
Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1)

{kind=link}
{kind=link}
Abstract
:1. Introduction
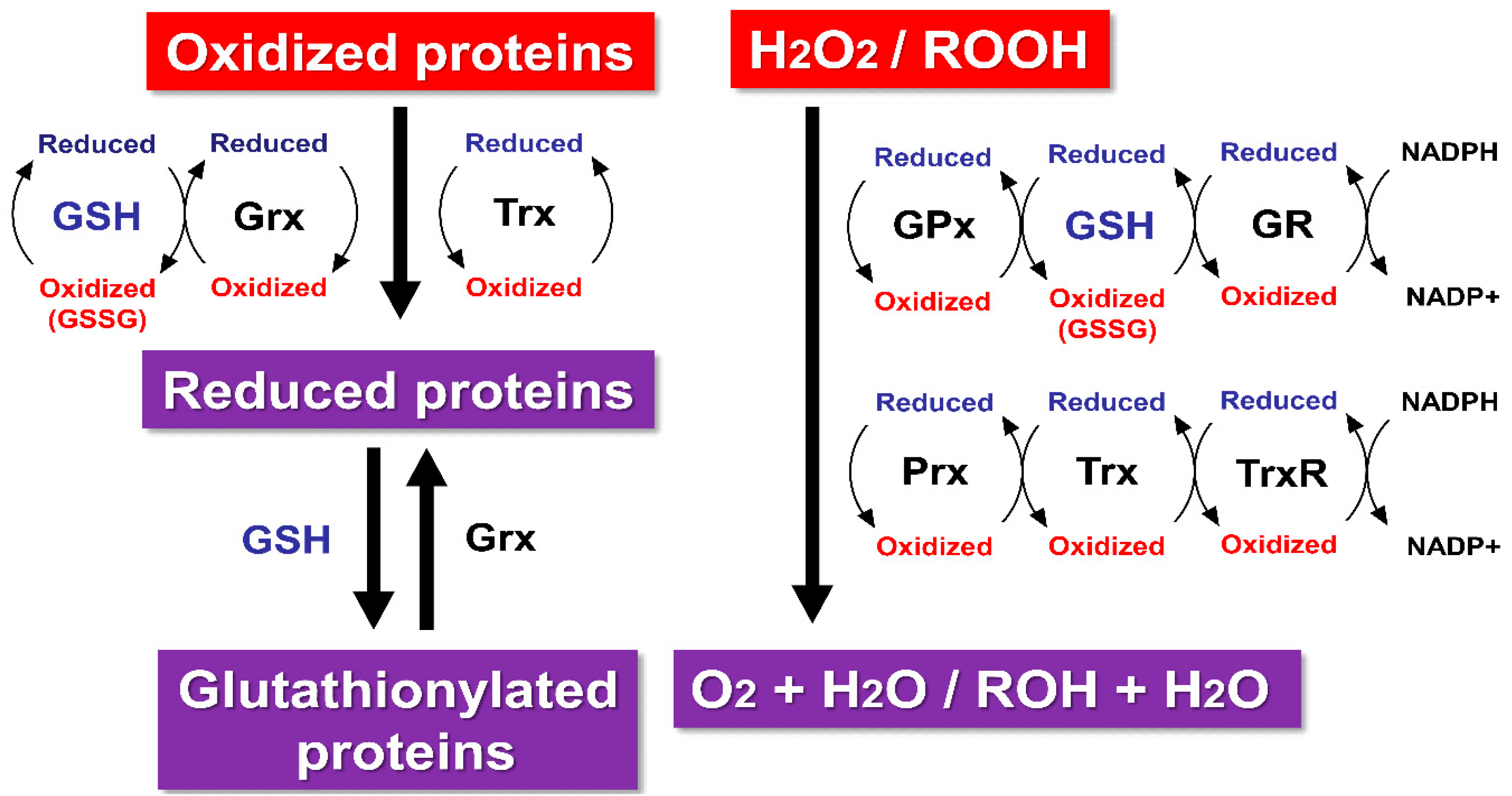
2. Glutathione as a Redox Buffer
3. Antioxidant Defense System

4. ROS/RNS Generation
5. Glutathione as a Regulator of Redox Signal Transduction
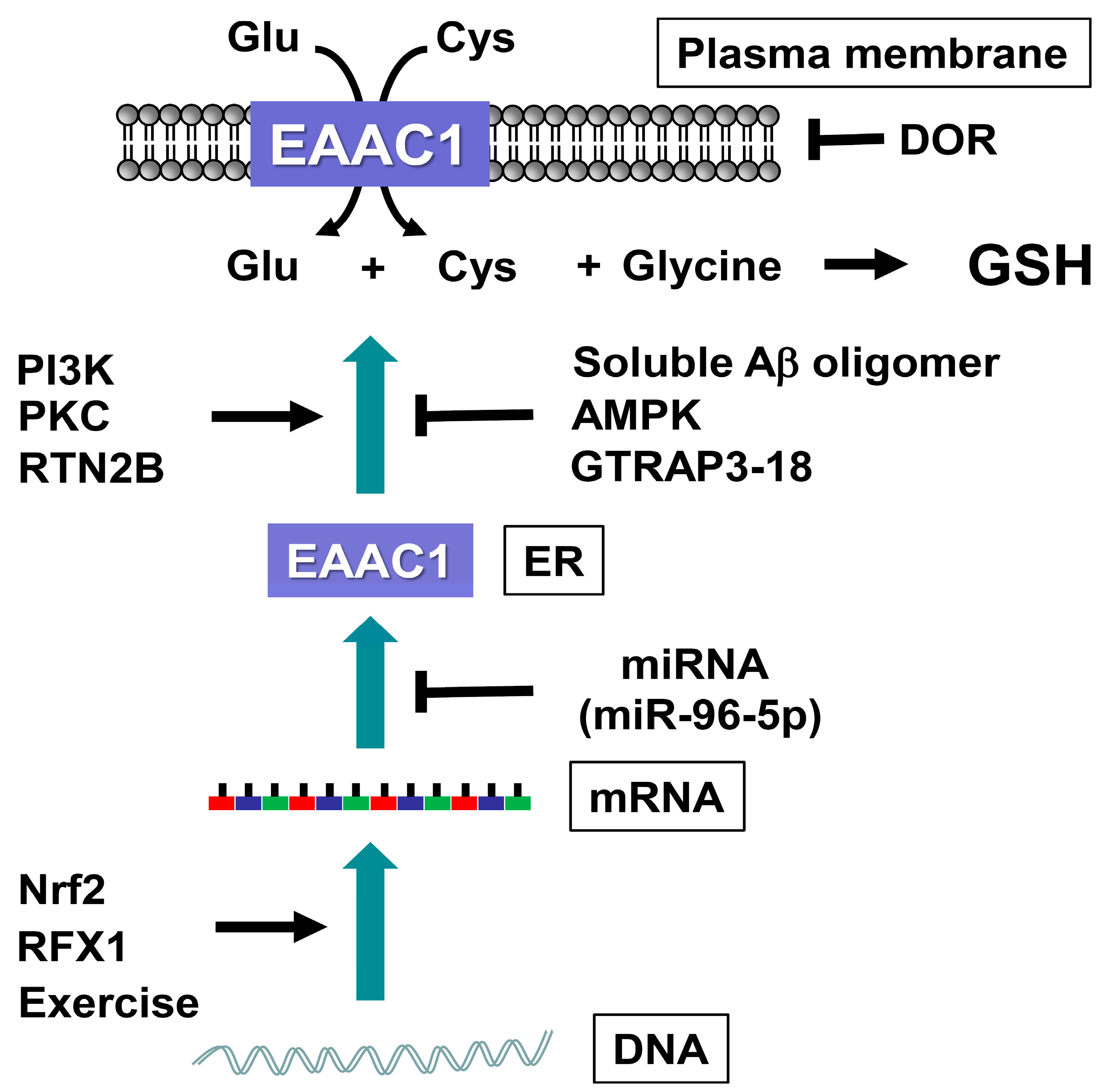
6. EAAC1 Dysfunction Leading to Neurodegeneration
7. Regulation of EAAC1

8. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Harman, D. Aging: A theory based on free radical and radiation chemistry. J. Gerontol. 1956, 11, 298–300. [Google Scholar] [CrossRef] [PubMed]
- Imlay, J.A. Cellular defenses against superoxide and hydrogen peroxide. Annu. Rev. Biochem. 2008, 77, 755–776. [Google Scholar] [CrossRef] [PubMed]
- Brosnan, J.T.; Brosnan, M.E. The sulfur-containing amino acids: An overview. J. Nutr. 2006, 136, 1636s–1640s. [Google Scholar] [PubMed]
- Dickinson, D.A.; Forman, H.J. Glutathione in defense and signaling: Lessons from a small thiol. Ann. N. Y. Acad. Sci. 2002, 973, 488–504. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Watabe, M.; Nakaki, T. Regulation of neuronal glutathione synthesis. J. Pharmacol. Sci. 2008, 108, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Impaired glutathione synthesis in neurodegeneration. Int. J. Mol. Sci. 2013, 14, 21021–21044. [Google Scholar] [CrossRef] [PubMed]
- Commandeur, J.N.; Stijntjes, G.J.; Vermeulen, N.P. Enzymes and transport systems involved in the formation and disposition of glutathione S-conjugates. Role in bioactivation and detoxication mechanisms of xenobiotics. Pharmacol. Rev. 1995, 47, 271–330. [Google Scholar] [PubMed]
- McBean, G.J. The transsulfuration pathway: A source of cysteine for glutathione in astrocytes. Amino Acids 2012, 42, 199–205. [Google Scholar] [CrossRef] [PubMed]
- Schafer, F.Q.; Buettner, G.R. Redox environment of the cell as viewed through the redox state of the glutathione disulfide/glutathione couple. Free Radic. Biol. Med. 2001, 30, 1191–1212. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, H.F. Redox control of enzyme activities by thiol/disulfide exchange. Methods Enzymol. 1984, 107, 330–351. [Google Scholar] [PubMed]
- Gilbert, H.F. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995, 251, 8–28. [Google Scholar] [PubMed]
- Poot, M.; Teubert, H.; Rabinovitch, P.S.; Kavanagh, T.J. De novo synthesis of glutathione is required for both entry into and progression through the cell cycle. J. Cell. Physiol. 1995, 163, 555–560. [Google Scholar] [CrossRef] [PubMed]
- Arrigo, A.P. Gene expression and the thiol redox state. Free Radic. Biol. Med. 1999, 27, 936–944. [Google Scholar] [CrossRef] [PubMed]
- Voehringer, D.W. Bcl-2 and glutathione: Alterations in cellular redox state that regulate apoptosis sensitivity. Free Radic. Biol. Med. 1999, 27, 945–950. [Google Scholar] [CrossRef] [PubMed]
- Filomeni, G.; Rotilio, G.; Ciriolo, M.R. Glutathione disulfide induces apoptosis in U937 cells by a redox-mediated p38 MAP kinase pathway. FASEB J. 2003, 17, 64–66. [Google Scholar] [PubMed]
- Markovic, J.; Borras, C.; Ortega, A.; Sastre, J.; Vina, J.; Pallardo, F.V. Glutathione is recruited into the nucleus in early phases of cell proliferation. J. Biol. Chem. 2007, 282, 20416–20424. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Protein glutathionylation in health and disease. Biochim. Biophys. Acta 2013, 1830, 3165–3172. [Google Scholar] [CrossRef] [PubMed]
- Klatt, P.; Lamas, S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur. J. Biochem. 2000, 267, 4928–4944. [Google Scholar] [CrossRef] [PubMed]
