
Triel Bonds, π-Hole-π-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene


Abstract
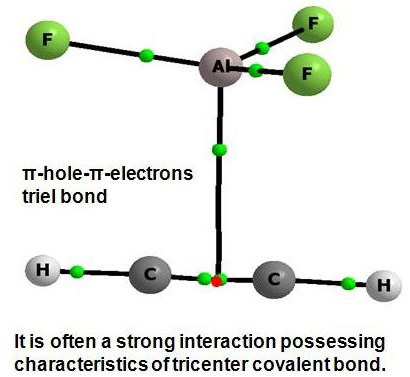
:
1. Introduction

2. Results and Discussion
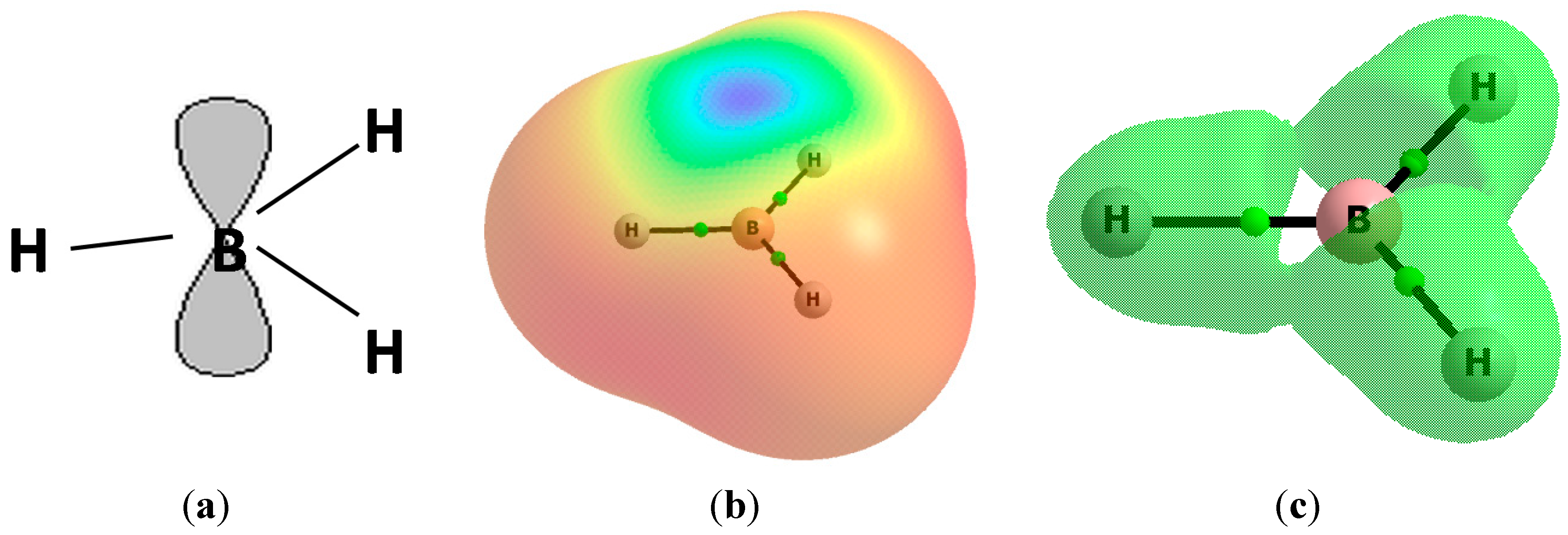
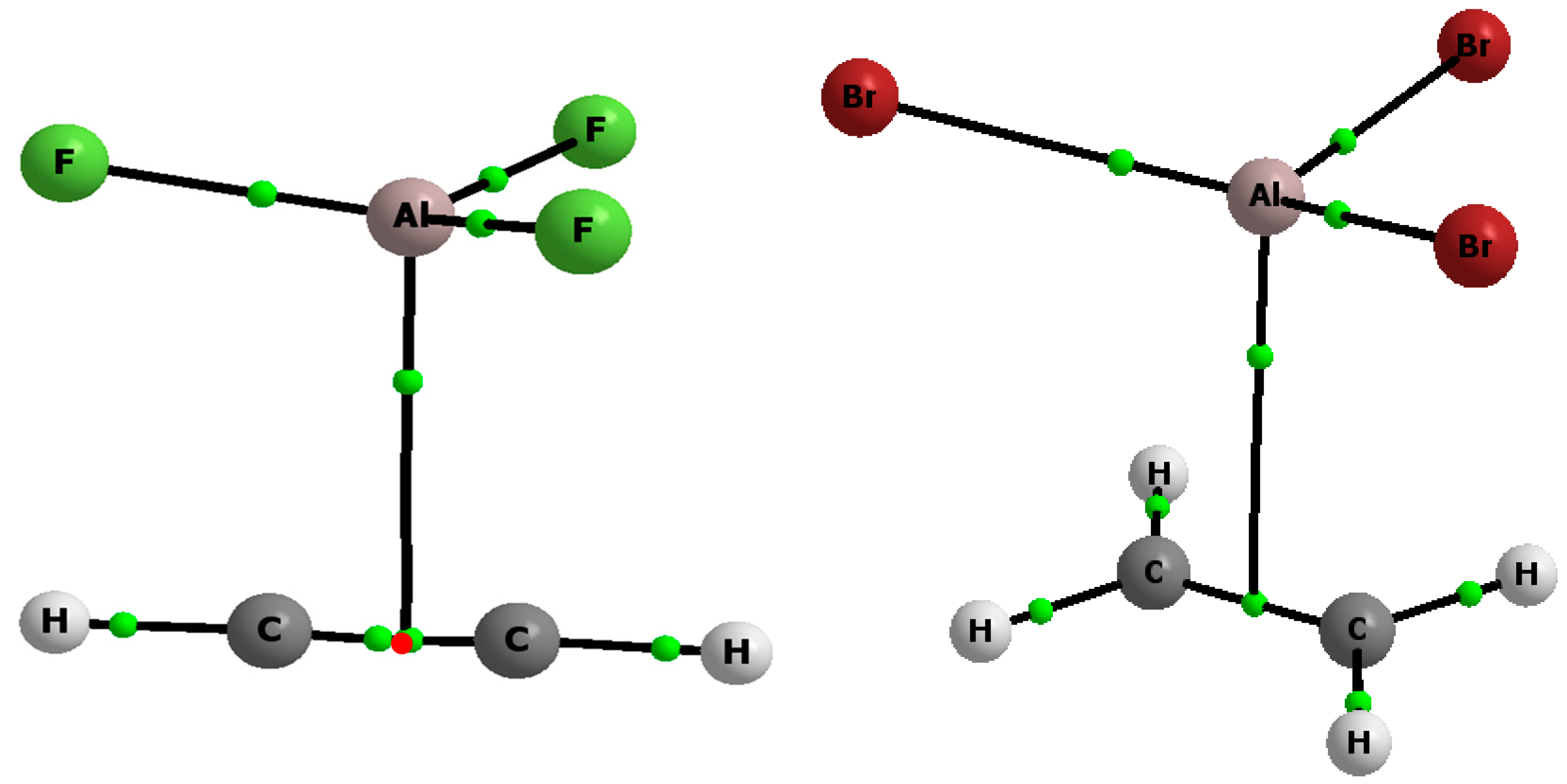
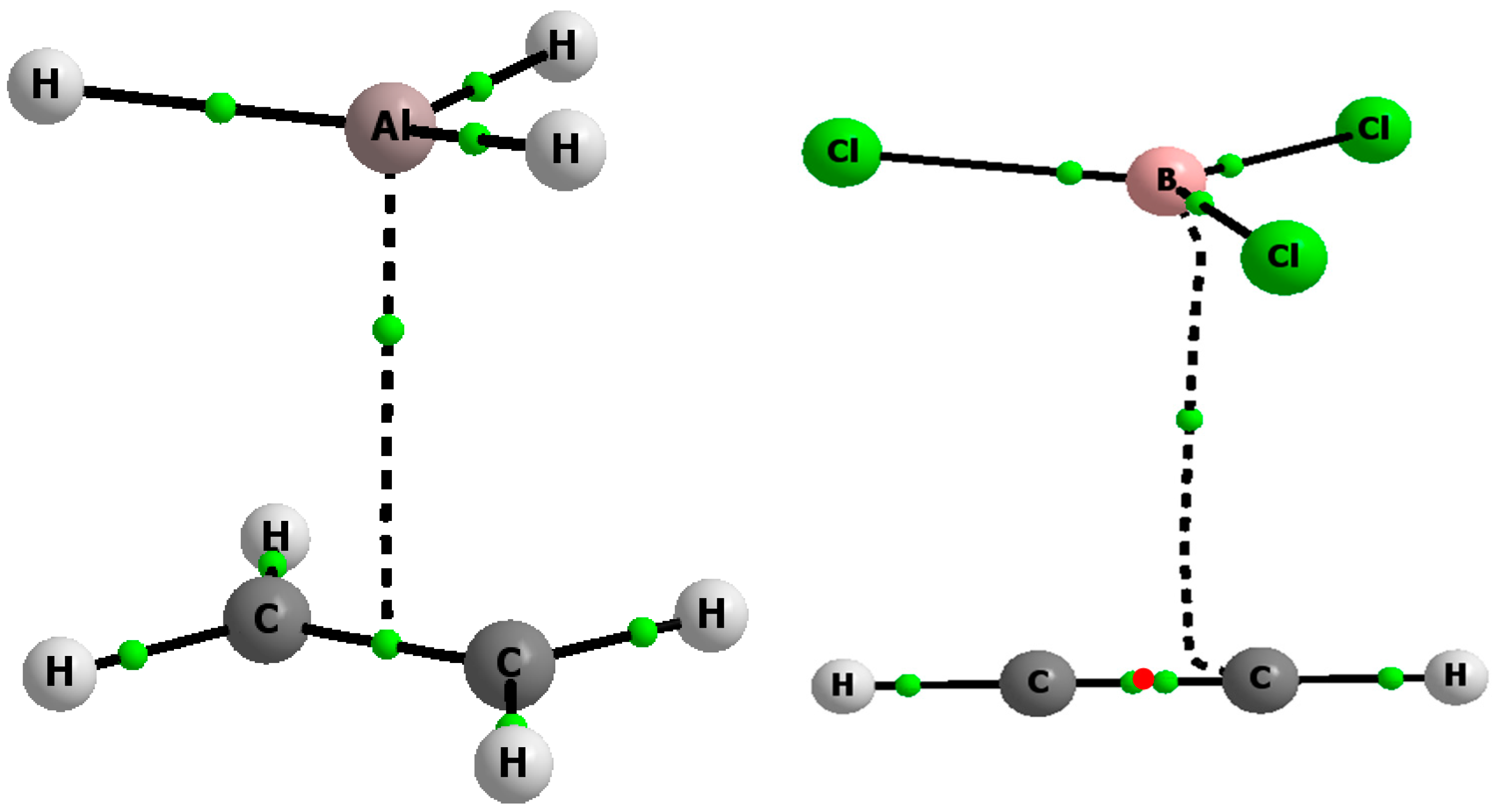
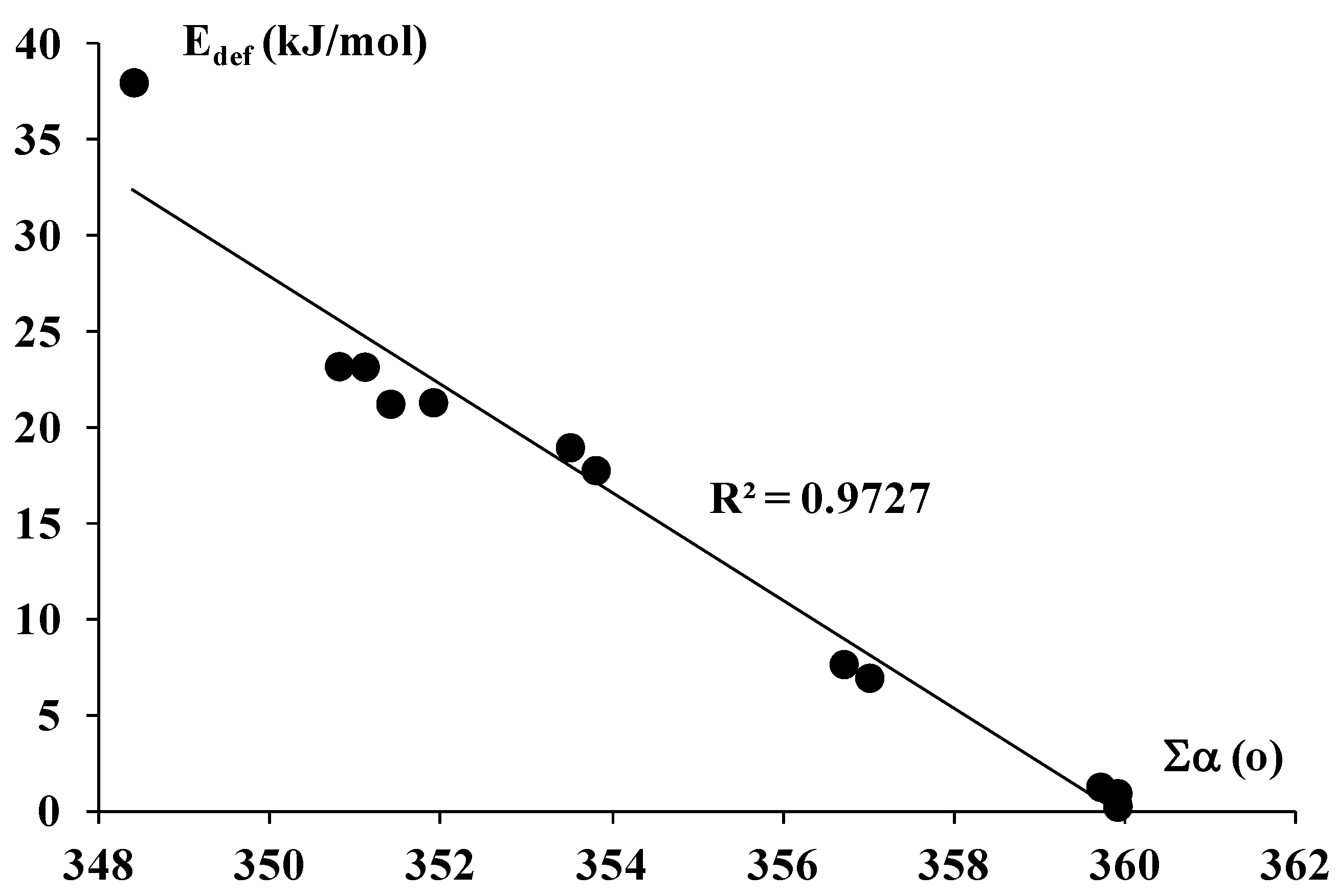
2.1. The Strength of π-Hole-π Electrons Triel Bonds



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
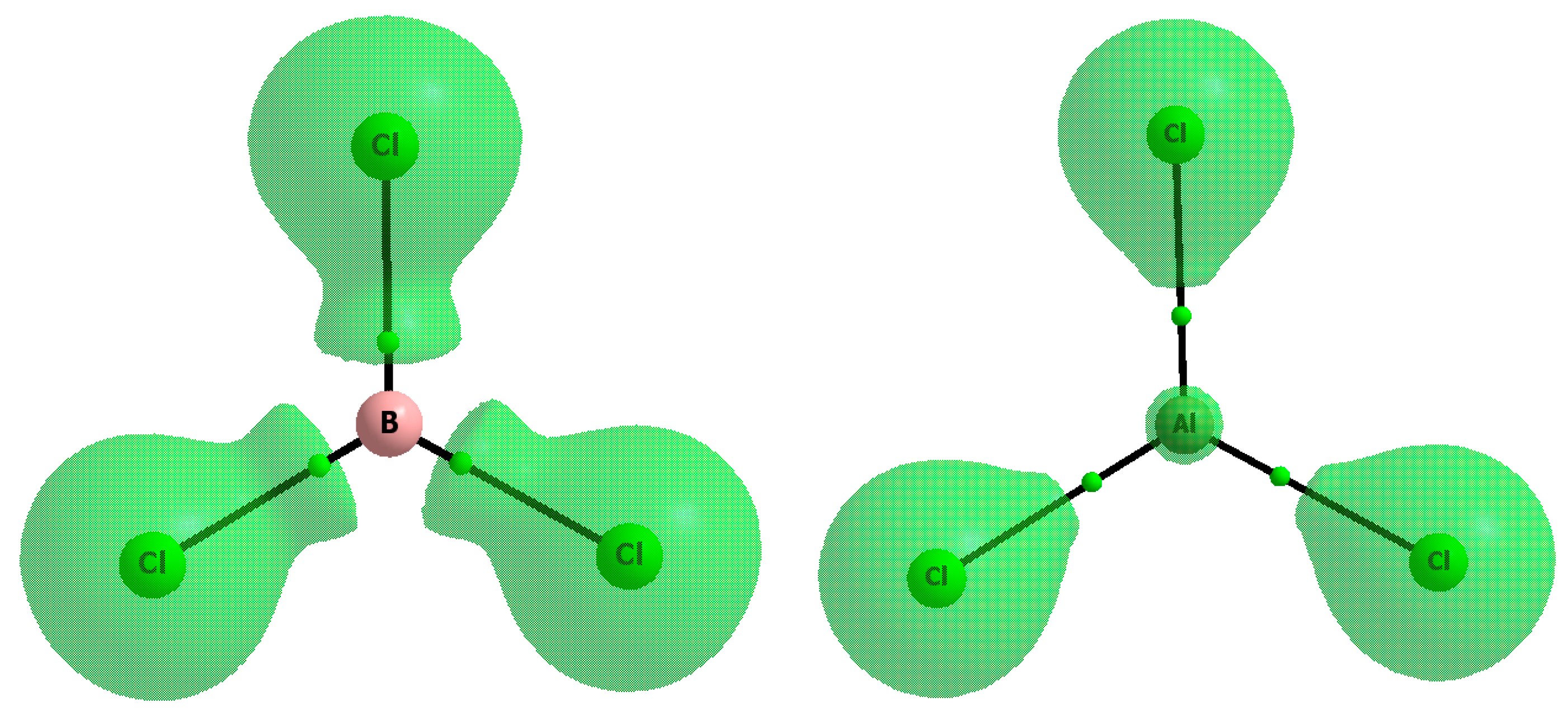{kind=link}
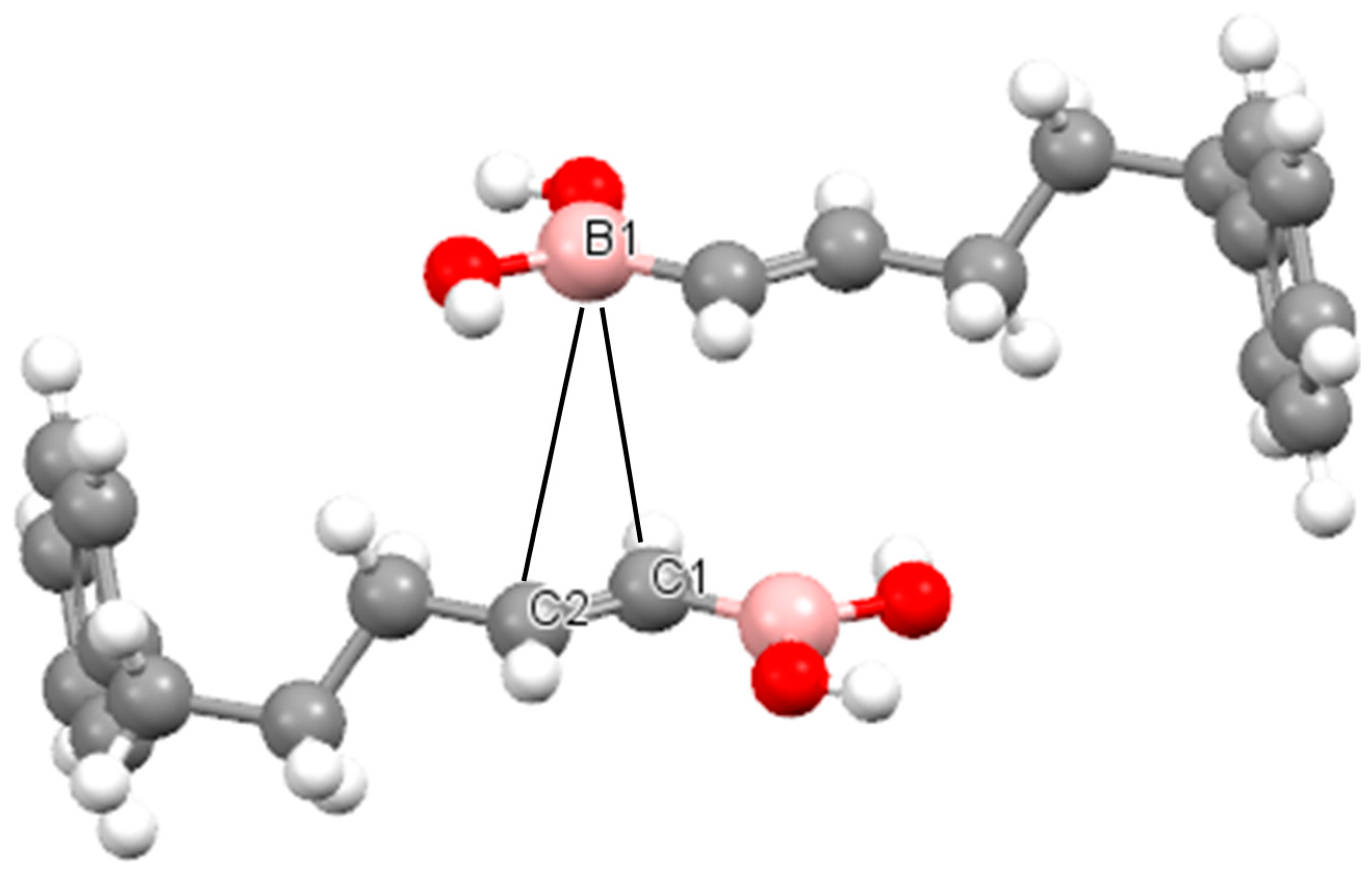{kind=link}
| Complex | EP | Eint | EintBSSE | Ebin | EbinBSSE | Edef | ∑α | R |
|---|---|---|---|---|---|---|---|---|
| AlBr3-C2H2 | −81.8 | −68.9 | −60.6 | −47.4 | 21.2 | 351.4 | 2.482 | |
| AlBr3-C2H4 | 0.103 | −94.3 | −77.9 | −71.1 | −54.7 | 23.2 | 350.8 | 2.497 |
| AlCl3-C2H2 | −79.6 | −73.2 | −58.2 | −51.9 | 21.3 | 351.9 | 2.479 | |
| AlCl3-C2H4 | 0.122 | −89.5 | −81.6 | −66.3 | −58.4 | 23.2 | 351.1 | 2.502 |
| AlF3-C2H2 | −84.7 | −78.3 | −66.9 | −60.5 | 17.8 | 353.8 | 2.437 | |
| AlF3-C2H4 | 0.202 | −91.6 | −83.9 | −72.6 | −64.9 | 19.0 | 353.5 | 2.467 |
| AlH3-C2H2 | −50.2 | −47.7 | −43.3 | −40.7 | 7.0 | 357.0 | 2.562 | |
| AlH3-C2H4 | 0.130 | −58.1 | −55.1 | −50.5 | −47.4 | 7.7 | 356.7 | 2.552 |
| BBr3-C2H2 | −18.6 | −11.0 | −18.4 | −10.8 | 0.2 | 359.9 | 3.325 | |
| BBr3-C2H4 | 0.034 | −22.8 | −12.2 | −22.5 | −11.9 | 0.3 | 359.9 | 3.288 |
| BCl3-C2H2 | −14.2 | −11.3 | −14.0 | −11.0 | 0.3 | 359.9 | 3.312 | |
| BCl3-C2H4 | 0.043 | −16.5 | −12.6 | −16.1 | −12.3 | 0.3 | 359.9 | 3.297 |
| BF3-C2H2 | −16.4 | −12.9 | −15.4 | −12.0 | 0.9 | 359.9 | 2.994 | |
| BF3-C2H4 | 0.090 | −18.2 | −14.0 | −17.0 | −12.7 | 1.3 | 359.7 | 2.990 |
| BH3-C2H2 | 0.070 | −74.2 | −69.2 | −36.2 | −31.2 | 38.0 | 348.4 | 2.014 |

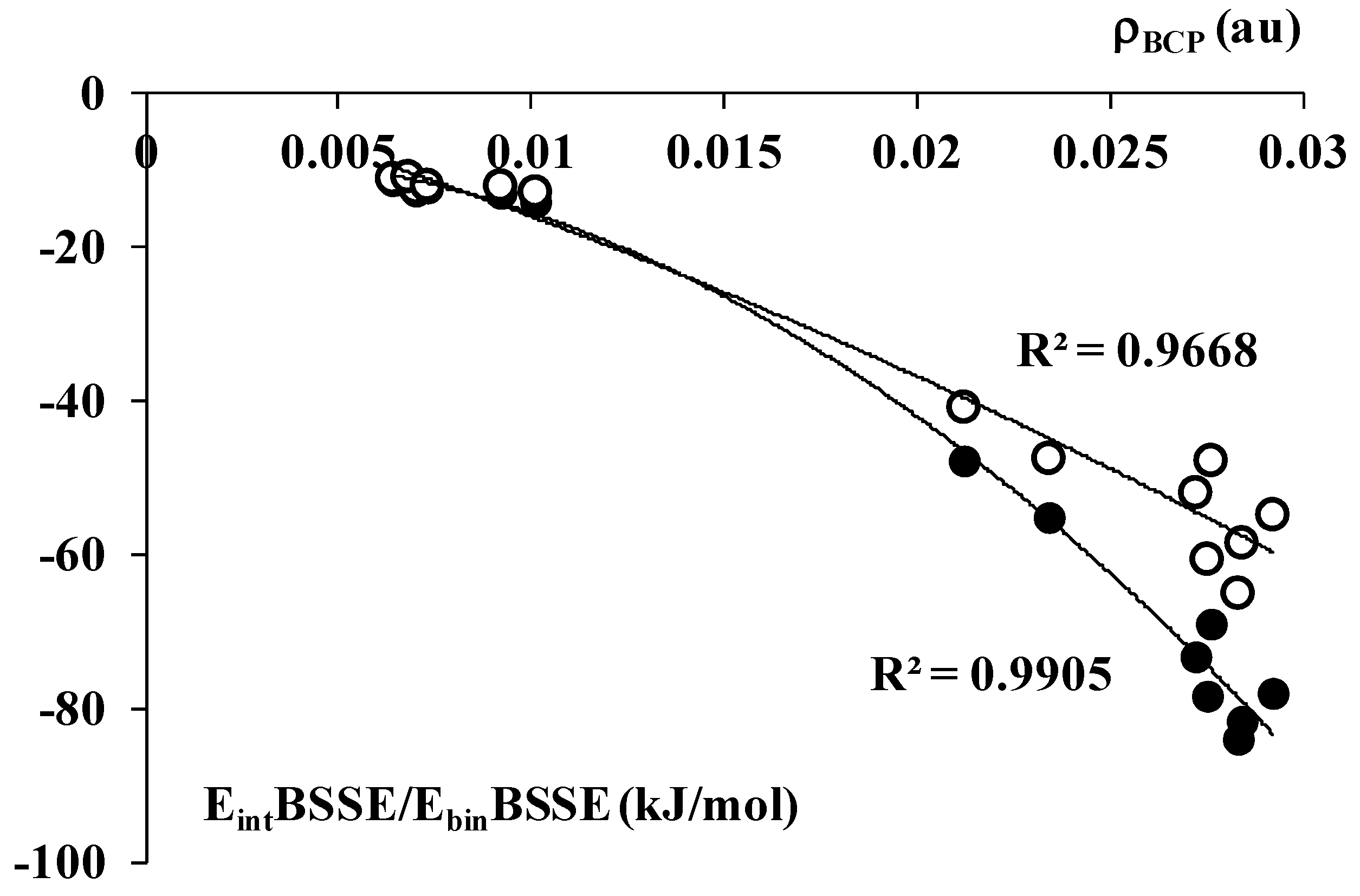
2.2. The Analysis of QTAIM Parameters
| Complex | ρBCP | ∇2ρBCP | HBCP |
|---|---|---|---|
| AlBr3-C2H2 | 0.0276 | 0.0818 | −0.0035 |
| AlBr3-C2H4 | 0.0292 | 0.0725 | −0.0049 |
| AlCl3-C2H2 | 0.0272 | 0.0847 | −0.0031 |
| AlCl3-C2H4 | 0.0284 | 0.0735 | −0.0044 |
| AlF3-C2H2 | 0.0275 | 0.1036 | −0.0017 |
| AlF3-C2H4 | 0.0283 | 0.0903 | −0.0030 |
| AlH3-C2H2 | 0.0212 | 0.0683 | −0.0014 |
| AlH3-C2H4 | 0.0234 | 0.0687 | −0.0023 |
