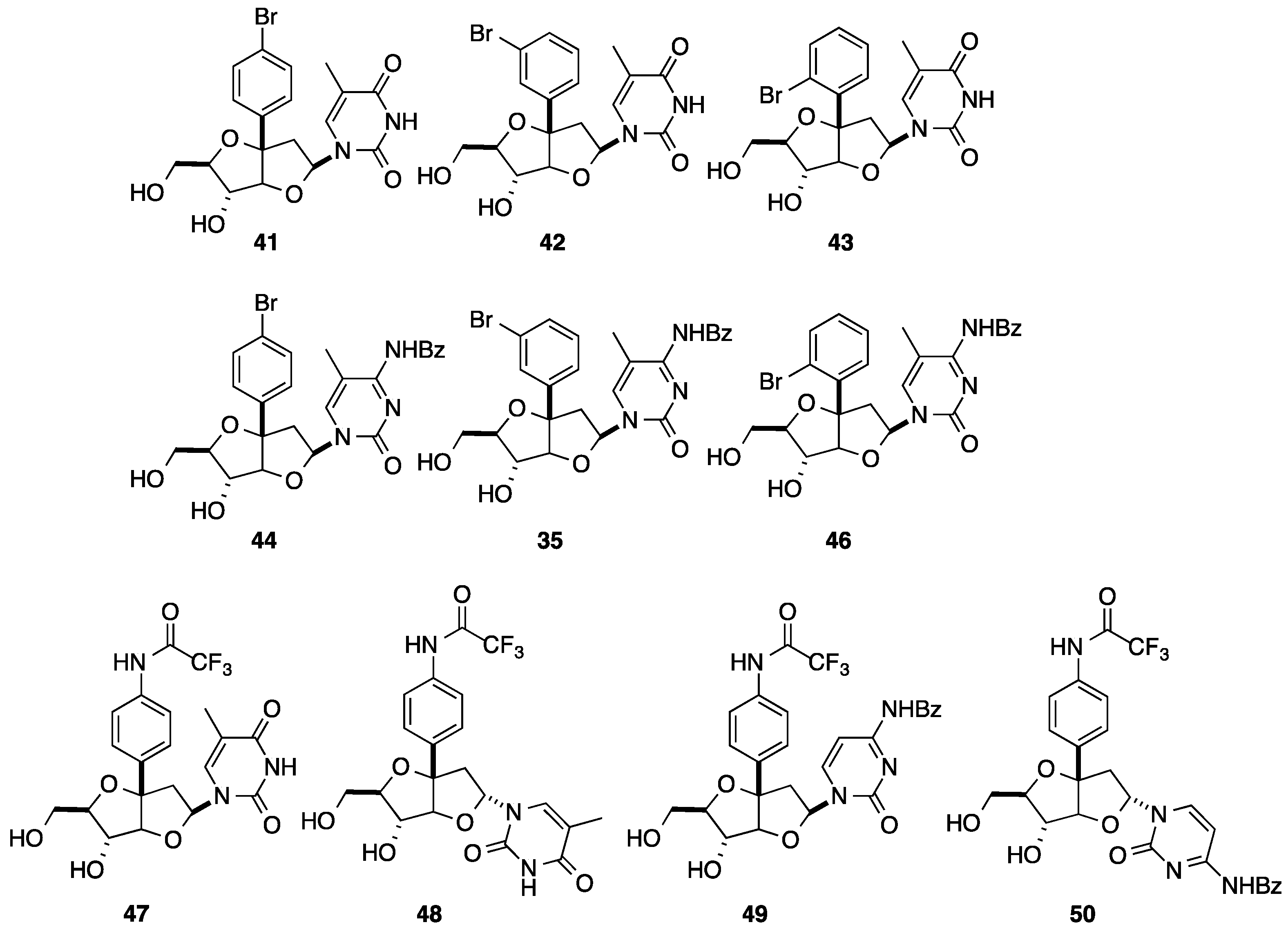{kind=link}
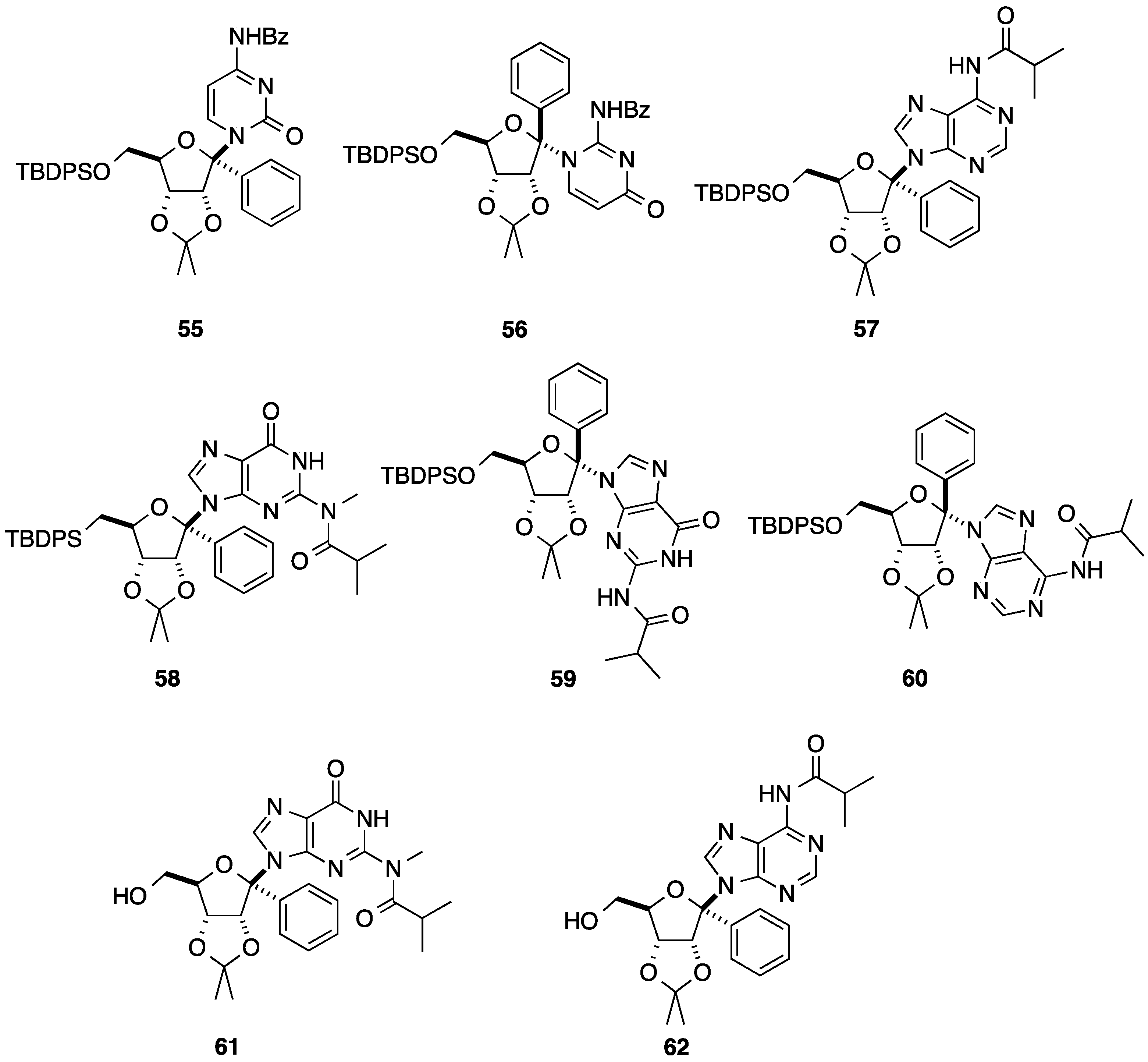{kind=link}
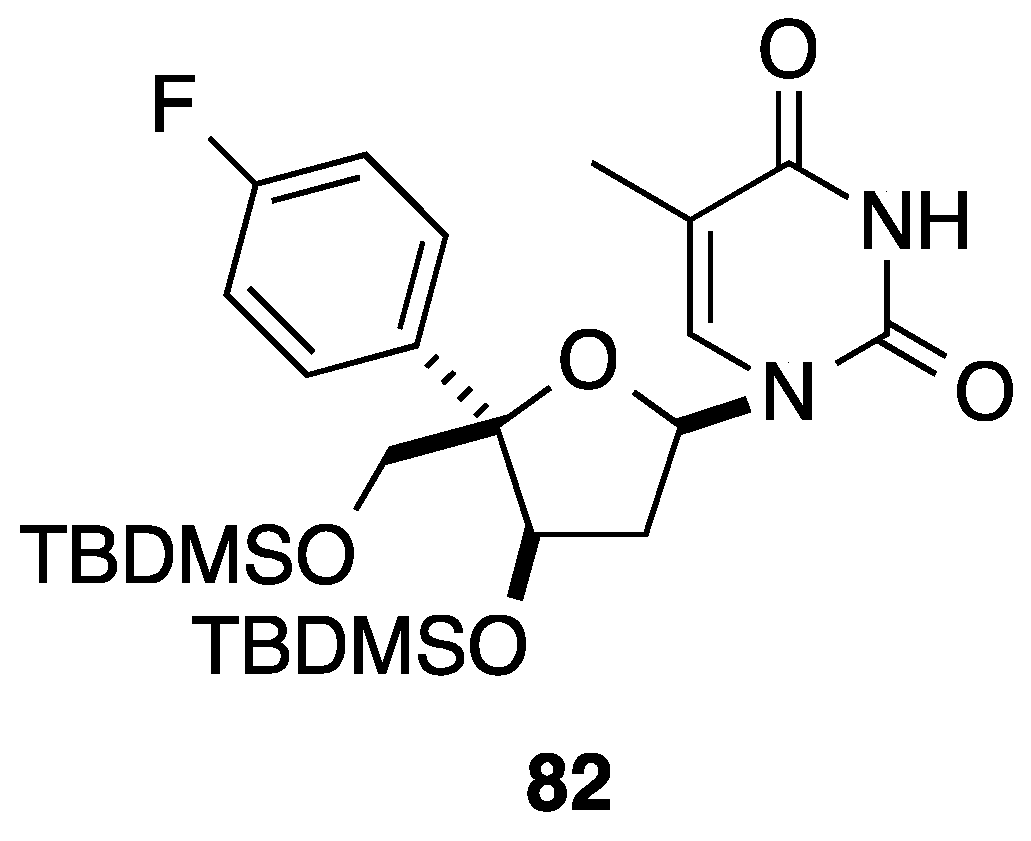{kind=link}
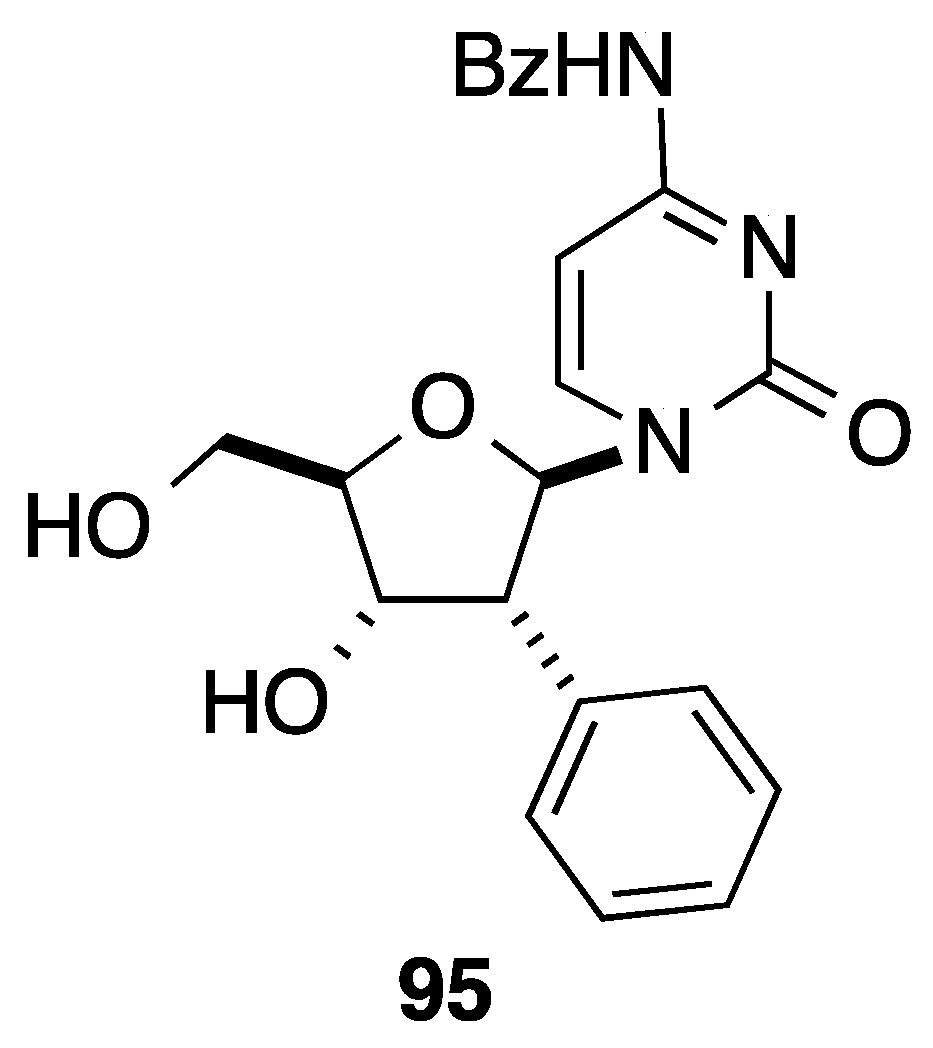{kind=link}
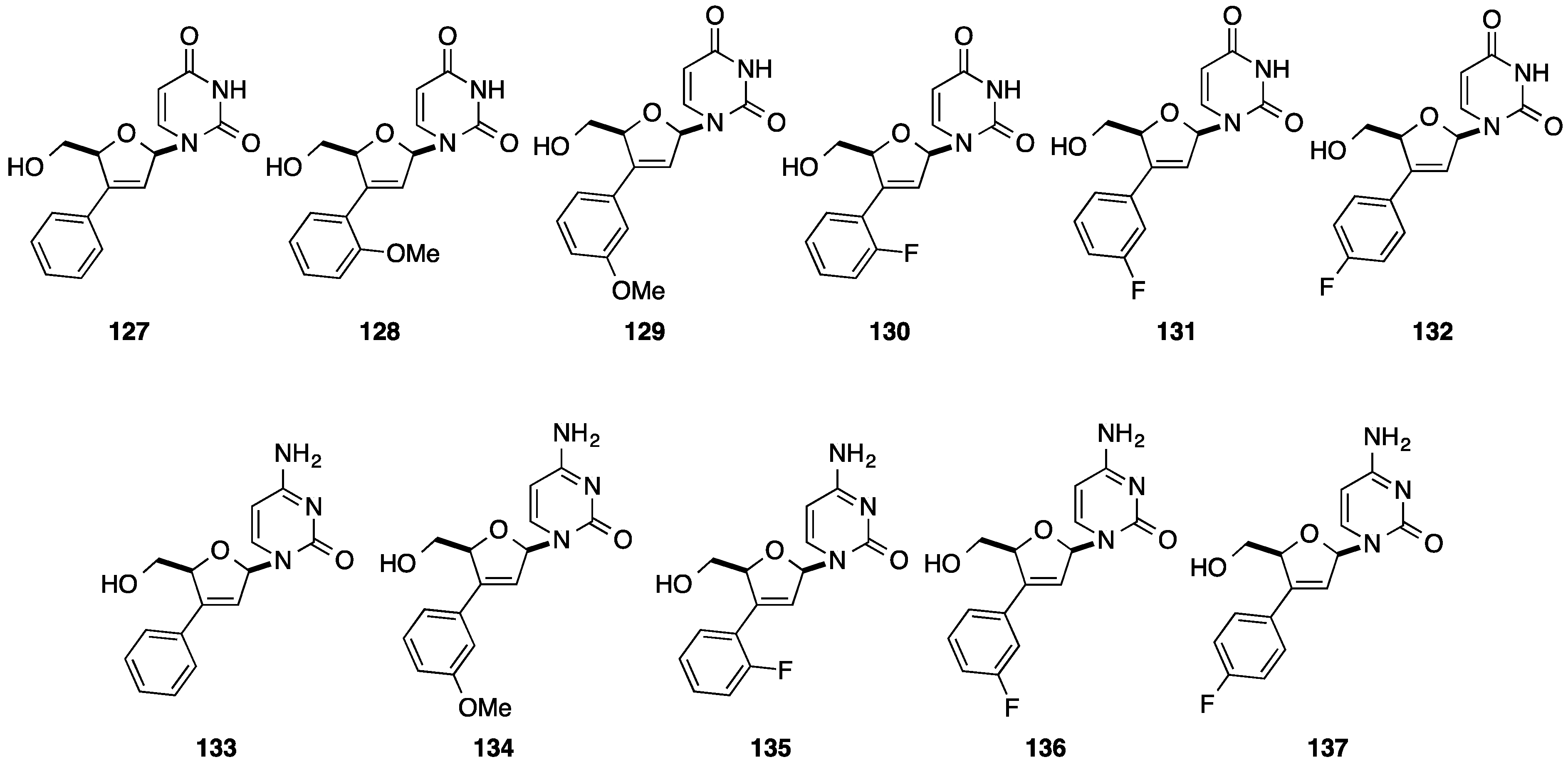{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
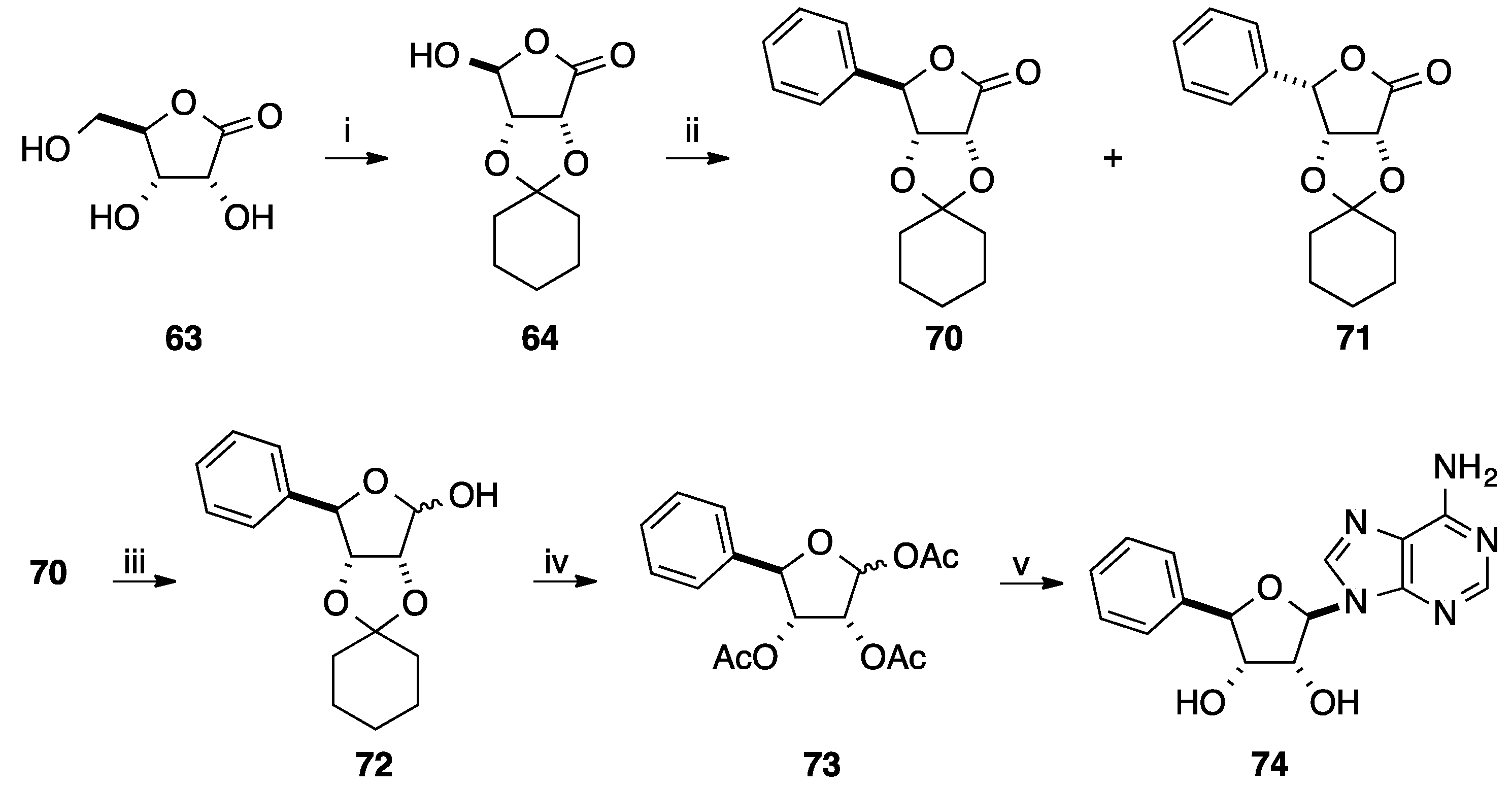{kind=link}
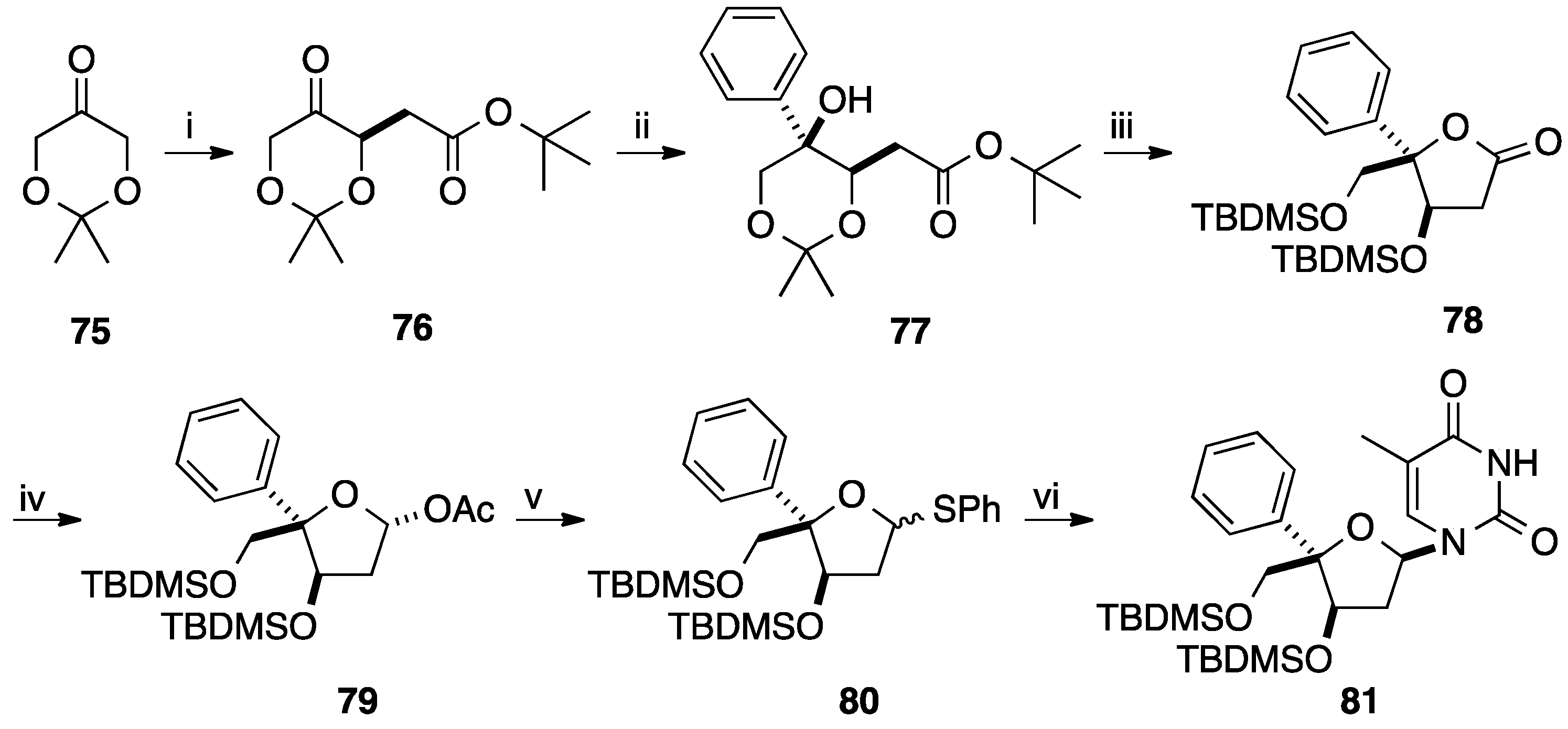{kind=link}
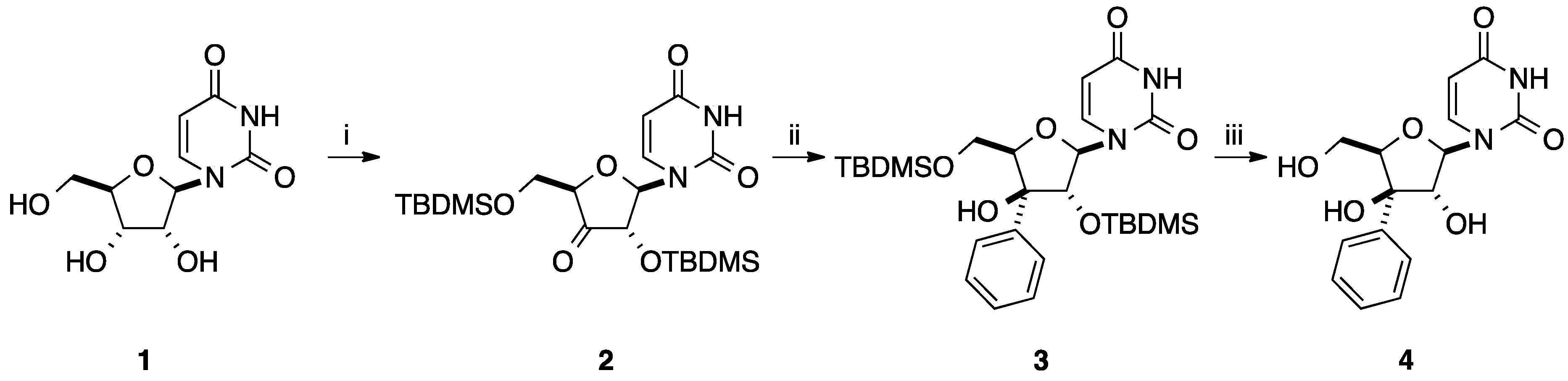{kind=link}
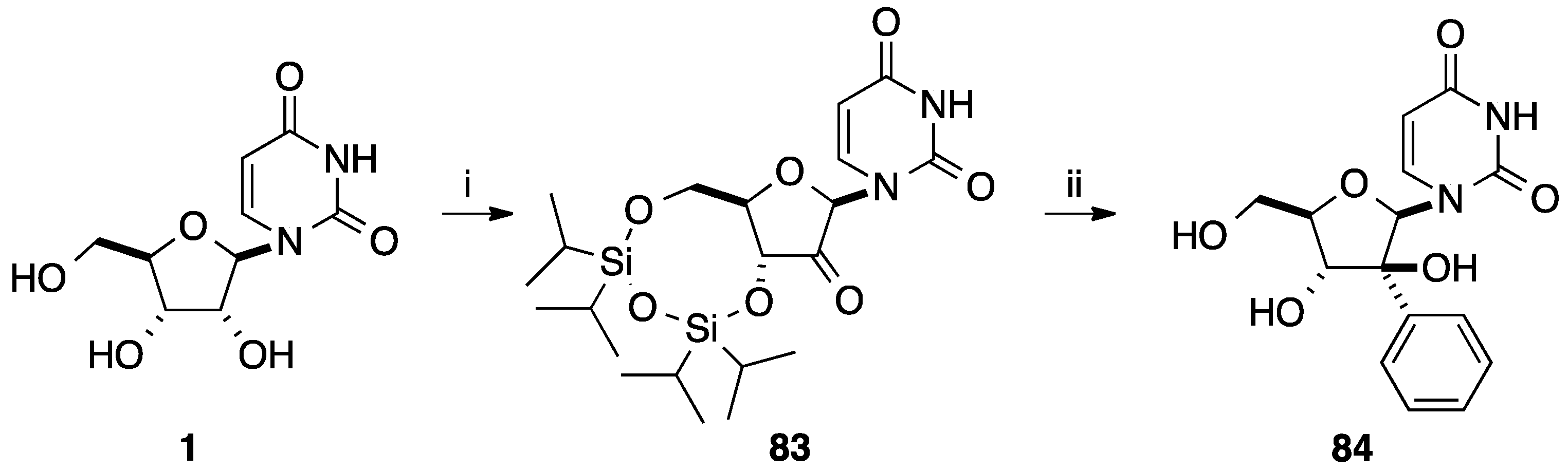{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}




2.2. Addition of Aromatic Organomagnesium Reagents to Carbonyl Groups or Analogues



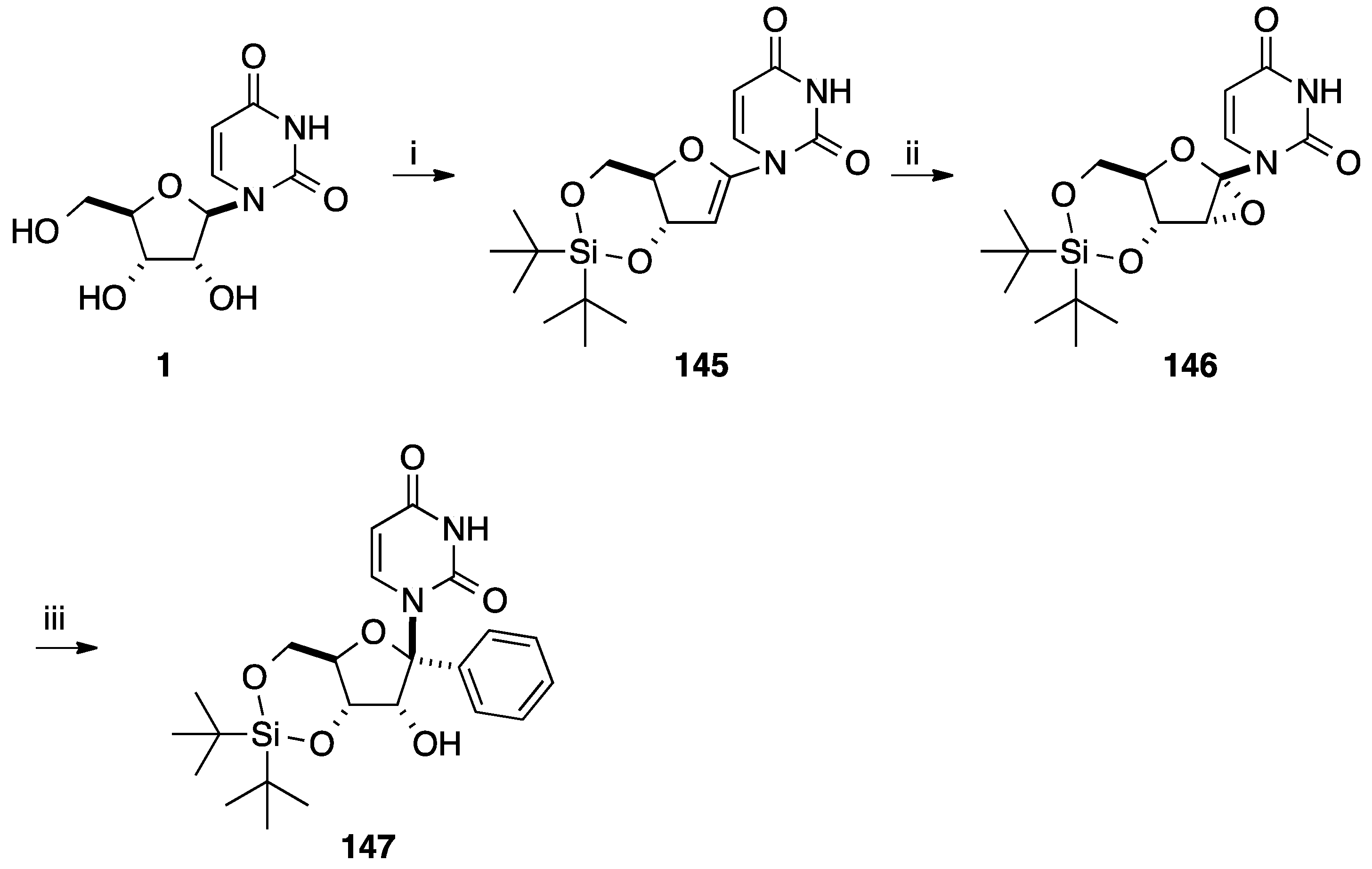
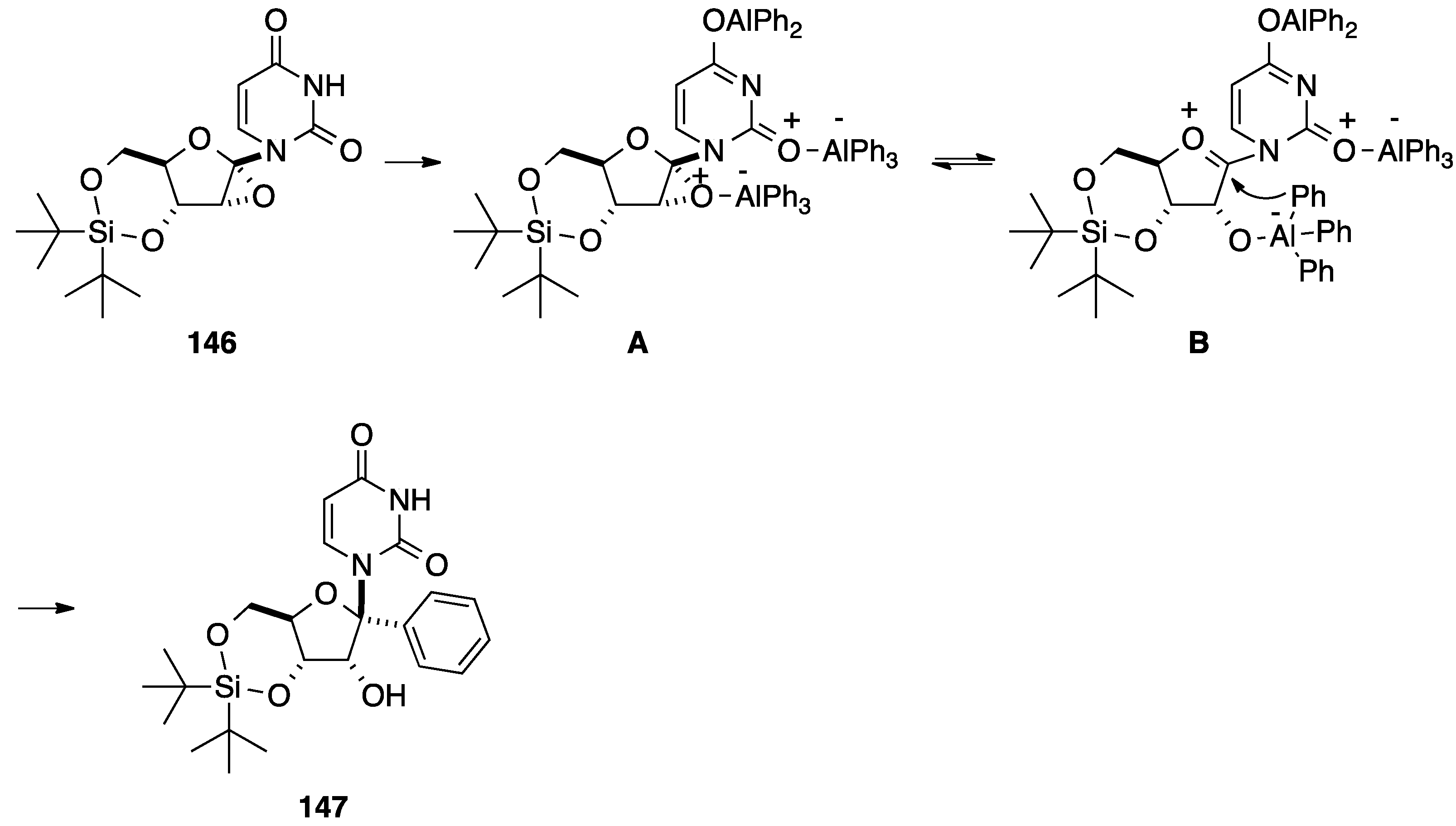
2.3. Addition of Aromatic Organoaluminium Reagents to Carbonyl Groups


2.4. Addition of Aromatic Organotitanium Reagents to Carbonyl Groups or Analogues


3. Cross-Coupling Reactions







4. Addition of Aromatic Rings to an Epoxide


5. Cyclization



6. Construction of the Glycone Part Starting from an Aromatic Moiety







7. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Len, C.; Mackenzie, G. Synthesis of 2',3'-didehydro-2',3'-dideoxynucleosides having variations at either or both of the 2'- and 3'-positions. Tetrahedron 2006, 62, 9085–9107. [Google Scholar] [CrossRef]
- Len, C.; Postel, D. Synthesis of 2',3'-didehydro-2',3'-dideoxynucleosides via nucleoside route. Curr. Org. Synth. 2006, 21, 261–281. [Google Scholar] [CrossRef]
- Lebreton, J.; Escudier, J.M.; Arzel, L.; Len, C. Synthesis of bicyclonucleosides having a C-C bridge. Chem. Rev. 2010, 110, 3371–3418. [Google Scholar] [CrossRef] [PubMed]
- Len, C.; Mondon, M.; Lebreton, J. Synthesis of cyclonucleosides having a C-C bridge. Tetrahedron 2008, 64, 7453–7475. [Google Scholar] [CrossRef]
