
Synthesis of Bicyclic Isoxazoles and Isoxazolines via Intramolecular Nitrile Oxide Cycloaddition

Abstract
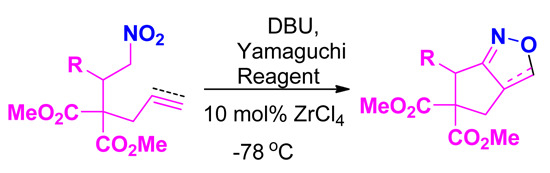
:
1. Introduction
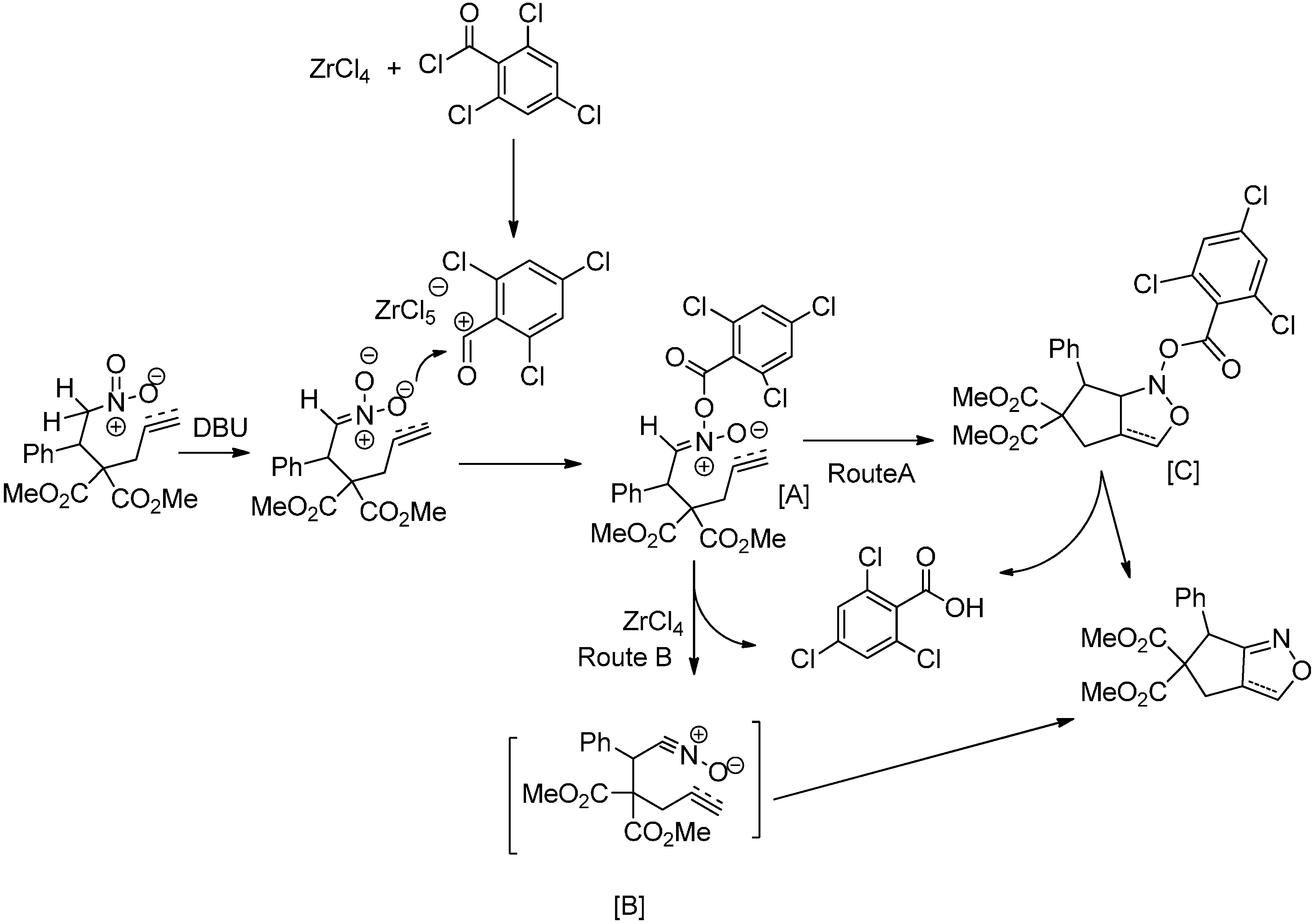
2. Results and Discussion


| Entry | Base (equiv.) | Dehydrating Agent (equiv.) | Lewis Acid | Temp. (°C) | Time (h) | Yield a, b | ||
|---|---|---|---|---|---|---|---|---|
| 1 | K t-BuO | (2.5) | 2,4,6-Trichlorobenzoyl chloride | (3) | — | ‒78 | 24 | 65 |
| 2 | TEA | (2.5) | 2,4,6-Trichlorobenzoyl chloride | (3) | — | ‒78 | 24 | 50 |
| 3 | DMAP | (2.5) | 2,4,6-Trichlorobenzoyl chloride | (3) | — | ‒78 | 24 | 67 |
| 4 | DBU | (2.5) | 2,4,6-Trichlorobenzoyl chloride | (3) | — | ‒78 | 24 | 73 |
| 5 | DBU | (2.5) | Benzoyl chloride | (3) | — | ‒78 | 48 | 62 |
| 6 | DBU | (2.5) | 2-Chlorobenzoyl; chloride | (3) | — | ‒78 | 35 | 65 |
| 7 | DBU | (2.5) | 3,4-Dichlorobenzoyl chloride | (3) | — | ‒78 | 24 | 65 |
| 8 | DBU | (2.5) | 2,4,6-Trichlorobenzoyl chloride | (1.5) | ZnCl2 | ‒78 | 3 | 82 |
| 9 | DBU | (2.5) | 2,4,6-Trichlorobenzoyl chloride | (1.5) | ZnCl4 | ‒78 | 4 | 87 |
| 10 | DBU | (2.5) | 2,4,6-Trichlorobenzoyl chloride | (1.5) | SnCl4 | ‒78 | 5 | 60 |
| 11 | DBU | (2.0) | 2,4,6-Trichlorobenzoyl chloride | (1.5) | ZnCl4 | ‒78 | 4 | 90 |
| 12 | DBU | (1.5) | 2,4,6-Trichlorobenzoyl chloride | (1.5) | ZnCl4 | ‒78 | 4 | 95 |
| 13 | DBU | (1.0) | 2,4,6-Trichlorobenzoyl chloride | (1.5) | ZnCl4 | ‒78 | 7 | 85 |
| 14 | DBU | (1.5) | 2,4,6-Trichlorobenzoyl chloride | (1.5) | ZnCl4 | 0 | 2 | 40 |
| 15 | DBU | (1.5) | 2,4,6-Trichlorobenzoyl chloride | (1.5) | ZnCl4 | 25 | 2 | 20 |

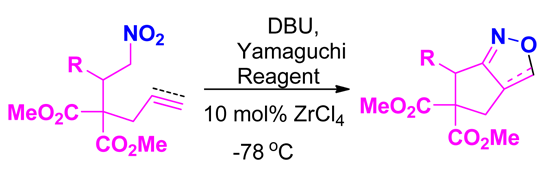
| Entry | Substrate | Product | Time (h) | Yield% a,b |
|---|---|---|---|---|
| 1 |  1a |  2a | 4 | 95 |
| 2 |  1b |  2b | 2 | 86 |
| 3 |  1c |  2c | 3 | 89 |
| 4 |  1d |  2d | 3 | 92 |
| 5 |  1e |  2e | 5 | 90 |
| 6 |  1f |  2f | 5 | 87 |
| 7 |  1g |  2g | 3 | 84 |
| 8 |  1h |  2h | 3 | 86 |
| 9 |  1i |  2i | 3 | 84 |
| 10 |  1j |  2j | 15 | 79 |
| 11 |  1k |  2k | 7 | 92 |
| 12 |  1l |  2l | 12 | 80 |
| 13 |  1m |  2m | 5 | 87 |


{kind=link}
{kind=link}
{kind=link}
| Entry | Substrate | Product | Time (h) | Yield% a,b | cis:trans c |
|---|---|---|---|---|---|
| 1 |  3a |  4a | 3 | 88 | 5.3:1 |
| 2 |  3b |  4b | 4 | 83 | 4.6:1 |
| 3 |  3c |  4c | 5 | 81 | 3.6:1 |
| 4 |  3d |  4d | 5 | 85 | 5.4:1 |
| 5 |  3e |  4e | 12 | 78 | 4.8:1 |