| BBr3-C2H2 | 0.0068 | 0.0191 | 0.0008 |
| BBr3-C2H4 | 0.0073 | 0.0195 | 0.0007 |
| BCl3-C2H2 | 0.0064 | 0.0183 | 0.0007 |
| BCl3-C2H4 | 0.0070 | 0.0182 | 0.0007 |
| BF3-C2H2 | 0.0092 | 0.0273 | 0.0010 |
| BF3-C2H4 | 0.0101 | 0.0264 | 0.0008 |
| BH3-C2H2 | 0.0620 | 0.0325 | −0.0392 |

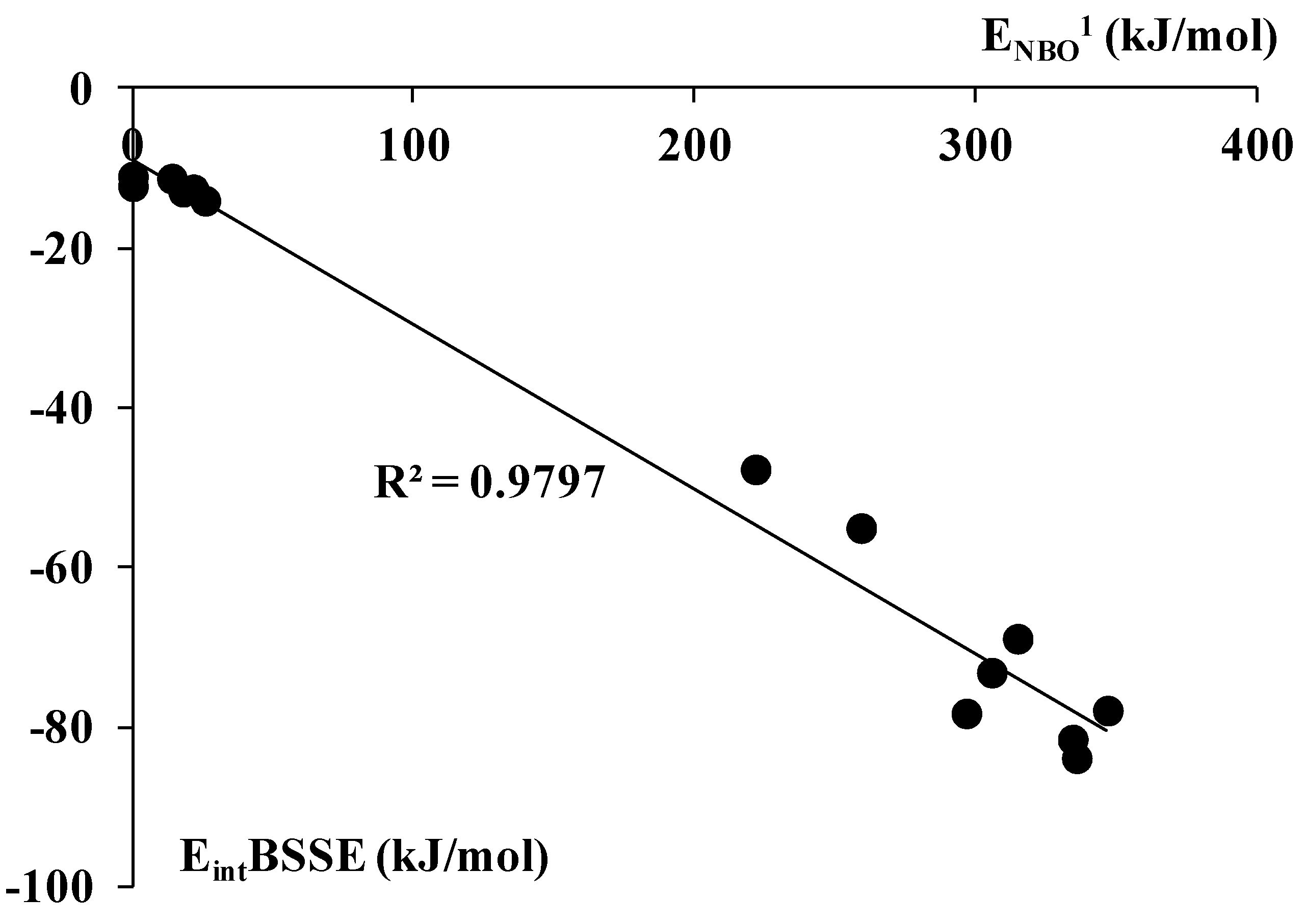
2.3. The NBO Analysis and the Electron Charge Redistributions being the Result of Complexation
| Complex | ENBO1 | ENBO2 | Zcom | Zmon | X/Hcom | X/Hmon | ET |
|---|---|---|---|---|---|---|---|
| AlBr3-C2H2 | 314.6 | 77.8 | 1.230 | 1.333 | −0.458 | −0.444 | −0.145 |
| AlBr3-C2H4 | 346.7 | 79.7 | 1.210 | −0.460 | −0.169 | ||
| AlCl3-C2H2 | 305.4 | 73.4 | 1.479 | 1.591 | −0.540 | −0.530 | −0.142 |
| AlCl3-C2H4 | 334.2 | 75.2 | 1.461 | −0.542 | −0.164 | ||
| AlF3-C2H2 | 296.4 | 0 | 2.273 | 2.381 | −0.791 | −0.794 | −0.101 |
| AlF3-C2H4 | 335.6 | 0 | 2.258 | −0.792 | −0.117 | ||
| AlH3-C2H2 | 221.5 | 34.0 | 1.161 | 1.315 | −0.422 | −0.438 | −0.104 |
| AlH3-C2H4 | 259.0 | 40.7 | 1.121 | −0.415 | −0.124 | ||
| BBr3-C2H2 | 0 | 17.7 | 0.153 | 0.127 | −0.053 | −0.043 | −0.008 |
| BBr3-C2H4 | 0 | 22.9 | 0.151 | −0.054 | −0.011 | ||
| BCl3-C2H2 | 13.8 | 0 | 0.465 | 0.442 | −0.177 | −0.147 | −0.006 |
| BCl3-C2H4 | 21.5 | 0 | 0.464 | −0.158 | −0.010 | ||
| BF3-C2H2 | 17.9 | 1.6 | 1.582 | 1.569 | −0.530 | −0.523 | −0.007 |
| BF3-C2H4 | 25.7 | 2.9 | 1.580 | −0.530 | −0.012 | ||
| BH3-C2H2 | 997.9 | 34.7 | −0.066 | 0.418 | −0.069 | −0.139 | −0.273 |


3. Experimental Section
4. Conclusions

Acknowledgments
Conflicts of Interest
References
- Jeffrey, G.A.; Saenger, W. Hydrogen Bonding in Biological Structures; Springer-Verlag: Berlin, Germany, 1991. [Google Scholar]
- Jeffrey, G.A. An Introduction to Hydrogen Bonding; Oxford University Press: New York, NY, USA, 1997. [Google Scholar]
- Grabowski, S.J. What is the covalency of hydrogen bonding? Chem. Rev. 2011, 11, 2597–2625. [Google Scholar] [CrossRef] [PubMed]
- Desiraju, G.R.; Steiner, T. The Weak Hydrogen Bond in Structural Chemistry and Biology; Oxford University Press: New York, NY, USA, 1999. [Google Scholar]
- Kollman, P. A General analysis of noncovalent intermolecular interactions. J. Am. Chem. Soc. 1977, 99, 4875–4894. [Google Scholar] [CrossRef]