- Giustarini, D.; Rossi, R.; Milzani, A.; Colombo, R.; Dalle-Donne, I. S-glutathionylation: From redox regulation of protein functions to human diseases. J. Cell. Mol. Med. 2004, 8, 201–212. [Google Scholar] [CrossRef] [PubMed]
- Hwang, C.; Sinskey, A.J.; Lodish, H.F. Oxidized redox state of glutathione in the endoplasmic reticulum. Science 1992, 257, 1496–1502. [Google Scholar] [CrossRef] [PubMed]
- Bass, R.; Ruddock, L.W.; Klappa, P.; Freedman, R.B. A major fraction of endoplasmic reticulum-located glutathione is present as mixed disulfides with protein. J. Biol. Chem. 2004, 279, 5257–5262. [Google Scholar] [CrossRef] [PubMed]
- Banhegyi, G.; Lusini, L.; Puskas, F.; Rossi, R.; Fulceri, R.; Braun, L.; Mile, V.; di Simplicio, P.; Mandl, J.; Benedetti, A. Preferential transport of glutathione versus glutathione disulfide in rat liver microsomal vesicles. J. Biol. Chem. 1999, 274, 12213–12216. [Google Scholar] [CrossRef] [PubMed]
- Banhegyi, G.; Csala, M.; Nagy, G.; Sorrentino, V.; Fulceri, R.; Benedetti, A. Evidence for the transport of glutathione through ryanodine receptor channel type 1. Biochem. J. 2003, 376, 807–812. [Google Scholar] [CrossRef] [PubMed]
- Thannickal, V.J.; Fanburg, B.L. Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell. Mol. Physiol. 2000, 279, L1005–L1028. [Google Scholar] [PubMed]
- Robertson, G.; Leclercq, I.; Farrell, G.C. Nonalcoholic steatosis and steatohepatitis. II. Cytochrome P-450 enzymes and oxidative stress. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G1135–G1139. [Google Scholar] [PubMed]
- Jezek, P.; Hlavata, L. Mitochondria in homeostasis of reactive oxygen species in cell, tissues, and organism. Int. J. Biochem. Cell Biol. 2005, 37, 2478–2503. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Sies, H.; Boveris, A. Hydroperoxide metabolism in mammalian organs. Physiol. Rev. 1979, 59, 527–605. [Google Scholar] [PubMed]
- St-Pierre, J.; Buckingham, J.A.; Roebuck, S.J.; Brand, M.D. Topology of superoxide production from different sites in the mitochondrial electron transport chain. J. Biol. Chem. 2002, 277, 44784–44790. [Google Scholar] [CrossRef] [PubMed]
- Cadenas, E.; Davies, K.J. Mitochondrial free radical generation, oxidative stress, and aging. Free Radic. Biol. Med. 2000, 29, 222–230. [Google Scholar] [CrossRef] [PubMed]
- Tyler, D.D. Polarographic assay and intracellular distribution of superoxide dismutase in rat liver. Biochem. J. 1975, 147, 493–504. [Google Scholar] [PubMed]
- Singh, I. Biochemistry of peroxisomes in health and disease. Mol. Cell. Biochem. 1997, 167, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Antonenkov, V.D.; Grunau, S.; Ohlmeier, S.; Hiltunen, J.K. Peroxisomes are oxidative organelles. Antioxid. Redox Signal. 2010, 13, 525–537. [Google Scholar] [CrossRef] [PubMed]
- Giordano, C.R.; Terlecky, S.R. Peroxisomes, cell senescence, and rates of aging. Biochim. Biophys. Acta 2012, 1822, 1358–1362. [Google Scholar] [CrossRef] [PubMed]
- Rahman, K. Studies on free radicals, antioxidants, and co-factors. Clin. Interv. Aging 2007, 2, 219–236. [Google Scholar] [PubMed]
- Girotti, A.W. Lipid hydroperoxide generation, turnover, and effector action in biological systems. J. Lipid Res. 1998, 39, 1529–1542. [Google Scholar] [PubMed]
- Rhee, S.G.; Chae, H.Z.; Kim, K. Peroxiredoxins: A historical overview and speculative preview of novel mechanisms and emerging concepts in cell signaling. Free Radic. Biol. Med. 2005, 38, 1543–1552. [Google Scholar] [CrossRef] [PubMed]