| 6 |  3f |  4f | 7 | 84 | 5.6:1 |

3. Experimental Section
3.1. General Information
3.2. General Procedure for the Preparation of 1a–1o
3.3. General Procedure for the Synthesis of 3a–3f
3.4. General Procedure for the Preparation of 2a–2o and 4a–4f
Dimethyl-6-(o-tolyl)-4H-cyclopenta[c]isoxazole-5,5(6H)-dicarboxylate (2c): White solid; m.p. 117–118 °C; 1H-NMR (CDCl3) δ 8.08 (s, 1H), 7.16–7.08 (m, 2H), 7.03 (t, J = 7.4 Hz, 1H), 6.97 (d, J = 7.4 Hz, 1H), 5.63 (s, 1H), 3.63 (d, J = 17.4, 1.5 Hz, 1H), 3.79 (s, 3H), 3.25 (d, J = 17.4, 1.4Hz, 1H), 3.19 (s, 3H), 2.49 (s, 3H); 13C-NMR (CDCl3) δ 173.5, 171.5, 168.5, 150.5, 137.1, 135.8, 130.6, 128.3, 127.9, 126.3, 121.4, 72.9, 53.6, 52.4, 44.0, 29.8, 20.2. HRMS (EI) m/z calc. for C17H17NO5Na (M+ + Na) 338.1005, found 338.1015.
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kamal, A.; Bharathi, E.V.; Reddy, J.S.; Ramaiah, M.J.; Dastagiri, D.; Reddy, M.K.; Viswanath, A.; Reddy, T.L.; Shaik, T.B.; Pushpavalli, S.N.; et al. Synthesis and biological evaluation of 3,5-diaryl isoxazoline/isoxazole linked 2,3-dihydroquinazolinone hybrids as anticancer agents. Eur. J. Med. Chem. 2011, 46, 691–703. [Google Scholar] [CrossRef] [PubMed]
- Sperry, J.B.; Wright, D. Furans, thiophenes and related heterocycles in drug discovery. Curr. Opin. Drug Discov. Dev. 2005, 8, 723–740. [Google Scholar] [CrossRef]
- Sammelson, R.E.; Ma, T.; Galietta, L.J.V.; Verkman, A.S.; Kurth, M.J. 3-(2-Benzyloxyphenyl)isoxazoles and isoxazolines: Synthesis and evaluation as CFTR activators. Bioorg. Med. Chem. Lett. 2003, 13, 2509–2512. [Google Scholar] [CrossRef]
- Kozikowski, A.P. The isoxazoline route to the molecules of nature. Acc. Chem. Res. 1984, 17, 410–416. [Google Scholar] [CrossRef]
- Torssell, K.B.G. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; VCH Publishers: New York, NY, USA, 1988. [Google Scholar]
- Tang, S.; He, J.; Sun, Y.; He, L.; She, X. Efficient and regioselective synthesis of 5-hydroxy-2-isoxazolines: Versatile synthons for isoxazoles, β-lactams, and γ-amino alcohols. J. Org. Chem. 2010, 75, 1961–1966. [Google Scholar] [CrossRef] [PubMed]
- Kleinbeck, F.; Carreira, E.M. Total Synthesis of Bafilomycin A1. Angew. Chem. Int. Ed. 2009, 48, 578–581. [Google Scholar] [CrossRef] [PubMed]
- Fuller, A.A.; Chen, B.; Minter, A.R.; Mapp, A.K. Succinct synthesis of β-amino acids via chiral isoxazolines. J. Am. Chem. Soc. 2005, 127, 5376–5383. [Google Scholar] [CrossRef] [PubMed]