- Schneider, H.J. Binding mechanisms in supramolecular complexes. Angew. Chem. Int. Ed. 2009, 48, 3924–3977. [Google Scholar] [CrossRef] [PubMed]
- Hobza, P.; Müller-Dethlefs, K. Non-Covalent Interactions, Theory and Experiment; Royal Society of Chemistry: Cambridge, UK, 2010. [Google Scholar]
- Clark, T.; Hennemann, M.; Murray, J.S.; Politzer, P. Halogen bonding: The σ-hole. J. Mol. Model. 2007, 13, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Lane, P.; Concha, M.C.; Ma, Y.; Murray, J.S. An overview of halogen bonding. J. Mol. Model. 2007, 13, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Politzer, P.; Riley, K.E.; Bulat, F.A.; Murray, J.S. Perspectives on halogen bonding and other σ-hole interactions: Lex parsimoniae (Occam’s Razor). Comput. Theor. Chem. 2012, 998, 2–8. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding and other σ-hole interactions: A perspective. Phys. Chem. Chem. Phys. 2013, 15, 11178–11189. [Google Scholar] [CrossRef] [PubMed]
- Murray, J.S.; Riley, K.E.; Politzer, P.; Clark, T. Directional Weak Intermolecular Interactions: σ-Hole Bonding. Aust. J. Chem. 2010, 63, 1598–1607. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S.; Clark, T. Halogen bonding: An electrostatically driven highly directional noncovalent interaction. Phys. Chem. Chem. Phys. 2010, 12, 7748–7758. [Google Scholar] [CrossRef] [PubMed]
- Alkorta, I.; Rozas, I.; Elguero, J. Molecular complexes between silicon derivatives and electron-rich groups. J. Phys. Chem. A 2001, 105, 743–749. [Google Scholar] [CrossRef]
- Bundhun, A.; Ramasami, P.; Murray, J.S.; Politzer, P. Trends in σ-hole strengths and interactions of H3MX molecules (M = C, Si, Ge and X = F, Cl, Br, I). J. Mol. Model. 2013, 19, 2739–2746. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Tetrel-bonding interaction: Rediscovered supramolecular force? Angew. Chem. Int. Ed. 2013, 52, 12317–12321. [Google Scholar] [CrossRef] [PubMed]
- Mani, D.; Arunan, E. The X-C···Y (X = O/F, Y = O, S, F, Cl, Br, N, P,) Carbon bond and hydrophobic interactions. Phys. Chem. Chem. Phys. 2013, 15, 14377–14383. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Tetrel bond-σ-hole bond as a preliminary stage of the SN2 reaction. Phys. Chem. Chem. Phys. 2014, 16, 1824–1834. [Google Scholar] [CrossRef] [PubMed]