- Ahsan, M.K.; Lekli, I.; Ray, D.; Yodoi, J.; Das, D.K. Redox regulation of cell survival by the thioredoxin superfamily: An implication of redox gene therapy in the heart. Antioxid. Redox Signal. 2009, 11, 2741–2758. [Google Scholar] [CrossRef] [PubMed]
- Brigelius-Flohe, R.; Maiorino, M. Glutathione peroxidases. Biochim. Biophys. Acta 2013, 1830, 3289–3303. [Google Scholar] [CrossRef] [PubMed]
- Ritz, D.; Beckwith, J. Roles of thiol-redox pathways in bacteria. Annu. Rev. Microbiol. 2001, 55, 21–48. [Google Scholar] [CrossRef] [PubMed]
- Dringen, R.; Gutterer, J.M. Glutathione reductase from bovine brain. Methods Enzymol. 2002, 348, 281–288. [Google Scholar] [PubMed]
- Gallogly, M.M.; Mieyal, J.J. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr. Opin. Pharmacol. 2007, 7, 381–391. [Google Scholar] [CrossRef] [PubMed]
- Lillig, C.H.; Berndt, C.; Holmgren, A. Glutaredoxin systems. Biochim. Biophys. Acta 2008, 1780, 1304–1317. [Google Scholar] [CrossRef] [PubMed]
- Rhee, S.G.; Woo, H.A.; Kil, I.S.; Bae, S.H. Peroxiredoxin functions as a peroxidase and a regulator and sensor of local peroxides. J. Biol. Chem. 2012, 287, 4403–4410. [Google Scholar] [CrossRef] [PubMed]
- Arner, E.S.; Holmgren, A. Measurement of thioredoxin and thioredoxin reductase. Curr. Protoc. Toxicol. 2001, Chapter 7. Unit 7.4. [Google Scholar]
- Mari, M.; Morales, A.; Colell, A.; Garcia-Ruiz, C.; Fernandez-Checa, J.C. Mitochondrial glutathione, a key survival antioxidant. Antioxid. Redox Signal. 2009, 11, 2685–2700. [Google Scholar] [CrossRef] [PubMed]
- Stults, F.H.; Forstrom, J.W.; Chiu, D.T.; Tappel, A.L. Rat liver glutathione peroxidase: Purification and study of multiple forms. Arch. Biochem. Biophys. 1977, 183, 490–497. [Google Scholar] [CrossRef] [PubMed]
- Staniek, K.; Nohl, H. Are mitochondria a permanent source of reactive oxygen species? Biochim. Biophys. Acta 2000, 1460, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef] [PubMed]
- Turrens, J.F. Mitochondrial formation of reactive oxygen species. J. Physiol. 2003, 552, 335–344. [Google Scholar] [CrossRef] [PubMed]
- Townsend, D.M.; Tew, K.D.; Tapiero, H. The importance of glutathione in human disease. Biomed. Pharmacother. 2003, 57, 145–155. [Google Scholar] [CrossRef] [PubMed]
- Ristoff, E.; Larsson, A. Inborn errors in the metabolism of glutathione. Orphanet J. Rare Dis. 2007, 2, 16. [Google Scholar] [CrossRef] [PubMed]
- Lin, M.T.; Beal, M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006, 443, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.H.; Fountoulakis, M.; Cairns, N.; Lubec, G. Protein levels of human peroxiredoxin subtypes in brains of patients with Alzheimer’s disease and Down syndrome. J. Neural Transm. Suppl. 2001, 223–235. [Google Scholar]
- Krapfenbauer, K.; Engidawork, E.; Cairns, N.; Fountoulakis, M.; Lubec, G. Aberrant expression of peroxiredoxin subtypes in neurodegenerative disorders. Brain Res. 2003, 967, 152–160. [Google Scholar] [CrossRef] [PubMed]
- Kato, S.; Kato, M.; Abe, Y.; Matsumura, T.; Nishino, T.; Aoki, M.; Itoyama, Y.; Asayama, K.; Awaya, A.; Hirano, A.; et al. Redox system expression in the motor neurons in amyotrophic lateral sclerosis (ALS): Immunohistochemical studies on sporadic ALS, superoxide dismutase 1 (SOD1)-mutated familial ALS, and SOD1-mutated ALS animal models. Acta Neuropathol. 2005, 110, 101–112. [Google Scholar] [CrossRef] [PubMed]