- Namboothiri, I.N.N.; Rastogi, N. Synthesis of Heterocycles via Cycloadditions I. In Topics in Heterocyclic Chemistry; Hassner, A., Ed.; Springer: Berlin, Germany, 2008; volume 12, p. 1. [Google Scholar]
- Bode, J.W.; Carreira, E.M. A Mild and Chemoselective Method for the Reduction of Conjugated Isoxazolines to β-Hydroxy Ketones. Org. Lett. 2001, 3, 1587–1590. [Google Scholar] [CrossRef] [PubMed]
- Yermolina, M.V.; Wang, J.; Caffrey, M.; Rong, L.L.; Wardrop, D.J. Discovery, synthesis, and biological evaluation of a novel group of selective inhibitors of filoviral entry. J. Med. Chem. 2011, 54, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Conti, P.; Amici, M.D.; di Ventimiglia, S.J.; Stensbøl, T.B.; Madsen, U.; Bräuner-Osborne, H.; Russo, E.; Sarro, G.D.; Bruno, G.; Micheli, C.D. Synthesis and anticonvulsant activity of novel bicyclic acidic amino acids. J. Med. Chem. 2003, 46, 3102–3108. [Google Scholar] [CrossRef] [PubMed]
- Kozikowski, A.P.; Park, P.U. Synthesis of streptazolin: Use of the aza-Ferrier reaction in conjunction with the INOC process to deliver a unique but sensitive natural product. J. Org. Chem. 1990, 55, 4668–4682. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Stein, P.D. The INOC route to carbocyclics: A formal total synthesis of (+)-sarkomycin. J. Am. Chem. Soc. 1982, 104, 4023–4024. [Google Scholar] [CrossRef]
- Okamoto, S.; Kobayashi, Y.; Kato, H.; Hotri, K.; Takahashi, T.; Tsuji, J.; Sato, F. Prostaglandin synthesis via two-component coupling. Highly efficient synthesis of chiral prostaglandin intermediates 4-alkoxy-2-alkyl-2-cyclopenten-1-one and 4-alkoxy-3-alkenyl-2-methylenecyclopentan-1-one. J. Org. Chem. 1988, 53, 5590–5592. [Google Scholar] [CrossRef]
- Trogu, E.; Vinattieri, C.; de Sarlo, F.; Machetti, F. Acid-Base-Catalysed Condensation Reaction in Water: Isoxazolines and Isoxazoles from Nitroacetates and Dipolarophiles. Chem. Eur. J. 2012, 18, 2081–2093. [Google Scholar] [CrossRef] [PubMed]
- Raihan, M.J.; Kavala, V.; Kuo, C.W.; Raju, B.R.; Yao, C.F. “On-water” synthesis of chromeno-isoxazoles mediated by [hydroxy (tosyloxy) iodo] benzene (HTIB). Green Chem. 2010, 12, 1090–1096. [Google Scholar] [CrossRef]
- Cecchi, L.; de Sarlo, F.; Machetti, F. Synthesis of 4,5-Dihydroisoxazoles by Condensation of Primary Nitro Compounds with Alkenes by Using a Copper/Base Catalytic System. Chem. Eur. J. 2008, 14, 7903–7912. [Google Scholar] [CrossRef] [PubMed]