- Sundberg, M.R.; Uggla, R.; Viñas, C.; Teixidor, F.; Paavola, S.; Kivekäs, R. Nature of intramolecular interactions in hypercoordinate C-substituted 1,2-dicarba-closo-dodecaboranes with short P···P distances. Inorg. Chem. Commun. 2007, 10, 713–716. [Google Scholar] [CrossRef]
- Tschirschwitz, S.; Lönnecke, P.; Hey-Hawkins, E. Aminoalkylferrocenyldichlorophosphanes: Facile synthesis of versatile chiral starting materials. Dalton Trans. 2007, 1377–1382. [Google Scholar] [CrossRef] [PubMed]
- Del Bene, J.E.; Alkorta, I.; Sanchez-Sanz, G.; Elguero, J. Structures, energies, bonding, and NMR properties of pnicogen complexes H2XP:NXH2 (X=H, CH3, NH2, OH, F, Cl). J. Phys. Chem. A 2011, 115, 13724–13731. [Google Scholar] [CrossRef] [PubMed]
- Scheiner, S. Can two trivalent n atoms engage in a direct N···N noncovalent interaction? Chem. Phys. Lett. 2011, 514, 32–35. [Google Scholar] [CrossRef]
- Minyaev, R.M.; Minkin, V.I. Theoretical study of O→X (S, Se, Te) coordination in organic compounds. Can. J. Chem. 1998, 76, 776–788. [Google Scholar] [CrossRef]
- Sanz, P.; Mó, O.; Yañez, M. Competition between X···H···Y intramolecular hydrogen bonds and X···Y (X = O, S, and Y = Se, Te) chalcogen-chalcogen interactions. J. Phys. Chem. A 2002, 106, 4661–4668. [Google Scholar] [CrossRef]
- Wang, W.; Ji, B.; Zhang, Y. Chalcogen bond: A sister noncovalent bond to halogen bond. J. Phys. Chem. A 2009, 113, 8132–8135. [Google Scholar] [CrossRef] [PubMed]
- Alikhani, E.; Fuster, F.; Madebene, B.; Grabowski, S.J. Topological reaction sites—Very strong chalcogen bonds. Phys. Chem. Chem. Phys. 2014, 16, 2430–2442. [Google Scholar] [CrossRef] [PubMed]
- Metrangolo, P.; Resnati, G. Halogen bonding: A paradigm in supramolecular chemistry. Chem. Eur. J. 2001, 7, 2511–2519. [Google Scholar] [CrossRef]
- Cavallo, G.; Metrangolo, P.; Pilati, T.; Resnati, G.; Sansotera, M.; Terraneo, G. Halogen bonding: A general route in anion recognition and coordination. Chem. Soc. Rev. 2010, 39, 3772–3783. [Google Scholar] [CrossRef] [PubMed]
- Bauzá, A.; Mooibroek, T.J.; Frontera, A. Directionality of π-holes in nitro compounds. Chem. Commun. 2015, 51, 1491–1493. [Google Scholar] [CrossRef] [PubMed]
