
Plasmodium falciparum Thioredoxin Reductase (PfTrxR) and Its Role as a Target for New Antimalarial Discovery

Abstract
:1. Introduction
2. Key Aspects of PfTrxR Active Sites for Enzyme Inhibition
2.1. N-Terminal Redox Center
2.2. C-Terminal Redox Center
2.3. Enzyme Catalytic Cycle
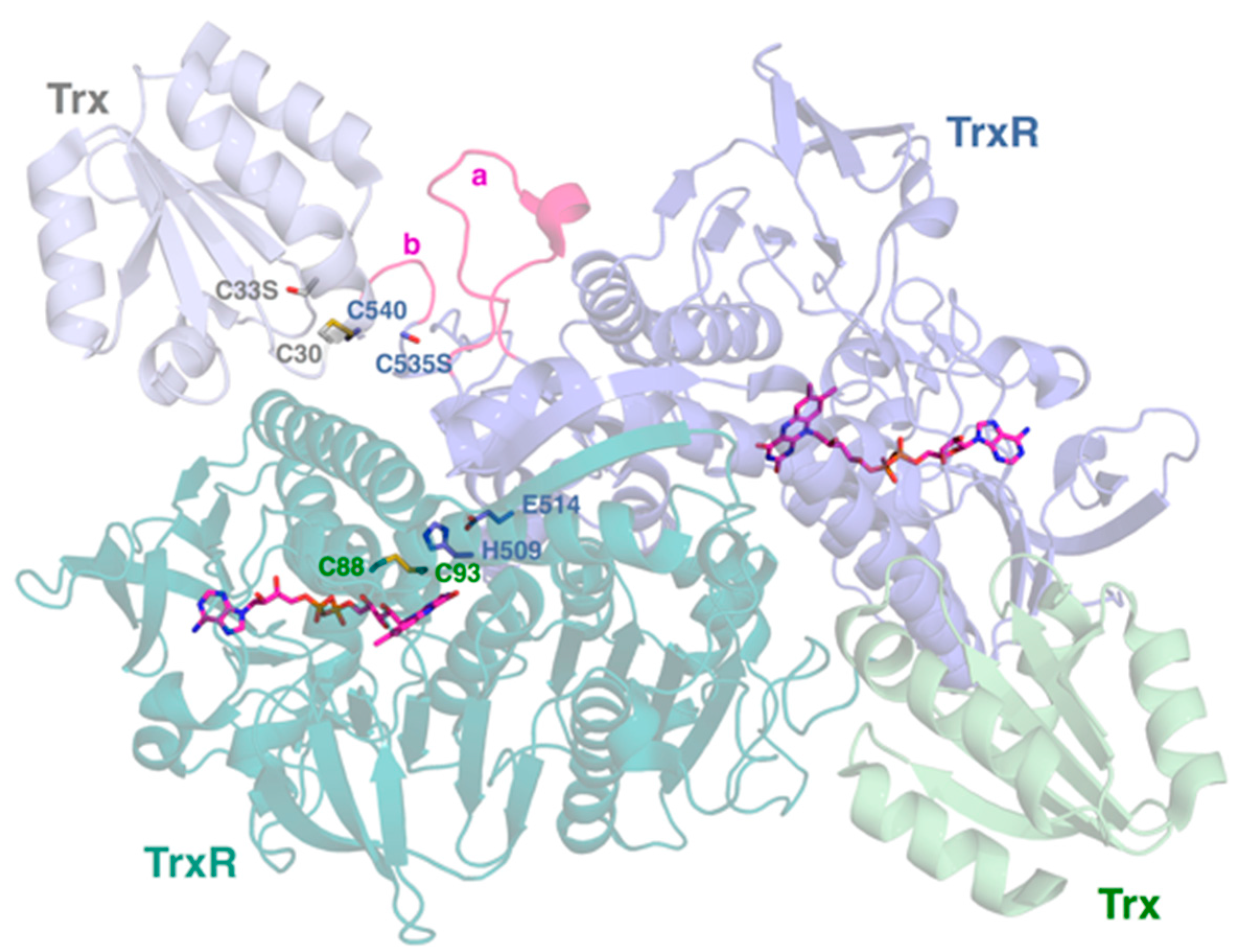
2.4. Interactive Cavity and Interface
2.5. Insertion Loop
3. TrxR: Human and Plasmodium Species
3.1. TrxR Isoenzymes
3.2. Identities in the Sequences of Human and Plasmodium Species TrxRs


{kind=link}
{kind=link}
| H. sapiens (hTrxR1) | H. sapiens (hTrxR2) | P. falciparum | P. vivax | P. yoelii yoelii | P. berghei ANKA | P. knowlesi Strain H | P. cynomolgi Strain B | |
|---|---|---|---|---|---|---|---|---|
| Accession Number | AAB35418 | AAD19597 | CAA60574 | EDL45043 | EAA21839 | XP_679935 | XP_002258509 | XP_004221759 |
| Number of amino acids | 497 | 524 | 541 | 546 | 638 | 542 | 623 | 628 |
| Identity to P. falciparum (%) | 42 | 40 | 100 | 77 | 79 | 79 | 79 | 80 |
| Identity to H. sapiens (hTrxR2) (%) | 54 | 100 | 40 | 41 | 41 | 40 | 41 | 41 |
| Identity to H. sapiens (hTrxR1) (%) | 100 | 54 | 42 | 41 | 42 | 42 | 41 | 41 |

3.3. Correlation of Enzymatic Inhibitory Activity between PfTrxR and the TrxRs of Other Plasmodium Species
3.3.1. Selective Inhibitory Activity toward PfTrxR versus hTrxR
3.3.2. Selective Inhibitory Activity toward PfTrxR versus Other Plasmodium Species TrxRs