- Belenkii, L.D.; Zelinsky, N.D. Nitrile Oxides. In Nitrile Oxides, Nitrones and Nitronates in Organic Synthesis, 2nd edition; Feuer, H., Ed.; John Willey and Sons: Hoboken, NJ, USA, 2008; p. 1. [Google Scholar]
- Kankala, S.; Kankala, R.K.; Gundepaka, P.; Thota, N.; Nerella, S.; Gangula, M.R.; Guguloth, H.; Kagga, M.; Vadde, R.; Vasam, C.S. Regioselective synthesis of isoxazole-mercaptobenzimidazole hybrids and their in vivo analgesic and anti-inflammatory activity studies. Bioorg. Med. Chem. Lett. 2013, 23, 1306–1309. [Google Scholar] [CrossRef] [PubMed]
- Vitale, P.; Nunno, L.D. A novel synthesis of N-unsubstituted β-enamino thioesters from 3-arylisoxazoles and 3-aryl-5-phenylthio-2-isoxazolines. Synthesis 2010, 3195–3203. [Google Scholar] [CrossRef]
- Domingo, L.R.; Chamorro, E.; Perez, P. An Analysis of the Regioselectivity of 1,3-Dipolar Cycloaddition Reactions of Benzonitrile N-Oxides Based on Global and Local Electrophilicity and Nucleophilicity Indices. Eur. J. Org. Chem. 2009, 2009, 3036–3044. [Google Scholar] [CrossRef]
- Grecian, S.; Fokin, V.V. Ruthenium-Catalyzed Cycloaddition of Nitrile Oxides and Alkynes: Practical Synthesis of Isoxazoles. Angew. Chem. Int. Ed. 2008, 47, 8285–8287. [Google Scholar] [CrossRef] [PubMed]
- Barr, L.; Lincoln, S.F.; Easton, C.J. Reversal of regioselectivity and enhancement of rates of nitrile oxide cycloadditions through transient attachment of dipolarophiles to cyclodextrins. Chem. Eur. J. 2006, 12, 8571–8580. [Google Scholar] [CrossRef] [PubMed]
- Han, L.; Zhang, B.; Xiang, C.; Yan, J. One-Pot Synthesis of Isoxazolines from Aldehydes Catalyzed by Iodobenzene. Synthesis 2014, 46, 503–509. [Google Scholar]
- Xiang, C.; Li, T.; Yan, J. Hypervalent Iodine-Catalyzed Cycloaddition of Nitrile Oxides to Alkenes. Synth. Commun. 2014, 44, 682–688. [Google Scholar] [CrossRef]
- Chau, J.; Xu, S.; Ciufolini, M.A. Assembly of a Key Dienic Intermediate for Tetrodotoxin via a Machetti-DeSarlo Reaction. J. Org. Chem. 2013, 78, 11901–11910. [Google Scholar] [CrossRef] [PubMed]
- Mendelsohn, B.A.; Lee, S.; Kim, S.; Teyssier, F.; Aulakh, V.S.; Ciufolini, M.A. Oxidation of oximes to nitrile oxides with hypervalent iodine reagents. Org. Lett. 2009, 11, 1539–1542. [Google Scholar] [CrossRef] [PubMed]