- Herve, G.; Sartori, G.; Enderlin, G.; Mackenzie, G.; Len, C. Palladium-catalysed Suzuki reaction in aqueous solvents applied to unprotected nucleosides and nucleotides. RSC Adv. 2014, 4, 18558–18594. [Google Scholar] [CrossRef]
- Stambasky, J.; Hocek, M.; Kocovsky, P. C-Nucleosides: Synthetic strategies and biological applications. Chem. Rev. 2009, 109, 6729–6764. [Google Scholar] [CrossRef] [PubMed]
- Ferrero, M.; Gotor, V. Biocatalytic selective modifications of conventional nucleosides, carbocyclic nucleosides and C-nucleosides. Chem. Rev. 2000, 100, 4319–4348. [Google Scholar] [CrossRef] [PubMed]
- Hayakawa, H.; Tanaka, H.; Itoh, N.; Nakajima, M.; Miyasaka, T.; Yamaguchi, K.; Iitaka, Y. Reaction of organometallic reagents with 2'- and 3'-ketouridine derivatives: Synthesis of uracil nucleosides branched at the 2'- and 3'-positions. Chem. Pharm. Bull. 1987, 35, 2605–2608. [Google Scholar] [CrossRef]
- Hansske, F.; Robins, M.J. Nucleic acid related compounds. 43. A convenient procedure for the synthesis of 2' and 3'-ketonucleosides. Tetrahedron Lett. 1983, 24, 1589–1583. [Google Scholar] [CrossRef]
- Yang, C.O.; Wu, H.Y.; Fraser-Smith, E.B.; Walker, K.A.M. Synthesis of 4'-cyanothymidine and analogs as potent inhibitors of HIV. Tetrahedron Lett. 1992, 33, 37–40. [Google Scholar] [CrossRef]
- Dussy, A.; Meyer, C.; Quennet, E.; Bickle, T.A.; Giese, B.; Marx, A. New light-sensitive nucleosides for caged DNA strand breaks. ChemBioChem 2002, 3, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Thoeni, S.; Kressierer, C.J.; Giese, B. Site-specific DNA cleavage on a solid support: A method for mismatch detection. Angew. Chem. Int. Ed. 2007, 46, 2112–2114. [Google Scholar] [CrossRef]
- Sasaki, S.; Yamauchi, H.; Nagatsugi, F.; Takahashi, R.; Taniguchi, Y.; Maeda, M. W-shape nucleic acid (WNA) for selective formation of non-natural anti-parallel triplex including a TA interrupting site. Tetrahedron Lett. 2001, 42, 6915–6918. [Google Scholar] [CrossRef]