- Mitra, M.; Manna, P.; Bauzá, A.; Ballester, P.; Seth, S.K.; Choudhury, S.R.; Frontera, A.; Mukhopadhyay, S. 3-Picoline mediated self-assembly of M(II)-malonate complexes (M = Ni/Co/Mn/Mg/Zn/Cu) assisted by various weak forces involving lone pair—π, π-π, and Anion…π-hole interactions. J. Phys. Chem. B 2014, 118, 14713–14726. [Google Scholar] [CrossRef] [PubMed]
- Pal, R.; Nagendra, G.; Samarasimhareddy, M.; Sureshbabu, V.V.; Guru Row, T.N. Observation of a reversible isomorphous phase transition and an interplay of “σ-holes” and “π-holes” in Fmoc-Leu-ψ[CH2-NCS]. Chem. Commun. 2015, 51, 933–936. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. Boron and other triel lewis acid centers: From hypovalency to hypervalency. Chem. Phys. Chem. 2014, 15, 2985–2993. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J. π-hole bonds: Boron and aluminium lewis acid centers. Chem. Phys. Chem. 2015, 16, 1470–1479. [Google Scholar] [CrossRef] [PubMed]
- Weinhold, F.; Landis, C. Valency and Bonding, a Natural Bond Orbital Donor—Acceptor Perspective; Cambridge University Press: Cambridge, UK, 2005. [Google Scholar]
- Phillips, J.A.; Cramer, C.J. Quantum chemical characterization of the structural properties of HCN–BF3. J. Chem. Theory Comput. 2005, 1, 827–833. [Google Scholar] [CrossRef]
- Smith, E.L.; Sadowsky, D.; Cramer, C.J.; Phillips, J.A. Structure, bonding, and energetic properties of nitrile-Borane complexes: RCN-BH3. J. Phys. Chem. A 2011, 115, 1955–1963. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Hirota, M.; Umezawa, Y. The CH/π Interaction, Evidence, Nature, and Consequences; Wiley-VCH: New York, NY, USA, 1998. [Google Scholar]
- Bauzá, A.; Quiñonero, D.; Deyà, P.M.; Frontera, A. Pnicogen-π complexes: Theoretical study and biological implications. Phys. Chem. Chem. Phys. 2012, 14, 14061–14066. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Liu, R.; Li, P.; Zhang, H. On the properties of S…O and S…π noncovalent interactions: The analysis of geometry, interaction energy and electron density. New J. Chem. 2015, 39, 1611–1618. [Google Scholar] [CrossRef]
- Nziko, V.P.; Scheiner, S. S…π Chalcogen Bonds between SF2 or SF4 and C-C multiple bonds. J. Chem. Phys. A 2015, 119, 5889–5897. [Google Scholar] [CrossRef] [PubMed]