- Becker, N.; Carreira, E.M. Hydroxyl-Directed Nitrile Oxide Cycloaddition Reactions with Cyclic Allylic Alcohols. Org. Lett. 2007, 9, 3857–3858. [Google Scholar] [CrossRef] [PubMed]
- Perez, L.; Khouly, M.E.E.; Cruz, P.D.L.; Araki, Y.; Ito, O.; Langa, F. Comparison between the Photophysical Properties of Pyrazolo- and Isoxazolo[60]fullerenes with Dual Donors (Ferrocene, Aniline and Alkoxyphenyl). Eur. J. Org. Chem. 2007, 2007, 2175–2185. [Google Scholar] [CrossRef]
- Hansen, T.V.; Wu, P.; Fokin, V.V. One-pot copper (I)-catalyzed synthesis of 3,5-disubstituted isoxazoles. J. Org. Chem. 2005, 70, 7761–7764. [Google Scholar] [CrossRef] [PubMed]
- Cha, M.Y.; Choi, B.C.; Kang, K.H.; Pae, A.N.; Choi, K.I.; Cho, Y.S.; Koh, H.Y.; Lee, H.Y.; Jung, D.; Kong, J.Y. Design and synthesis of a piperazinylalkylisoxazole library for subtype selective dopamine receptor ligands. Bioorg. Med. Chem. Lett. 2002, 12, 1327–1331. [Google Scholar] [CrossRef]
- Nonn, M.; Kiss, L.; Forró, E.; Mucsi, Z.; Fülöp, F. Synthesis of novel isoxazoline-fused cyclic β-amino esters by regio-and stereo-selective 1,3-dipolar cycloaddition. Tetrahedron 2011, 67, 4079–4085. [Google Scholar] [CrossRef]
- Tu, Z.; Jang, Y.; Lin, C.; Liu, J.T.; Hsu, J.; Sastry, M.N.V.; Yao, C.F. The study of reaction mechanism for the transformation of nitronate into nitrile by phosphorus trichloride. Tetrahedron 2005, 61, 10541–10551. [Google Scholar] [CrossRef]
- Liu, J.T.; Lin, W.W.; Jang, J.J.; Liu, J.Y.; Yan, M.C.; Hung, C.; Kao, K.H.; Wang, Y.; Yao, C.F. One-pot synthesis of five- or six-membered carbocycles through intramolecular cycloadditions by the use of ethyl chloroformate. Tetrahedron 1999, 55, 7115–7128. [Google Scholar] [CrossRef]
- Yan, M.C.; Liu, J.Y.; Lin, W.W.; Kao, K.H.; Liu, J.T.; Jang, J.J.; Yao, C.F. One-pot synthesis of five-membered cyclic thioethers or ethers via intramolecular nitrile oxide-olefin cycloaddition (INOC) or intramolecular alkoxycarbonyl nitronate-olefin cycloaddition (IAOC) by the use of methyl chloroformate. Tetrahedron 1999, 55, 12493–12514. [Google Scholar] [CrossRef]
- Butler, J.D.; Donald, M.B.; Ding, Z.; Fettinger, J.C.; Kurth, M.J. Phenylsulfonyl as a directing group for nitrile oxide cycloadditions and mCPBA epoxidations. Tetrahedron Lett. 2009, 50, 5110–5112. [Google Scholar] [CrossRef]