- Sasaki, S.; Taniguchi, Y.; Takahashi, R.; Senko, Y.; Kodama, K.; Nagatsugi, F.; Maeda, M. Selective formation of stable triplexes including a TA or a CG interrupting site with new bicyclic nucleoside analogues (WNA). J. Am. Chem. Soc. 2004, 126, 516–528. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Nakamura, A.; Senko, Y.; Nagatsuji, F.; Sasaki, S. Effects of halogenated WNA derivatives on sequence dependency for expansion of recognition sequences in non-natural-type triplexes. J. Org. Chem. 2006, 71, 2115–2122. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, Y.; Togo, M.; Aoki, E.; Uchida, Y.; Sasaki, S. Synthesis of p-amino-WNA derivatives to enhance the stability of the anti-parallel triplex. Tetrahedron 2008, 64, 7184–7170. [Google Scholar]
- Nasr, T.; Taniguchi, Y.; Sasaki, S. Synthesis of 1'-phenyl substituted nucleoside analogs. Heterocycles 2007, 71, 2659–2668. [Google Scholar] [CrossRef]
- Hayakawa, H.; Miyazawa, M.; Tanaka, H.; Miyasaka, T. A ribonolactone-based approach to the synthesis of 1'-carbon-substituted thymine ribonucleosides. Nucleosides Nucleotides 1994, 12, 297–308. [Google Scholar] [CrossRef]
- Beer, D.; Mewly, R.; Vasella, A. l-Erythruronic acid derivatives as building blocks for nucleoside analogs. Synthesis of 4'-C-aryl-D-ribonucleosides. Helv. Chim. Acta 1982, 65, 2570–2582. [Google Scholar] [CrossRef]
- Enders, D.; Voith, M.; Lenzen, A. The dihydroxyacetone unit—A versatile C3 building bloc in organic synthesis. Angew. Chem. Int. Ed. 2005, 44, 1304–1325. [Google Scholar] [CrossRef]
- Enders, D.; Hieronymi, A.; Raabe, G. Asymmetric synthesis of 4'-quaternary 2'-deoxy-3'- and -4'-epi-β-C- and -N-nucleosides. Synthesis 2008, 10, 1545–1558. [Google Scholar] [CrossRef]
- Cook, A.F.; Moffatt, J.G. Sulfoxide-carbodiimide reactions. VI. Synthesis of 2'- and 3'-ketouridines. J. Am. Chem. Soc. 1967, 89, 2697–2705. [Google Scholar] [CrossRef]