- Brown, H.C. Hydroboration—A powerful synthetic tool. Tetrahedron 1961, 12, 117–138. [Google Scholar] [CrossRef]
- Brown, H.C.; Zaidlewicz, M. Organic Syntheses via Boranes; Aldrich Chemical Co.: Milwaukee, WI, USA, 2001; Volume 2. [Google Scholar]
- Blackborow, J.R. Stereoselectivity of halogenoboronation of phenylacetylene. J. Chem. Soc. Perkin II 1973, 1989–1993. [Google Scholar] [CrossRef]
- Uhl, W. Hydroalumination and hydrogallation of alkynes: New insights into the course of well-known reactions. Coord. Chem. Rev. 2008, 252, 1540–1563. [Google Scholar] [CrossRef]
- Hugas, D.; Simon, S.; Duran, M.; Guerra, C.F.; Bickelhaupt, F.M. Dihydrogen bonding: Donor-acceptor bonding (AH...HX) versus the H2 molecule (A-H2-X). Chem. Eur. J. 2009, 15, 5814–5822. [Google Scholar] [CrossRef] [PubMed]
- Wolstenholme, D.J.; Dobson, J.L.; McGrady, G.S. Homopolar dihydrogen bonding in main group hydrides: Discovery, consequences, and applications. Dalton Trans. 2015, 44, 9718–9731. [Google Scholar] [CrossRef] [PubMed]
- Clark, T.; Schleyer, P.V.R. Hydroboration: An ab initio study of the reaction of BH3 with ethylene. J. Organomet. Chem. 1978, 156, 191–202. [Google Scholar] [CrossRef]
- Nagase, S.; Ray, N.K.; Morokuma, K. Reaction mechanism of hydroboration, Ab initio MO study on the C2H4 + BH3 reaction. J. Am. Chem. Soc. 1980, 102, 4536–4537. [Google Scholar] [CrossRef]
- Fau, S.; Frenking, G. Theoretical investigation of the weakly bonded donor-acceptor complexes X3B-H2, X3B-C2H4, and X3B-C2H2 (X = H, F, Cl). Mol. Phys. 1999, 96, 519–527. [Google Scholar]
- Hirao, H.; Omoto, K.; Fujimoto, H. Lewis acidity of boron trihalides. J. Phys. Chem. A 1999, 103, 5807–5811. [Google Scholar] [CrossRef]
- Herrebout, W.A.; van der Veken, B.J. Van der Waals complexes between unsaturated hydrocarbons and boron trifluoride: An infrared and ab initio study of ethane·BF3 and propene·BF3. J. Am. Chem. Soc. 1997, 119, 10446–10454. [Google Scholar] [CrossRef]
- Lipscomb, W.N. Boron Hydrides; W. A. Benjamin: New York, NY, USA, 1963. [Google Scholar]
- NIST Chemistry WebBook. NIST Standard Reference Database Number 69; Linstrom, P.J., Mallard, W.G., Eds.; National Institute of Standards and Technology: Gaithersburg, MD, USA, 2010. [Google Scholar]