- Roda, G.; Conti, P.; Amici, M.D.; He, J.; Polavarapu, P.L.; Micheli, C.D. Enantiopure stereoisomeric homologues of glutamic acid: Chemoenzymatic synthesis and assignment of their absolute configurations. Tetrahedron Asymmetry 2004, 15, 3079–3090. [Google Scholar] [CrossRef]
- Akritopoulou-Zanze, I.; Gracias, V.; Moore, J.D.; Djuric, S.W. Synthesis of novel fused isoxazoles and isoxazolines by sequential Ugi/INOC reactions. Tetrahedron Lett. 2004, 45, 3421–3423. [Google Scholar] [CrossRef]
- Giorgi, G.; Lampariello, L.R.; Minetto, G.; Paoli, M.L.; Riello, V.; Rodriquez, M.; Sega, A. Developing Molecular Diversity in the Construction of a Family of Bicyclic Isoxazolines Scaffolds: Control of Regio- and Diastereoselectivities. Eur. J. Org. Chem. 2003, 4777–4785. [Google Scholar]
- Park, K.H.; Olmstead, M.M.; Kurth, M.J. Diastereoselective synthesis of cyclopentanoids with hydantoin and isoxazoline substituents. J. Org. Chem. 1998, 63, 113–117. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.E.; Vu, B.T. Substituent effects on intramolecular dipolar cycloadditions: The gem-dicarboalkoxy effect. Tetrahedron Lett. 1996, 37, 451–454. [Google Scholar] [CrossRef]
- Gao, S.; Tu, Z.; Kuo, C.W.; Liu, J.T.; Chu, C.M.; Yao, C.F. Efficient conversion of nitronate into nitrile oxide using cyanuric chloride. One-pot synthesis of bicyclic isoxazolines and isoxazoles from nitroalkenes. Org. Biomol. Chem. 2006, 4, 2851–2857. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Q.; Oritani, T.; Horiguchi, Y.; Shi, Q. High Stereoselectivity in One-Pot Intramolecular Cycloadditions of Olefinic Silyl Nitronates. Eur. J. Org. Chem. 1999, 1999, 2689–2694. [Google Scholar] [CrossRef]
- Namboothiri, I.N.N.; Hassner, A. A Highly Stereoselective One-Pot Tandem Consecutive 1,4-Addition-Intramolecular 1,3-Dipolar Cycloaddition Strategy for the Construction of Functionalized Five- and Six-Membered Carbocycles. J. Org. Chem. 1997, 62, 485–492. [Google Scholar] [CrossRef]
- Basel, Y.; Hassner, A. An improved method for preparation of nitrile oxides from nitroalkanes for in situ dipolar cycloadditions. Synthesis 1997, 1997, 309–312. [Google Scholar] [CrossRef]
- Mukaiyama, T.; Hoshino, T. The Reactions of Primary Nitroparaffins with Isocyanates1. J. Am. Chem. Soc. 1960, 82, 5339–5342. [Google Scholar] [CrossRef]
- Glorius, F.; Grohmann, C.; Wang, H. Rh [III]-Catalyzed C-H Amidation Using Aroyloxycarbamates to Give N-Boc Protected Arylamines. Org. Lett. 2013, 15, 3014–3017. [Google Scholar]