- Duthaler, R.O.; Hafner, A.; Alsters, P.L.; Rothe-Streit, P.; Rihs, G. Stetreoselective transformations mediated by chiral monocyclo-pentadienyl titanium, zirconium, and hafnium complexes. Pure Appl. Chem. 1992, 64, 1897–1910. [Google Scholar] [CrossRef]
- Schmidt, C. Efficient synthesis of 2'-deoxy-2'-α-C-substituted nucleosides. Synlett 1994, 238–240. [Google Scholar] [CrossRef]
- Schmidt, C.; Bevierre, M.O.; De Mesmaeker, A.; Altmann, K.H. The effects of 2'- and 3'-alkyl substituents on oligonucleotide hybridization and stability. Bioorg. Med. Chem. Lett. 1994, 4, 1969–1974. [Google Scholar] [CrossRef]
- Haraguchi, K.; Itoh, Y.; Tanaka, H.; Miyasaka, T. Preparation and reactions of 2'- and 3'-vinyl bromides of uracil-nucleosides: Versatile synthons for anti-HIV agents. Tetrahedron Lett. 1991, 32, 3391–3394. [Google Scholar] [CrossRef]
- Haraguchi, K.; Itoh, Y.; Tanaka, H.; Akita, M.; Miyasaka, T. Uracil and adenine nucleosides having a 2',3'-bromovinyl structure: Highly versatile synthons for the synthesis of 2'-C- and 3'-C-branched 2',3'-unsaturated derivatives. Tetrahedron 1993, 49, 1371–1390. [Google Scholar] [CrossRef]
- Kamaike, K.; Uemura, F.; Yamakage, S.; Nishino, S.; Ishido, Y. Partial protection of carbohydrate derivatives. Part 23. Simple, efficient procedure for the preparation of 3'- and 2'-O-(tetrahydropyran-2-yl)ribonucleoside derivatives involving highly regioselective 2',5'-di-O-acylation or that followed by acyl migration on silica gel and subsequent O-(tetrahydropyran-2-yl)ation. Nucleosides Nucleotides 1987, 6, 699–736. [Google Scholar] [CrossRef]
- Onuma, S.; Kumamoto, H.; Kawato, M.; Tanaka, H. A versatile intermediate for the synthesis of 3'-substituted 2',3'-didehydro-2',3'-dideoxyadenosine (d4A): Preparation of 3'-C-stannyl-d4A via radical-mediated desulfonylative stannylation. Tetrahedron 2002, 58, 2497–2503. [Google Scholar] [CrossRef]