- Grabowski, S.J.; Ugalde, J.M. Bond paths show preferable interactions: Ab initio and QTAIM studies on the X-H···π hydrogen bond. J. Phys. Chem. A 2010, 114, 7223–7229. [Google Scholar] [CrossRef] [PubMed]
- Grabowski, S.J.; Sokalski, W.A. Different types of hydrogen bonds: Correlation analysis of interaction energy components. J. Phys. Org. Chem. 2005, 18, 779–784. [Google Scholar] [CrossRef]
- Cremer, D.; Kraka, E. A description of the chemical bond in terms of local properties of electron density and energy. Croat. Chem. Acta 1984, 57, 1259–1281. [Google Scholar]
- Jenkins, S.; Morrison, I. The chemical character of the intermolecular bonds of seven phases of ice as revealed by ab initio calculation of electron densities. Chem. Phys. Lett. 2000, 317, 97–102. [Google Scholar] [CrossRef]
- Pauling, L. Nature of the Chemical Bond; Cornell University Press: Ithaca, NY, USA, 1960; pp. 88–107. [Google Scholar]
- Könczöl, N.; Turczel, G.; Szpisjak, T.; Szieberth, D. The stability of η2-H2 borane complexes: A theoretical investigation. Dalton Trans. 2014, 43, 13571–13577. [Google Scholar] [CrossRef] [PubMed]
- Hiberty, P.C.; Ohanessian, G. Comparison of minimal and extended basis sets in terms of resonant formulas. Application to 1,3 dipoles. J. Am. Chem. Soc. 1982, 104, 66–70. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Revision A.1; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Peterson, K.A.; Figgen, D.; Goll, E.; Stoll, H.; Dolg, M. Systematically convergent basis sets with relativistic pseudopotentials. II. Small-core pseudopotentials and correlation consistent basis sets for the post-d group 16–18 elements. J. Chem. Phys. 2003, 119, 11113–11123. [Google Scholar] [CrossRef]
- Piela, L. Ideas of Quantum Chemistry; Elsevier Science Publishers: Amsterdam, The Netherlands, 2007; pp. 684–691. [Google Scholar]
- Boys, S.F.; Bernardi, F. The calculation of small molecular interactions by the differences of separate total energies: Some procedures with reduced errors. Mol. Phys. 1970, 19, 553–561. [Google Scholar] [CrossRef]