- Fang, J.; Nakamura, T.; Cho, D.H.; Gu, Z.; Lipton, S.A. S-nitrosylation of peroxiredoxin 2 promotes oxidative stress-induced neuronal cell death in Parkinson’s disease. Proc. Natl. Acad. Sci. USA 2007, 104, 18742–18747. [Google Scholar] [CrossRef] [PubMed]
- Sorolla, M.A.; Reverter-Branchat, G.; Tamarit, J.; Ferrer, I.; Ros, J.; Cabiscol, E. Proteomic and oxidative stress analysis in human brain samples of Huntington disease. Free Radic. Biol. Med. 2008, 45, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Ferri, A.; Fiorenzo, P.; Nencini, M.; Cozzolino, M.; Pesaresi, M.G.; Valle, C.; Sepe, S.; Moreno, S.; Carri, M.T. Glutaredoxin 2 prevents aggregation of mutant SOD1 in mitochondria and abolishes its toxicity. Hum. Mol. Genet. 2010, 19, 4529–4542. [Google Scholar] [CrossRef] [PubMed]
- Sabens Liedhegner, E.A.; Gao, X.H.; Mieyal, J.J. Mechanisms of altered redox regulation in neurodegenerative diseases-focus on S-glutathionylation. Antioxid. Redox Signal. 2012, 16, 543–566. [Google Scholar] [CrossRef] [PubMed]
- Arodin, L.; Lamparter, H.; Karlsson, H.; Nennesmo, I.; Bjornstedt, M.; Schroder, J.; Fernandes, A.P. Alteration of thioredoxin and glutaredoxin in the progression of Alzheimer’s disease. J. Alzheimers Dis. 2014, 39, 787–797. [Google Scholar] [PubMed]
- Johnson, W.M.; Yao, C.; Siedlak, S.L.; Wang, W.; Zhu, X.; Caldwell, G.A.; Wilson-Delfosse, A.L.; Mieyal, J.J.; Chen, S.G. Glutaredoxin deficiency exacerbates neurodegeneration in C. elegans models of Parkinson’s disease. Hum. Mol. Genet. 2015, 24, 1322–1335. [Google Scholar] [CrossRef] [PubMed]
- Casado, A.; Encarnacion Lopez-Fernandez, M.; Concepcion Casado, M.; de La Torre, R. Lipid peroxidation and antioxidant enzyme activities in vascular and Alzheimer dementias. Neurochem. Res. 2008, 33, 450–458. [Google Scholar] [CrossRef] [PubMed]
- Paz-y-Mino, C.; Carrera, C.; Lopez-Cortes, A.; Munoz, M.J.; Cumbal, N.; Castro, B.; Cabrera, A.; Sanchez, M.E. Genetic polymorphisms in apolipoprotein e and glutathione peroxidase 1 genes in the ecuadorian population affected with Alzheimer’s disease. Am. J. Med. Sci. 2010, 340, 373–377. [Google Scholar] [CrossRef] [PubMed]
- Kish, S.J.; Morito, C.; Hornykiewicz, O. Glutathione peroxidase activity in Parkinson’s disease brain. Neurosci. Lett. 1985, 58, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Albers, D.S.; Augood, S.J. New insights into progressive supranuclear palsy. Trends Neurosci. 2001, 24, 347–353. [Google Scholar] [CrossRef] [PubMed]
- Fitzmaurice, P.S.; Ang, L.; Guttman, M.; Rajput, A.H.; Furukawa, Y.; Kish, S.J. Nigral glutathione deficiency is not specific for idiopathic Parkinson’s disease. Mov. Disord. 2003, 18, 969–976. [Google Scholar] [CrossRef] [PubMed]
- Kinter, M.; Roberts, R.J. Glutathione consumption and glutathione peroxidase inactivation in fibroblast cell lines by 4-hydroxy-2-nonenal. Free Radic. Biol. Med. 1996, 21, 457–462. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Matsubara, K.; Kobayashi, S. Aging and oxidative stress in progressive supranuclear palsy. Eur. J. Neurol. 2006, 13, 89–92. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative damage in neurodegenerative disease. Lancet 1994, 344, 796–798. [Google Scholar] [CrossRef] [PubMed]
- Barnham, K.J.; Masters, C.L.; Bush, A.I. Neurodegenerative diseases and oxidative stress. Nat. Rev. Drug Discov. 2004, 3, 205–214. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Oxidative stress and neurodegeneration: Where are we now? J. Neurochem. 2006, 97, 1634–1658. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Matsubara, K.; Fujikawa, Y.; Nagahiro, Y.; Shimizu, K.; Umegae, N.; Hayase, N.; Shiono, H.; Kobayashi, S. Nitration of manganese superoxide dismutase in cerebrospinal fluids is a marker for peroxynitrite-mediated oxidative stress in neurodegenerative diseases. Ann. Neurol. 2000, 47, 524–527. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev. 2007, 87, 315–424. [Google Scholar] [CrossRef] [PubMed]