- Robins, M.J.; Fouron, Y.; Mengel, R. Nucleic acid related compounds. II. Adenosine 2',3'-ribo-epoxide. Synthesis, intramolecular degradation, and transformation into 3'-substituted xylofuranosyl nucleosides and the lyxo-epoxide. J. Org. Chem. 1974, 39, 1564–1570. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, H.; Onuma, S.; Tsuchiya, K.; Egusa, Y.; Tanaka, H.; Satoh, T. Sulfoxide-metal exchange for the synthesis of the 2'-tributylstannyl derivative of the 2',3'-didehydro-2',3'-dideoxyuridine (d4U): A general entry to 2'-carbon-substituted analogues of d4U. Nucleosides Nucleotides Nucleic Acids 2002, 21, 275–286. [Google Scholar] [CrossRef]
- Divakar, K.J.; Reese, C.B. Reaction between 2,2'-anhydro-1-β-D-arabinofuranosyluracil and thiolate ions. J. Chem. Soc. Perkin Trans. 1 1982, 1625–1628. [Google Scholar] [CrossRef]
- Kumamoto, H.; Tanaka, H. Simple entry to 3'-substituted analogues of anti-HIV agent Stavudine based on an anionic O-C stannyl migration. J. Org. Chem. 2002, 67, 3541–3547. [Google Scholar] [CrossRef] [PubMed]
- Kumamoto, H.; Onuma, S.; Tanaka, H.; Dutschman, G.E.; Cheng, Y.C. 3'-Carbon-substituted pyrimidine nucleosides having a 2',3'-dideoxy and 2',3'-didehydro-2'-3'-dideoxy structure: Synthesis and antiviral evaluation. Antivir. Chem. Chemother. 2006, 17, 225–234. [Google Scholar] [CrossRef] [PubMed]
- Haraguchi, K.; Kubota, Y.; Tanaka, H. Ring opening of nucleoside 1',2'-epoxides with organoaluminium reagents: Stereoselective entry to ribonucleosides branched at the anomeric position. J. Org. Chem. 2004, 69, 1831–1836. [Google Scholar] [CrossRef]