3.4. Essentiality of TrxR in Plasmodium falciparum and Other Plasmodium Species
3.5. Animal Models Available to Test PfTrxR Inhibitors
Primate Models
3.6. PfTrxR and Antimalarial Drug Resistance
4. Future Studies
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Malaria. Available online: http://www.cdc.gov/malaria/about/faqs.html (accessed on 16 February 2015).
- Malaria. Available online: http://www.who.int/mediacentre/factsheets/fs094/en/ (accessed on 2 February 2015).
- World Malaria Report 2014. Available online: http://apps.who.int/iris/bitstream/10665/144852/2/9789241564830_eng.pdf?ua=1 (accessed on 24 February 2015).
- Tachibana, S.; Sullivan, S.A.; Kawai, S.; Nakamura, S.; Kim, H.R.; Goto, N.; Arisue, N.; Palacpac, N.M.; Honma, H.; Yagi, M.; et al. Plasmodium cynomolgi genome sequences provide insight into Plasmodium vivax and the monkey malaria clade. Nat. Genet. 2012, 44, 1051–1055. [Google Scholar] [CrossRef] [PubMed]
- Treatment of Malaria: Guideline for Clinicians (United States). Available online: http://www.cdc.gov/malaria/diagnosis_treatment/clinicians2.html (accessed on 3 March 2015).
- Life Cycle. Available online: http://www.bio.davidson.edu/people/sosarafova/Assets/Bio307/ruturakhia/page01.html (accessed on 24 February 2015).
- Crutcher, J.M.; Hoffman, S.L. Medical Microbiology, 4th ed.University of Texas Medical Branch: Galveston, TX, USA, 1996; Chapter 83. Available online: http://www.ncbi.nlm.nih.gov/books/NBK8584/ (accessed on 5 May 2015).