- Mentel, L.M.; Baerends, E.J. Can the counterpoise correction for basis set superposition effect be justified? J. Chem. Theory Comput. 2014, 10, 252–267. [Google Scholar] [CrossRef]
- Alkorta, I.; Trujillo, C.; Elguero, J.; Solimannejad, M. A theoretical study of the hydrogen bonding properties of H2BNH2: Some considerations on the basis set superposition error issue. Comput. Theor. Chem. 2011, 967, 147–151. [Google Scholar] [CrossRef]
- Reed, E.; Curtiss, L.A.; Weinhold, F. Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev. 1988, 88, 899–926. [Google Scholar] [CrossRef]
- Glendening, E.D.; Badenhoop, J.K.; Reed, A.E.; Carpenter, J.E.; Bohmann, J.A.; Morales, C.M.; Weinhold, F. NBO 5.0; Theoretical Chemistry Institute, University of Wisconsin: Madison, WI, USA, 2001. [Google Scholar]
- Schmidt, M.W.; Baldridge, K.K.; Boatz, J.A.; Elbert, S.T.; Gordon, M.S.; Jensen, J.H.; Koseki, S.; Matsunaga, N.; Nguyen, K.A.; Su, S.J.; et al. General atomic and molecular electronic structure system. J. Comput. Chem. 1993, 14, 1347–1363. [Google Scholar] [CrossRef]
- Bader, R.F.W. Atoms in Molecules, a Quantum Theory; Oxford University Press: Oxford, UK, 1990. [Google Scholar]
- Keith, T.A. AIMAll; (Version 11.08.23); TK Gristmill Software: Overland Park, KS, USA, 2011. [Google Scholar]
- Wong, R.; Allen, F.H.; Willett, P. The scientific impact of the Cambridge Structural Database: A citation-based study. J. Appl. Cryst. 2010, 43, 811–824. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Grabowski, S.J. Triel Bonds, π-Hole-π-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene. Molecules 2015, 20, 11297-11316. https://doi.org/10.3390/molecules200611297
Grabowski SJ. Triel Bonds, π-Hole-π-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene. Molecules. 2015; 20(6):11297-11316. https://doi.org/10.3390/molecules200611297
Chicago/Turabian StyleGrabowski, Sławomir J. 2015. "Triel Bonds, π-Hole-π-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene" Molecules 20, no. 6: 11297-11316. https://doi.org/10.3390/molecules200611297
APA StyleGrabowski, S. J. (2015). Triel Bonds, π-Hole-π-Electrons Interactions in Complexes of Boron and Aluminium Trihalides and Trihydrides with Acetylene and Ethylene. Molecules, 20(6), 11297-11316. https://doi.org/10.3390/molecules200611297