- Haraguchi, K.; Itoh, Y.; Matsumoto, K.; Hashimoto, K.; Nakamuta, K.T.; Yanaka, H. Stetreoselective synthesis of 1'-C-branched arabinofuranosyl nucleosides via anomeric radicals generated by 1,2-acyloxy migration. J. Org. Chem. 2003, 68, 2006–2009. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y. Recent advances in intramolecular alkyne cyclotrimerization and its application. Curr. Org. Chem. 2005, 9, 503–519. [Google Scholar] [CrossRef]
- Ramana, C.V.; Suryawanshi, S.B. A [2+2+2]-cyclotrimerization approach for the synthesis of enantiopure isochromans using carbohydrate derived dialkyne template. Tetrahedron Lett. 2008, 49, 445–448. [Google Scholar] [CrossRef]
- Suryawanshi, S.B.; Dushing, M.P.; Gonnade, R.G.; Ramana, C.V. The isochroman and 1,3-dihydroisobenzofuran-annulation on carbohydrate templates via [2+2+2]-cyclotrimerization and synthesis of some tricyclic nucleosides. Tetrahedron 2010, 66, 6085–6096. [Google Scholar] [CrossRef]
- Adhavana, M.; Chari, K.S. Polymer-supported ferric chloride as a heterogeneous catalyst for chemoselective deprotection of acetonides. Synthesis 2005, 708–710. [Google Scholar]
- Dushing, M.P.; Ramana, C.V. Target cum flexibility: synthesis of C(3')-spiroannulated nucleosides. Tetrahedron Lett. 2011, 52, 4627–4630. [Google Scholar] [CrossRef]
- Hattori, H.; Tanaka, M.; Fukushima, M.; Sasaki, T.; Matsuda, A. J. Med. Chem. 1996, 39, 5005–5011. [CrossRef]
- Jogi, A.; Paju, A.; Pehk, T.; Kailas, T.; Muurisepp, A.M.; Lopp, M. Synthesis of 4'-aryl-2',3'-dideoxynucleoside analogues. Tetrahedron 2009, 65, 2959–2965. [Google Scholar] [CrossRef]
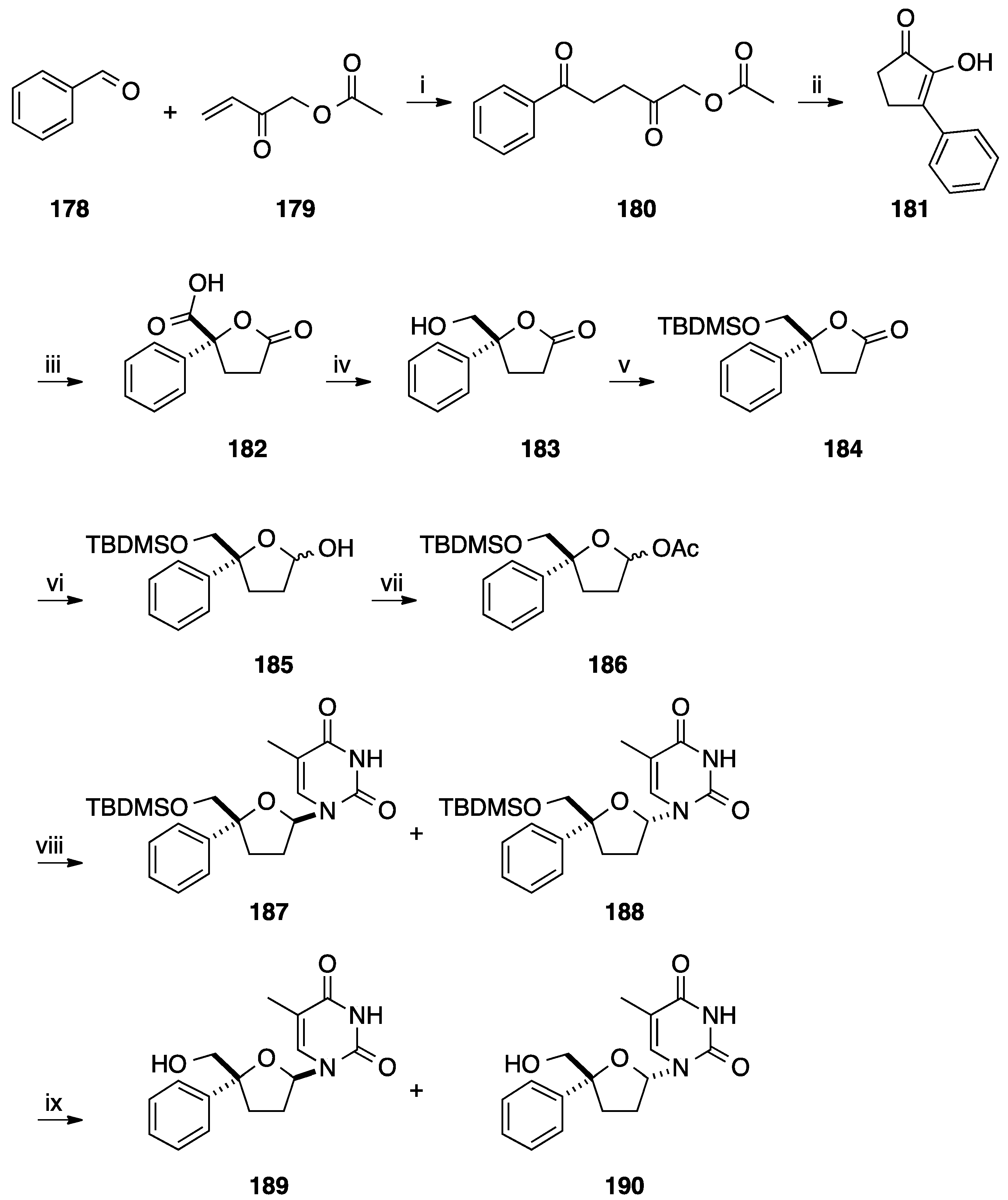
- Jogi, A.; Paju, A.; Pehk, T.; Kailas, T.; Muurisepp, A.M.; Kanger, T.; Lopp, M. Asymmetric synthesis of 2-aryl-5-oxotetrahydrofuran-2-carboxylic acids. Synthesis 2006, 18, 3031–3036. [Google Scholar]
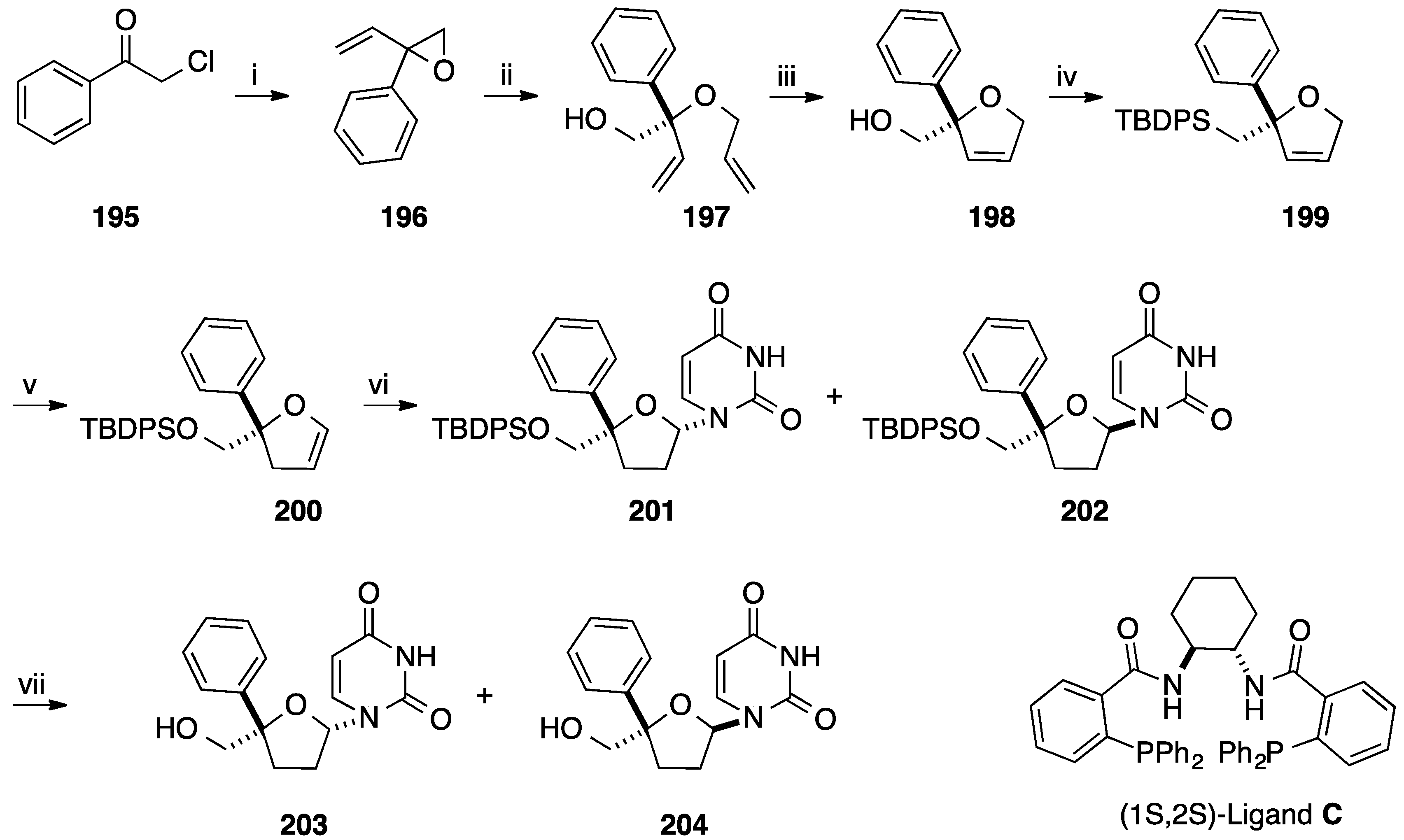
- Trost, B.M.; Brown, B.S.; McEachern, E.J.; Kuhn, O. Asymmetric synthesis of oxygen heterocycles via Pd-catalyzed dynamic kinetic asymmetric transformations: Application to nucleosides. Chem. Eur. J. 2003, 9, 4442–4451. [Google Scholar] [CrossRef] [PubMed]