- Jortzik, E.; Becker, K. Thioredoxin and glutathione systems in Plasmodium falciparum. Int. J. Med. Microbiol. 2012, 302, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Arnér, E.S.J.; Holmgren, A. Physiological functions of thioredoxin and thioredoxin reductase. Eur. J. Biochem. 2000, 267, 6102–6109. [Google Scholar] [CrossRef] [PubMed]
- Lakshminarayana, S.; Freymond, C.; Fischli, C.; Yu, J.; Weber, S.; Goh, A.; Yeung, B.K.S.; Ho, P.C.; Dartois, V.; Diagana, T.T.; et al. Pharmacokinetic-pharmacodynamic analysis of spiroindolone analogs and KAE609 in a murine malaria model. Antimicrob. Agents Chemother. 2015, 59, 1200–1210. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Gilberger, T.; Krnajski, Z.; Lüersen, K.; Meierjohann, S.; Walter, R. Thioredoxin and glutathione system of malaria parasite Plasmodium falciparum. Protoplasma 2001, 217, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Rahlfs, S.; Schirmer, R.; Becker, K. The thioredoxin system of Plasmodium falciparum and other parasites. Cell. Mol. Life Sci. 2002, 59, 1024–1041. [Google Scholar] [CrossRef] [PubMed]
- Rahlfs, S.; Nickel, C.; Deponte, M.; Schirmer, R.; Becker, K. Plasmodium falciparum thioredoxins and glutaredoxins as central players in redox metabolism. Redox Rep. 2003, 8, 246–250. [Google Scholar] [CrossRef] [PubMed]
- Mohring, F.; Pretzel, J.; Jortzik, E.; Becker, K. The redox systems of Plasmodium falciparum and Plasmodium vivax: Comparison, in silico analyses and inhibitor studies. Curr. Med. Chem. 2014, 15, 1728–1756. [Google Scholar] [CrossRef]
- Jaeger, T.; Flohé, L. The thiol-based redox networks of pathogens: Unexploited targets in the search for new drugs. BioFactors 2006, 27, 109–120. [Google Scholar] [CrossRef] [PubMed]
- Williams, C., Jr.; Arscott, D.; Müller, S.; Lennon, B.; Ludwig, M.; Veine, D.; Becker, K.; Schirmer, R. Thioredoxin reductase two modes of catalysis have evolved. Eur. J. Biochem. 2000, 267, 6110–6117. [Google Scholar] [CrossRef] [PubMed]
- Becker, K.; Gromer, S.; Schirmer, R.; Müller, S. Thioredoxin reductase as a pathophysiological factor and drug target. Eur. J. Biochem. 2000, 267, 6118–6125. [Google Scholar] [CrossRef] [PubMed]
- Müller, S.; Liebau, E.; Walter, R.; Krauth-Siegel, R. Thiol-based redox metabolism of protozoan parasites. Trends Parasitol. 2003, 19, 320–328. [Google Scholar] [CrossRef]
- Munigunti, R.; Gathiaka, S.; Acevedo, O.; Sahu, R.; Tekwani, B.; Calderon, A. Determination of antiplasmodial activity and binding affinity of curcumin and demethoxycurcumin towards PfTrxR. Nat. Prod. Res. 2014, 28, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Munigunti, R.; Becker, K.; Brun, R.; Calderon, A. Determination of antiplasmodial activity and binding affinity of selected natural products towards PfTrxR and PfGR. Nat. Prod. Commun. 2013, 8, 1135–1136. [Google Scholar] [PubMed]
- Andricopulo, A.; Akoachere, M.; Krogh, R.; Nickel, C.; McLeish, M.; Kenyon, G.; Arscott, L.; Williams, C.; Davioud-Charvet, E. Specific inhibitors of Plasmodium falciparum thioredoxin reductase as potential antimalarial agents. Bioorg. Med. Chem. Lett. 2006, 16, 2283–2292. [Google Scholar] [CrossRef] [PubMed]