- Szabo, C.; Ischiropoulos, H.; Radi, R. Peroxynitrite: Biochemistry, pathophysiology and development of therapeutics. Nat. Rev. Drug Discov. 2007, 6, 662–680. [Google Scholar] [CrossRef] [PubMed]
- Marletta, M.A. Nitric oxide synthase: Aspects concerning structure and catalysis. Cell 1994, 78, 927–930. [Google Scholar] [CrossRef] [PubMed]
- Daff, S. No synthase: Structures and mechanisms. Nitric Oxide 2010, 23, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schroder, N.W.; Opitz, B.; Lamping, N.; Michelsen, K.S.; Zahringer, U.; Gobel, U.B.; Schumann, R.R. Involvement of lipopolysaccharide binding protein, CD14, and Toll-like receptors in the initiation of innate immune responses by Treponema glycolipids. J. Immunol. 2000, 165, 2683–2693. [Google Scholar] [CrossRef] [PubMed]
- Ledo, A.; Barbosa, R.M.; Gerhardt, G.A.; Cadenas, E.; Laranjinha, J. Concentration dynamics of nitric oxide in rat hippocampal subregions evoked by stimulation of the NMDA glutamate receptor. Proc. Natl. Acad. Sci. USA 2005, 102, 17483–17488. [Google Scholar] [CrossRef] [PubMed]
- Marla, S.S.; Lee, J.; Groves, J.T. Peroxynitrite rapidly permeates phospholipid membranes. Proc. Natl. Acad. Sci. USA 1997, 94, 14243–14248. [Google Scholar] [CrossRef] [PubMed]
- Denicola, A.; Souza, J.M.; Radi, R. Diffusion of peroxynitrite across erythrocyte membranes. Proc. Natl. Acad. Sci. USA 1998, 95, 3566–3571. [Google Scholar] [CrossRef] [PubMed]
- Beckman, J.S. Peroxynitrite versus hydroxyl radical: The role of nitric oxide in superoxide-dependent cerebral injury. Ann. N. Y. Acad. Sci. 1994, 738, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Malinski, T.; Bailey, F.; Zhang, Z.G.; Chopp, M. Nitric oxide measured by a porphyrinic microsensor in rat brain after transient middle cerebral artery occlusion. J. Cereb. Blood Flow Metab. 1993, 13, 355–358. [Google Scholar] [CrossRef] [PubMed]
- Cherian, L.; Goodman, J.C.; Robertson, C.S. Brain nitric oxide changes after controlled cortical impact injury in rats. J. Neurophysiol. 2000, 83, 2171–2178. [Google Scholar] [PubMed]
- Torreilles, F.; Salman-Tabcheh, S.; Guerin, M.; Torreilles, J. Neurodegenerative disorders: The role of peroxynitrite. Brain Res. Rev. 1999, 30, 153–163. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Rossi, D.; Gjesdal, O.; Levy, L.M.; Racagni, G.; Danbolt, N.C.; Volterra, A. Peroxynitrite inhibits glutamate transporter subtypes. J. Biol. Chem. 1996, 271, 5976–5979. [Google Scholar] [CrossRef] [PubMed]
- Radi, R.; Rodriguez, M.; Castro, L.; Telleri, R. Inhibition of mitochondrial electron transport by peroxynitrite. Arch. Biochem. Biophys. 1994, 308, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Ischiropoulos, H.; Zhu, L.; Chen, J.; Tsai, M.; Martin, J.C.; Smith, C.D.; Beckman, J.S. Peroxynitrite-mediated tyrosine nitration catalyzed by superoxide dismutase. Arch. Biochem. Biophys. 1992, 298, 431–437. [Google Scholar] [CrossRef] [PubMed]
- Yamakura, F.; Taka, H.; Fujimura, T.; Murayama, K. Inactivation of human manganese-superoxide dismutase by peroxynitrite is caused by exclusive nitration of tyrosine 34 to 3-nitrotyrosine. J. Biol. Chem. 1998, 273, 14085–14089. [Google Scholar] [CrossRef] [PubMed]