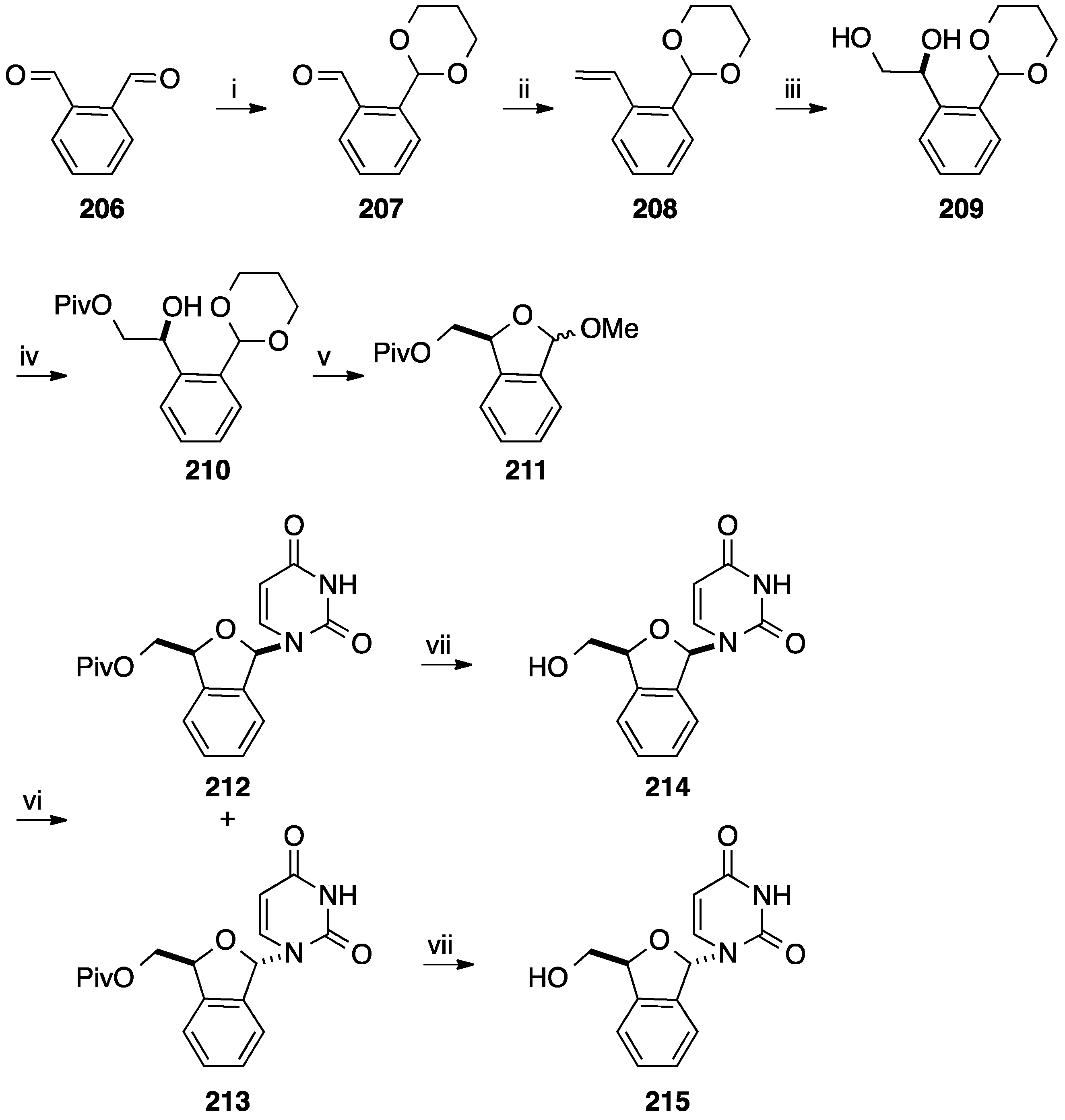
- Ewing, D.F.; Fahmi, N.; Len, C.; Mackenzie, G.; Ronco, G.; Villa, P.; Shaw, G. Nucleoside analogues: Glycones based on the benzo[c]furan core. Collect. Czechoslov. Chem. Commun. 1996, 61, 145–147. [Google Scholar]
- Ewing, D.F.; Fahmi, N.; Len, C.; Mackenzie, G.; Ronco, G.; Villa, P.; Shaw, G. Nucleoside analogues with a novel glycone based on the benzo[c]furan core. Nucleosides Nucleotides 1999, 18, 2613–2630. [Google Scholar] [CrossRef]
- Ewing, D.F.; Fahmi, N.; Len, C.; Mackenzie, G.; Pranzo, A. Stereoisomeric pyrimidine nucleoside analogues based on the benzo[c]furane core. J. Chem. Soc. Perkin Trans. 1 2000, 21, 3561–3565. [Google Scholar] [CrossRef]
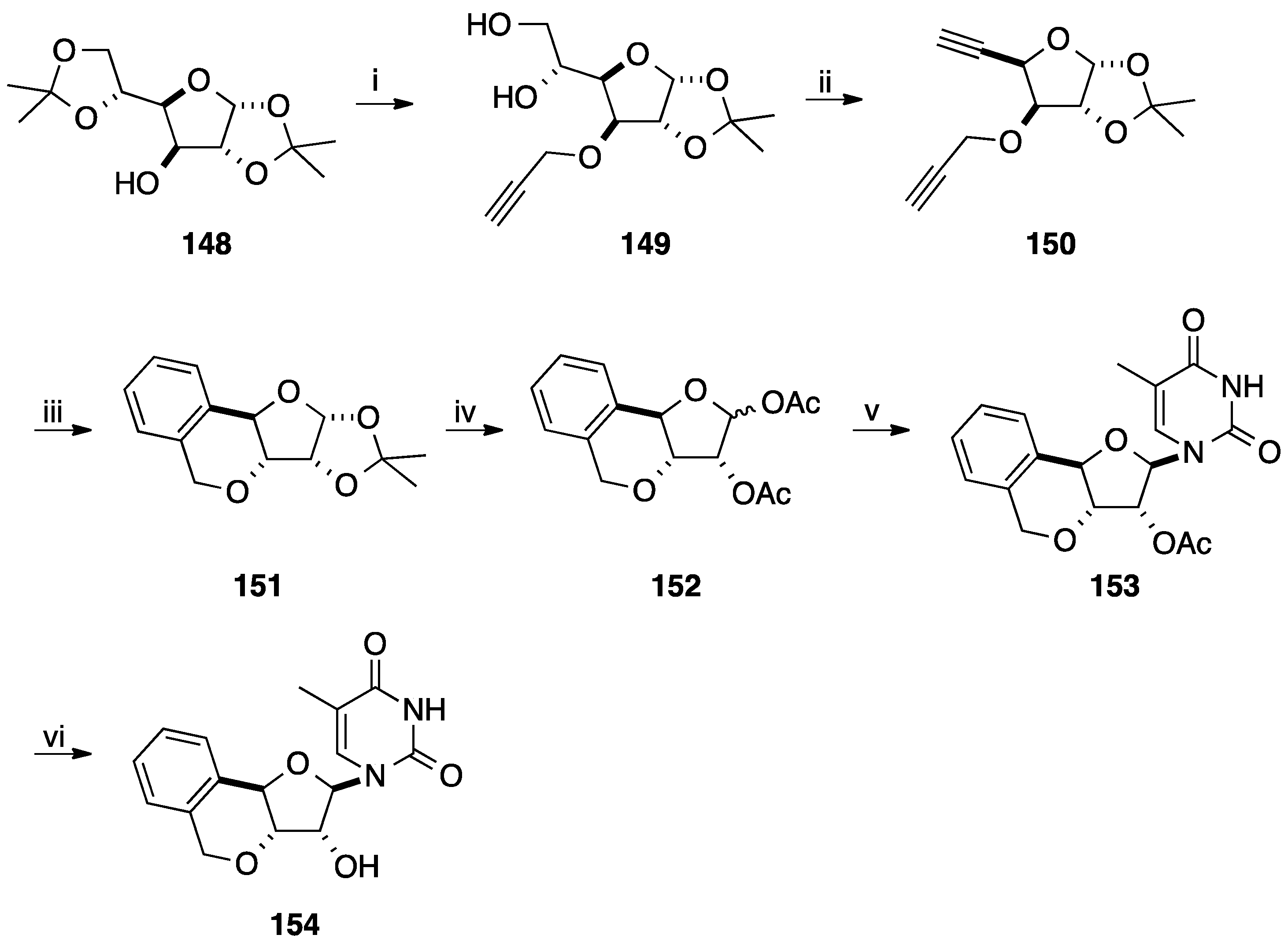
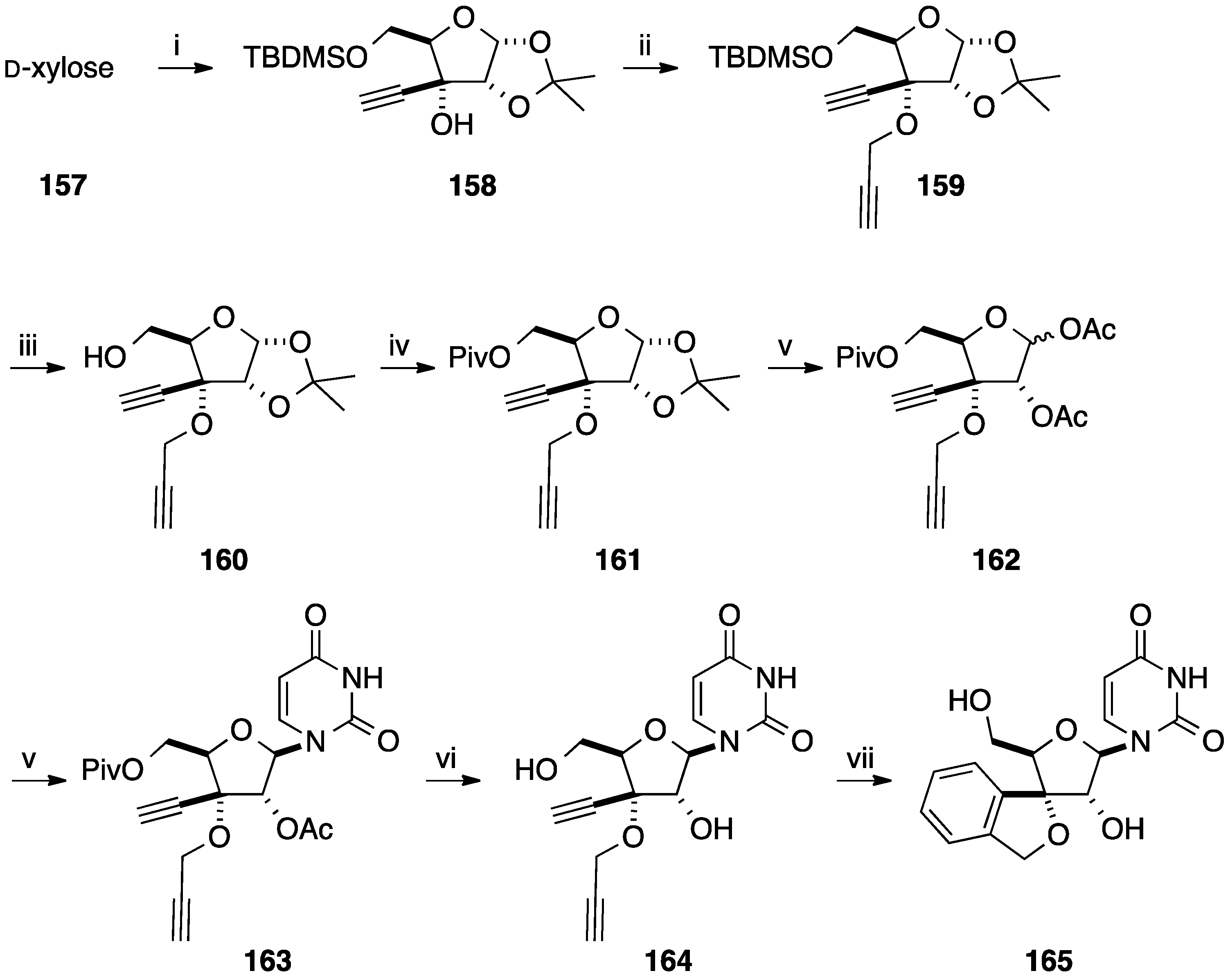
- Ewing, D.F.; Len, C.; Mackenzie, G.; Ronco, G.; Villa, P. Asymmetric synthesis of 1,3-dihydrobenzo[c]furan derivatives using a d-xylose moiety as a chiral auxiliary. Tetrahedron Asymmetry 2000, 11, 4995–5002. [Google Scholar] [CrossRef]
- Belloli, E.; Len, C.; Mackenzie, G.; Ronco, G.; Bonte, J.P.; Vaccher, C. Diastereomeric resolution of nucleoside analogues, new potential antiviral agents, using high-performance liquid chromatography on polysaccharide-type chiral stationary phases. J. Chromatogr. A 2001, 943, 91–100. [Google Scholar] [CrossRef]
- Pilard, S.; Riboul, D.; Glaçon, V.; Moitessier, N.; Chapleur, Y.; Postel, D.; Len, C. Asymmetric dihydroxylation of chiral styrene derivatives: Development of an analytical strategy for the determination of the diastereomeric excess. Tetrahedron Asymmetry 2002, 13, 529–537. [Google Scholar] [CrossRef]