- Boumis, G.; Giardina, G.; Angelucci, F.; Bellelli, A.; Brunori, M.; Dimastrogiovanni, D.; Saccoccia, F.; Miele, A.E. Crystal Structure of Plasmodium falciparum thioredoxin reductase, a validated drug target. Biochem. Biophys. Res. Commun. 2012, 425, 806–811. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Wolf, K.; Jortzik, E.; Stumpt, M.; Preuss, J.; Iozef, R.; Rahlfs, S.; Becker, K. Crystal structure of the Plasmodium falciparum thioredoxin reductase-thioredoxin complex. Mol. Biol. 2013, 425, 3446–3460. [Google Scholar] [CrossRef] [PubMed]
- Snider, G.; Dustin, C.; Ruggles, E.; Hondal, R. A mechanistic investigation of the c-terminal redox motif of thioredoxin reductase from Plasmodium falciparum. Biochemistry 2014, 53, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Gilberger, T.; Walter, R.; Muller, Sylke. Identification and characterization of the functional amino acids at the active site of the large thioredoxin reductase from Plasmodium falciparum. J. Biol. Chem. 1997, 272, 29584–29589. [Google Scholar] [CrossRef] [PubMed]
- Muller, S. Thioredoxin reductase and glutathione synthesis in Plasmodium falciparum. Redox Rep. 2003, 8, 251–255. [Google Scholar] [CrossRef] [PubMed]
- McMillan, P.; Arscott, L.; Ballou, D.; Becker, K.; Williams, C., Jr.; Müller, S. Identification of acid-base catalytic residues of high-Mr thioredoxin reductase from Plasmodium falciparum. J. Biol. Chem. 2006, 281, 32967–32977. [Google Scholar] [CrossRef] [PubMed]
- Bozdech, Z.; Ginsburg, H. Antioxidant defense in Plasmodium falciparum—Data mining of the transcriptome. Malar. J. 2004, 3, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miranda-Vizuete, A.; Damdimopoulos, A.; Pedrajas, J.R.; Gustafsson, J.; Spyrou, G. Human mitochondrial thioredoxin reductase. Eur. J. Biochem. 1999, 261, 405–412. [Google Scholar] [CrossRef] [PubMed]
- Su, D.; Novoselov, S.V.; Sun, Q.A.; Moustafa, M.E.; Zhou, Y.; Oko, R.; Hatfield, D.L.; Gladyshev, V.N. Mammalian selenoprotein thioredoxin-glutathione reductase. Roles in disulfide bond formation and sperm maturation. J. Biol. Chem. 2005, 280, 26491–26498. [Google Scholar] [CrossRef] [PubMed]
- Winkler, M.; Maynadier, M.; Wein, S.; Lespinasse, M.; Boumis, G.; Miele, A.; Vial, H.; Wong, Y. Uncovering new structural insights for antimalarial activity from cost-effective aculeatin-like derivatives. Org. Biomol. Chem. 2015, 13, 2064–2077. [Google Scholar] [CrossRef] [PubMed]
- Theobald, A.J.; Caballero, I.; Coma, I.; Colmenarejo, G.; Cid, C.; Gamo, F.; Hibbs, M.J.; Bass, A.L.; Thomas, D.A. Discovery and biochemical characterization of Plasmodium thioredoxin reductase inhibitor from an antimalarial set. Biochemistry 2012, 51, 4764–4771. [Google Scholar] [CrossRef] [PubMed]
- Buchholz, K.; Putrianti, E.D.; Rahlfs, S.; Schirmer, R.H.; Becker, K.; Matuschewski, K. Molecular genetics evidence for the in vivo roles of the two major NADPH-dependent disulfide reductases in the malaria parasite. J. Biol. Chem. 2010, 285, 37388–37395. [Google Scholar] [CrossRef] [PubMed]
- Patzewitz, E.M.; Wong, E.H.; Muller, S. Dissecting the role of glutathione biosynthesis in Plasmodium falciparum. Mol. Microbiol. 2012, 83, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Saccoccia, F.; Angelucci, F.; Boumis, G.; Carotti, D.; Desiato, G.; Miele, A.E.; Bellelli, A. Thioredoxin reductase and its inhibitors. Curr. Protein Pept. Sci. 2014, 15, 621–646. [Google Scholar] [CrossRef] [PubMed]