- Parenty, A.; Moreau, X.; Niel, G.; Campagne, J.M. Macrolactonizations in the Total Synthesis of Natural Products. Chem. Rev. 2013, 113, PR1–PR40. [Google Scholar] [CrossRef] [PubMed]
- Inanaga, J.; Hirata, K.; Saeki, H.; Katsuki, T.; Yamaguchi, M. A rapid esterification by means of mixed anhydride and its application to large-ring lactonization. Bull. Chem. Soc. Jpn. 1979, 52, 1989–1993. [Google Scholar] [CrossRef]
- Dehaen, W.; Hassner, A. Stereoselectivity in intramolecular 1,3-dipolar cycloadditions. Nitrile oxides versus silyl nitronates. Tetrahedron Lett. 1990, 31, 743–746. [Google Scholar] [CrossRef]
- Kao, K.H.; Yang, C.S.; Liu, J.T.; Lin, W.W.; Fang, H.Y.; Yao, C.F.; Chen, K. One-pot synthesis of the hydroximoyl chlorides and [3.3.0] bicyclic compounds from the reactions of β-nitrostyrenes with stabilized nucleophiles. Tetrahedron 1998, 54, 13997–14014. [Google Scholar] [CrossRef]
- McKillop, A.; Kobylecki, R.J. An investigation of the reaction of primary nitroalkanes with acetic anhydride/sodium acetate. Tetrahedron 1974, 30, 1365–1371. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2a–2o and 4a–4f are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, W.-C.; Kavala, V.; Shih, Y.-H.; Wang, Y.-H.; Kuo, C.-W.; Yang, T.-H.; Huang, C.-Y.; Chiu, H.-H.; Yao, C.-F. Synthesis of Bicyclic Isoxazoles and Isoxazolines via Intramolecular Nitrile Oxide Cycloaddition. Molecules 2015, 20, 10910-10927. https://doi.org/10.3390/molecules200610910
Chen W-C, Kavala V, Shih Y-H, Wang Y-H, Kuo C-W, Yang T-H, Huang C-Y, Chiu H-H, Yao C-F. Synthesis of Bicyclic Isoxazoles and Isoxazolines via Intramolecular Nitrile Oxide Cycloaddition. Molecules. 2015; 20(6):10910-10927. https://doi.org/10.3390/molecules200610910
Chicago/Turabian StyleChen, Wen-Chang, Veerababurao Kavala, Yu-Hsuan Shih, Yu-Hsuan Wang, Chun-Wei Kuo, Tang-Hao Yang, Chia-Yu Huang, Hao-Hsiang Chiu, and Ching-Fa Yao. 2015. "Synthesis of Bicyclic Isoxazoles and Isoxazolines via Intramolecular Nitrile Oxide Cycloaddition" Molecules 20, no. 6: 10910-10927. https://doi.org/10.3390/molecules200610910
APA StyleChen, W. -C., Kavala, V., Shih, Y. -H., Wang, Y. -H., Kuo, C. -W., Yang, T. -H., Huang, C. -Y., Chiu, H. -H., & Yao, C. -F. (2015). Synthesis of Bicyclic Isoxazoles and Isoxazolines via Intramolecular Nitrile Oxide Cycloaddition. Molecules, 20(6), 10910-10927. https://doi.org/10.3390/molecules200610910