- Halliwell, B. Reactive oxygen species and the central nervous system. J. Neurochem. 1992, 59, 1609–1623. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Gunn, C.; Beckman, J.S. Bactericidal activity of peroxynitrite. Arch. Biochem. Biophys. 1992, 298, 452–457. [Google Scholar] [CrossRef] [PubMed]
- Hutchinson, F. The distance that a radical formed by ionizing radiation can diffuse in a yeast cell. Radiat. Res. 1957, 7, 473–483. [Google Scholar] [CrossRef] [PubMed]
- Tabner, B.J.; Turnbull, S.; El-Agnaf, O.M.; Allsop, D. Formation of hydrogen peroxide and hydroxyl radicals from Aβ and α-synuclein as a possible mechanism of cell death in Alzheimer’s disease and Parkinson’s disease. Free Radic. Biol. Med. 2002, 32, 1076–1083. [Google Scholar] [CrossRef] [PubMed]
- Jones, C.M.; Lawrence, A.; Wardman, P.; Burkitt, M.J. Electron paramagnetic resonance spin trapping investigation into the kinetics of glutathione oxidation by the superoxide radical: Re-evaluation of the rate constant. Free Radic. Biol. Med. 2002, 32, 982–990. [Google Scholar] [CrossRef] [PubMed]
- Kharitonov, V.G.; Sundquist, A.R.; Sharma, V.S. Kinetics of nitrosation of thiols by nitric oxide in the presence of oxygen. J. Biol. Chem. 1995, 270, 28158–28164. [Google Scholar] [CrossRef] [PubMed]
- Quijano, C.; Alvarez, B.; Gatti, R.M.; Augusto, O.; Radi, R. Pathways of peroxynitrite oxidation of thiol groups. Biochem. J. 1997, 322, 167–173. [Google Scholar] [PubMed]
- Dalle-Donne, I.; Rossi, R.; Giustarini, D.; Colombo, R.; Milzani, A. S-glutathionylation in protein redox regulation. Free Radic. Biol. Med. 2007, 43, 883–898. [Google Scholar] [CrossRef] [PubMed]
- Aquilano, K.; Baldelli, S.; Ciriolo, M.R. Glutathione: New roles in redox signaling for an old antioxidant. Front. Pharmacol. 2014, 5, 196. [Google Scholar] [CrossRef] [PubMed]
- Ghezzi, P. Regulation of protein function by glutathionylation. Free Radic. Res. 2005, 39, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Biswas, S.; Chida, A.S.; Rahman, I. Redox modifications of protein-thiols: Emerging roles in cell signaling. Biochem. Pharmacol. 2006, 71, 551–564. [Google Scholar] [CrossRef] [PubMed]
- Jacob, C.; Knight, I.; Winyard, P.G. Aspects of the biological redox chemistry of cysteine: From simple redox responses to sophisticated signalling pathways. Biol. Chem. 2006, 387, 1385–1397. [Google Scholar] [CrossRef] [PubMed]
- Fratelli, M.; Goodwin, L.O.; Orom, U.A.; Lombardi, S.; Tonelli, R.; Mengozzi, M.; Ghezzi, P. Gene expression profiling reveals a signaling role of glutathione in redox regulation. Proc. Natl. Acad. Sci. USA 2005, 102, 13998–14003. [Google Scholar] [CrossRef] [PubMed]
- Pastore, A.; Piemonte, F. S-glutathionylation signaling in cell biology: Progress and prospects. Eur. J. Pharm. Sci. 2012, 46, 279–292. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.J.; Lu, C.T.; Lee, T.Y.; Chen, Y.J. Dbgsh: A database of S-glutathionylation. Bioinformatics 2014, 30, 2386–2388. [Google Scholar] [CrossRef] [PubMed]
- Danbolt, N.C. Glutamate uptake. Prog. Neurobiol. 2001, 65, 1–105. [Google Scholar] [CrossRef] [PubMed]
- Rothstein, J.D.; Dykes-Hoberg, M.; Pardo, C.A.; Bristol, L.A.; Jin, L.; Kuncl, R.W.; Kanai, Y.; Hediger, M.A.; Wang, Y.; Schielke, J.P.; et al. Knockout of glutamate transporters reveals a major role for astroglial transport in excitotoxicity and clearance of glutamate. Neuron 1996, 16, 675–686. [Google Scholar] [CrossRef] [PubMed]