- Len, C.; Sélouane, A.; Postel, D.; Villa, P.; Aubertin, A.M.; Egron, D.; Gosselin, G.; Périgaud, C. Synthesis, stability and biological evaluation of 1,3-dihydrobenzo[c]furan analogues of d4T and its SATE pronucleotide. Nucleosides Nucleotides Nucleic Acids 2003, 5–8, 943–946. [Google Scholar] [CrossRef]
- Egron, D.; Périgaud, C.; Gosselin, G.; Aubertin, A.M.; Faraj, A.; Sélouane, A.; Postel, D.; Len, C. 1,3-Dihydrobenzo[c]furan nucleoside analogues: Additional studies of the thymine derivative. Bioorg. Med. Chem. Lett. 2003, 13, 4473–4475. [Google Scholar] [CrossRef] [PubMed]
- Len, C.; Mackenzie, G.; Ewing, D.F.; Sheppard, G.; Banoub, J. Electrospray tandem mass spectrometric analysis of diastereo- and stereoisomeric pyrimidine nucleoside analogues based on the 1,3-dihydrobenzo[c]furan core. Carbohydr. Res. 2003, 338, 2311–2324. [Google Scholar] [CrossRef] [PubMed]
- Lipka, E.; Sélouane, A.; Postel, D.; Len, C.; Vaccher, M.P.; Bonte, J.P.; Vaccher, C. Enantioseparation of four cis and trans diastereoisomers of d4T analogs, by High Performance Liquid Chromaography and Capillary Electrophoresis. J. Chromatogr. A 2004, 1034, 161–167. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.P.; Meng, Z.L.; Wang, D.F. Synthesis of novel nucleoside analogues based on 1,3-dihydrobenzo[c]furan core. Chin. J. Chem. 2006, 24, 504–508. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Len, C.; Enderlin, G. Synthesis of C-Arylnucleoside Analogues. Molecules 2015, 20, 4967-4997. https://doi.org/10.3390/molecules20034967
Len C, Enderlin G. Synthesis of C-Arylnucleoside Analogues. Molecules. 2015; 20(3):4967-4997. https://doi.org/10.3390/molecules20034967
Chicago/Turabian StyleLen, Christophe, and Gérald Enderlin. 2015. "Synthesis of C-Arylnucleoside Analogues" Molecules 20, no. 3: 4967-4997. https://doi.org/10.3390/molecules20034967
APA StyleLen, C., & Enderlin, G. (2015). Synthesis of C-Arylnucleoside Analogues. Molecules, 20(3), 4967-4997. https://doi.org/10.3390/molecules20034967