- Boucher, I.W.; McMillan, P.J.; Gabrielsen, M.; Akerman, S.E.; Brannigan, J.A.; Schnick, C.; Brzozowski, A.M.; Wilkinson, A.J.; Muller, S. Structural and biochemical characterization of a mitochondrial peroxiredoxin from Plasmodium falciparum. Mol. Microbiol. 2006, 61, 948–959. [Google Scholar] [CrossRef] [PubMed]
- Krnajski, Z.; Gilberger, T.; Walter, R.D.; Cowman, A.F.; Muller, S. Thioredoxin reductase is essential for the survival of Plasmodium falciparum erythrocytic stages. J. Biol. Chem. 2002, 277, 25970–25975. [Google Scholar] [CrossRef] [PubMed]
- Kaira, B.; Chawla, S.; Gupta, P.; Valecha, N. Screening of antimalarial drugs: An overview. Indian J. Pharmacol. 2006, 38, 7–11. [Google Scholar]
- Badell, E.; Pasquetto, V.; Van Rooijen, N.; Druilhe, P. A mouse model for human malaria erythrocytic stages. Parasitol. Today 1995, 11, 235–237. [Google Scholar] [CrossRef]
- Fidock, D.; Rosenthal, P.; Croft, S.; Brun, R.; Nwaka, S. Antimalarial drug discovery: Efficacy models for compound screening. Nat. Rev. Drug Discov. 2004, 3, 509–520. [Google Scholar] [CrossRef] [PubMed]
- Moreno, A.; Badell, E.; Van Rooijen, N.; Druilhe, P. Human malaria n immunocompromised mice: New in vivo model for chemotherapy studies. Antimicrob. Agents Chemother. 2001, 45, 1847–1853. [Google Scholar] [CrossRef] [PubMed]
- Peters, W.; Howells, R.E.; Portus, J.; Robinson, B.L.; Thomas, S.; Warhurst, D.C. The chemotherapy of rodent malaria. XXVI. Studies on mefloquine (WR 142,490). Ann. Trop. Med. Parasitol. 1977, 71, 407–441. [Google Scholar] [PubMed]
- Peters, W.; Robinson, B.L.; Ellis, D.S. The chemotherapy of rodent malaria. XLII. Halofantrine and halofantrine resistance. Ann. Trop. Med. Parasitol. 1987, 81, 639–646. [Google Scholar] [PubMed]
- Vennerstrom, J.L.; Dong, Y.; Andersen, S.L.; Ager, A.L., Jr.; Fu, H.; Miller, R.E.; Wesche, D.L.; Kyle, D.E.; Gerena, L.; Walters, S.M.; et al. Synthesis and antimalarial activity of sixteen dispiro-1,2,4,5-tetraoxanes: Alkyl-substituted 7,8,15,16-tetraoxadispiro[5.2.5.2]hexadecanes. J. Med. Chem. 2000, 43, 2753–2758. [Google Scholar] [CrossRef] [PubMed]
- Moll, K.; Ljungstrӧm, I.; Perlmann, H.; Scherf, A.; Wahlgren, M. Methods in Malaria Research, 5th ed.; Malaria Research and Reference Reagent Resource Center: Manassas, VA, USA, 2008; pp. 141–152. [Google Scholar]
- Sahu, R.; Walker, L.A.; Tekwani, B.L. In vitro and in vivo anti-malarial activity of tigecycline, a glycylcycline antibiotic, in combination with chloroquine. Malar. J. 2014, 13, 414. [Google Scholar] [CrossRef] [PubMed]
- Deye, G.A.; Gettayacamin, M.; Hansukjariya, P.; Im-erbsin, R.; Sattabongkot, J.; Rothstein, Y.; Macareo, L.; Fracisco, S.; Bennett, K.; Magill, A.J.; et al. Use of a rhesus Plasmodium cynomolgi model to screen for anti-hypnozoite activity of pharmaceutical substances. Am. J. Trop. Med. Hyg. 2012, 86, 931–935. [Google Scholar] [CrossRef] [PubMed]
- Sinha, S.; Medhi, B.; Sehgal, R. Challenges of drug-resistant malaria. Parasite 2014, 21, 61. [Google Scholar] [CrossRef] [PubMed]
- Patzewitz, E.M.; Salcedo-Sora, J.E.; Wong, E.H.; Sethia, S.; Stocks, P.A.; Maughan, S.C.; Murray, J.A.; Krishna, S.; Bray, P.G.; Ward, S.A.; et al. Glutathione transport: A new role for PfCRT in chloroquine resistance. Antioxid Redox Signal. 2013, 19, 683–695. [Google Scholar] [CrossRef] [PubMed]