- Bendahan, A.; Armon, A.; Madani, N.; Kavanaugh, M.P.; Kanner, B.I. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J. Biol. Chem. 2000, 275, 37436–37442. [Google Scholar] [CrossRef] [PubMed]
- Griffith, O.W. Biologic and pharmacologic regulation of mammalian glutathione synthesis. Free Radic. Biol. Med. 1999, 27, 922–935. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Matsumura, N.; Watabe, M.; Nakaki, T. Oxidative stress on EAAC1 is involved in MPTP-induced glutathione depletion and motor dysfunction. Eur. J. Neurosci. 2008, 27, 20–30. [Google Scholar] [CrossRef] [PubMed]
- Berman, A.E.; Chan, W.Y.; Brennan, A.M.; Reyes, R.C.; Adler, B.L.; Suh, S.W.; Kauppinen, T.M.; Edling, Y.; Swanson, R.A. N-acetylcysteine prevents loss of dopaminergic neurons in the EAAC1−/− mouse. Ann. Neurol. 2011, 69, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimers Dis. 2013, 36, 197–209. [Google Scholar] [PubMed]
- Duerson, K.; Woltjer, R.L.; Mookherjee, P.; Leverenz, J.B.; Montine, T.J.; Bird, T.D.; Pow, D.V.; Rauen, T.; Cook, D.G. Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer’s disease patients. Brain Pathol. 2009, 19, 267–278. [Google Scholar] [CrossRef] [PubMed]
- Li, X.; Valencia, A.; Sapp, E.; Masso, N.; Alexander, J.; Reeves, P.; Kegel, K.B.; Aronin, N.; Difiglia, M. Aberrant Rab11-dependent trafficking of the neuronal glutamate transporter EAAC1 causes oxidative stress and cell death in Huntington’s disease. J. Neurosci. 2010, 30, 4552–4561. [Google Scholar] [CrossRef] [PubMed]
- Beckman, K.B.; Ames, B.N. The free radical theory of aging matures. Physiol. Rev. 1998, 78, 547–581. [Google Scholar] [PubMed]
- Maher, P. The effects of stress and aging on glutathione metabolism. Ageing Res. Rev. 2005, 4, 288–314. [Google Scholar] [CrossRef] [PubMed]
- Jenner, P. Oxidative stress in Parkinson’s disease. Ann. Neurol. 2003, 53, S26–S38. [Google Scholar] [CrossRef] [PubMed]
- Sultana, R.; Piroddi, M.; Galli, F.; Butterfield, D.A. Protein levels and activity of some antioxidant enzymes in hippocampus of subjects with amnestic mild cognitive impairment. Neurochem. Res. 2008, 33, 2540–2546. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Rizzini, B.L.; Rossi, D.; Haugeto, O.; Racagni, G.; Danbolt, N.C.; Volterra, A. Neuronal and glial glutamate transporters possess an SH-based redox regulatory mechanism. Eur. J. Neurosci. 1997, 9, 1236–1243. [Google Scholar] [CrossRef] [PubMed]
- Trotti, D.; Nussberger, S.; Volterra, A.; Hediger, M.A. Differential modulation of the uptake currents by redox interconversion of cysteine residues in the human neuronal glutamate transporter EAAC1. Eur. J. Neurosci. 1997, 9, 2207–2212. [Google Scholar] [CrossRef] [PubMed]
- Duan, S.; Anderson, C.M.; Stein, B.A.; Swanson, R.A. Glutamate induces rapid upregulation of astrocyte glutamate transport and cell-surface expression of GLAST. J. Neurosci. 1999, 19, 10193–10200. [Google Scholar] [PubMed]
- Kalandadze, A.; Wu, Y.; Fournier, K.; Robinson, M.B. Identification of motifs involved in endoplasmic reticulum retention-forward trafficking of the GLT-1 subtype of glutamate transporter. J. Neurosci. 2004, 24, 5183–5192. [Google Scholar] [CrossRef] [PubMed]
- Fournier, K.M.; Gonzalez, M.I.; Robinson, M.B. Rapid trafficking of the neuronal glutamate transporter, EAAC1: Evidence for distinct trafficking pathways differentially regulated by protein kinase C and platelet-derived growth factor. J. Biol. Chem. 2004, 279, 34505–34513. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.I.; Krizman-Genda, E.; Robinson, M.B. Caveolin-1 regulates the delivery and endocytosis of the glutamate transporter, excitatory amino acid carrier 1. J. Biol. Chem. 2007, 282, 29855–29865. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, M.I.; Susarla, B.T.; Fournier, K.M.; Sheldon, A.L.; Robinson, M.B. Constitutive endocytosis and recycling of the neuronal glutamate transporter, excitatory amino acid carrier 1. J. Neurochem. 2007, 103, 1917–1931. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Vidensky, S.; Ruggiero, A.M.; Maier, S.; Sitte, H.H.; Rothstein, J.D. Reticulon RTN2B regulates trafficking and function of neuronal glutamate transporter EAAC1. J. Biol. Chem. 2008, 283, 6561–6571. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Wang, F.; Matsumura, N.; Kiyonari, H.; Shioi, G.; Tanaka, K.; Kinoshita, C.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Increased neuronal glutathione and neuroprotection in GTRAP3-18-deficient mice. Neurobiol. Dis. 2012, 45, 973–982. [Google Scholar] [CrossRef] [PubMed]
- Xia, P.; Pei, G.; Schwarz, W. Regulation of the glutamate transporter EAAC1 by expression and activation of δ-opioid receptor. Eur. J. Neurosci. 2006, 24, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Watabe, M.; Nakaki, T. Modulation of neuronal glutathione synthesis by EAAC1 and its interacting protein GTRAP3-18. Amino Acids 2012, 42, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Inhibition of GTRAP3-18 may increase neuroprotective glutathione (GSH) synthesis. Int. J. Mol. Sci. 2012, 13, 12017–12035. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Nakaki, T. Neuroprotective properties of the excitatory amino acid carrier 1 (EAAC1). Amino Acids 2013, 45, 133–142. [Google Scholar] [CrossRef] [PubMed]
- Ma, K.; Zheng, S.; Zuo, Z. The transcription factor regulatory factor X1 increases the expression of neuronal glutamate transporter type 3. J. Biol. Chem. 2006, 281, 21250–21255. [Google Scholar] [CrossRef] [PubMed]
- Escartin, C.; Joon Won, S.; Malgorn, C.; Auregan, G.; Berman, A.E.; Chen, P.C.; Deglon, N.; Johnson, J.A.; Won Suh, S.; Swanson, R.A. Nuclear factor erythroid 2-related factor 2 facilitates neuronal glutathione synthesis by upregulating neuronal excitatory amino acid transporter 3 expression. J. Neurosci. 2011, 31, 7392–7401. [Google Scholar] [CrossRef] [PubMed]
- Molteni, R.; Ying, Z.; Gomez-Pinilla, F. Differential effects of acute and chronic exercise on plasticity-related genes in the rat hippocampus revealed by microarray. Eur. J. Neurosci. 2002, 16, 1107–1116. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, C.; Aoyama, K.; Matsumura, N.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Rhythmic oscillations of the microRNA miR-96-5p play a neuroprotective role by indirectly regulating glutathione levels. Nat. Commun. 2014, 5, 3823. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, C.; Aoyama, K.; Nakaki, T. microRNA as a new agent for regulating neuronal glutathione synthesis and metabolism. AIMS Mol. Sci. 2015, 2, 124–143. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742-8758. https://doi.org/10.3390/molecules20058742
Aoyama K, Nakaki T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules. 2015; 20(5):8742-8758. https://doi.org/10.3390/molecules20058742
Chicago/Turabian StyleAoyama, Koji, and Toshio Nakaki. 2015. "Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1)" Molecules 20, no. 5: 8742-8758. https://doi.org/10.3390/molecules20058742
APA StyleAoyama, K., & Nakaki, T. (2015). Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules, 20(5), 8742-8758. https://doi.org/10.3390/molecules20058742