- Raj, D.K.; Mu, J.; Jiang, H.; Kabat, J.; Singh, S.; Sullivan, M.; Fay, M.P.; McCutchan, T.F.; Su, X.Z. Disruption of a Plasmodium falciparum multidrug resistance-associated protein (PfMRP) alters its fitness and transport of antimalarial drugs and glutathione. J. Biol. Chem. 2009, 284, 7687–7696. [Google Scholar] [CrossRef] [PubMed]
- Zhao, L.; Li, W.; Zhou, Y.; Zhang, Y.; Huang, S.; Xu, X.; Li, Z.; Guo, Q. The overexpression and nuclear translocation of Trx-1 during hypoxia confers on HepG2 cells resistance to DDP, and GL-V9 reverses the resistance by suppressing the Trx-1/Ref-1 axis. Free Radic. Biol. Med. 2015, 82, 29–41. [Google Scholar] [CrossRef] [PubMed]
- Zhou, B.; Huang, J.; Zuo, Y.; Li, B.; Guo, Q.; Cui, B.; Shao, W.; Du, J.; Bu, X. 2a, a novel curcumin analog, sensitizes cisplatin-resistant A549 cells to cisplatin by inhibiting thioredoxin reductase concomitant oxidative stress damage. Eur. J. Pharmacol. 2013, 707, 130–139. [Google Scholar] [CrossRef] [PubMed]
- Sobhakumari, A.; Love-Homan, L.; Fletcher, E.V.; Martin, S.M.; Parsons, A.D.; Spitz, D.R.; Knudson, C.M.; Simons, A.L. Susceptibility of human head and neck cancer cells to combined inhibition of glutathione and thioredoxin metabolism. PLoS ONE 2012, 7, e48175. [Google Scholar] [CrossRef] [PubMed]
- Leitsch, D.; Burgess, A.G.; Dunn, L.A.; Krauer, K.G.; Tan, K.; Duchêne, M.; Upcroft, P.; Eckmann, L.; Upcroft, J.A. Pyruvate:ferredoxin oxidoreductase and thioredoxin reductase are involved in 5-nitroimidazole activation while flavin metabolism is linked to 5-nitroimidazole resistance in Giardia lamblia. J. Antimicrob. Chemother. 2011, 66, 1756–1765. [Google Scholar] [CrossRef] [PubMed]
- Eriksson, S.E.; Prast-Nielsen, S.; Flaberg, E.; Szekely, L.; Arnér, E.S. High levels of thioredoxin reductase 1 modulate drug-specific cytotoxic efficacy. Free Radic. Biol. Med. 2009, 47, 1661–1671. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
McCarty, S.E.; Schellenberger, A.; Goodwin, D.C.; Fuanta, N.R.; Tekwani, B.L.; Calderón, A.I. Plasmodium falciparum Thioredoxin Reductase (PfTrxR) and Its Role as a Target for New Antimalarial Discovery. Molecules 2015, 20, 11459-11473. https://doi.org/10.3390/molecules200611459
McCarty SE, Schellenberger A, Goodwin DC, Fuanta NR, Tekwani BL, Calderón AI. Plasmodium falciparum Thioredoxin Reductase (PfTrxR) and Its Role as a Target for New Antimalarial Discovery. Molecules. 2015; 20(6):11459-11473. https://doi.org/10.3390/molecules200611459
Chicago/Turabian StyleMcCarty, Sara E., Amanda Schellenberger, Douglas C. Goodwin, Ngolui Rene Fuanta, Babu L. Tekwani, and Angela I. Calderón. 2015. "Plasmodium falciparum Thioredoxin Reductase (PfTrxR) and Its Role as a Target for New Antimalarial Discovery" Molecules 20, no. 6: 11459-11473. https://doi.org/10.3390/molecules200611459
APA StyleMcCarty, S. E., Schellenberger, A., Goodwin, D. C., Fuanta, N. R., Tekwani, B. L., & Calderón, A. I. (2015). Plasmodium falciparum Thioredoxin Reductase (PfTrxR) and Its Role as a Target for New Antimalarial Discovery. Molecules, 20(6), 11459-11473. https://doi.org/10.3390/molecules200611459