
Recent Advances in the Synthesis of Carotenoid-Derived Flavours and Fragrances


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction

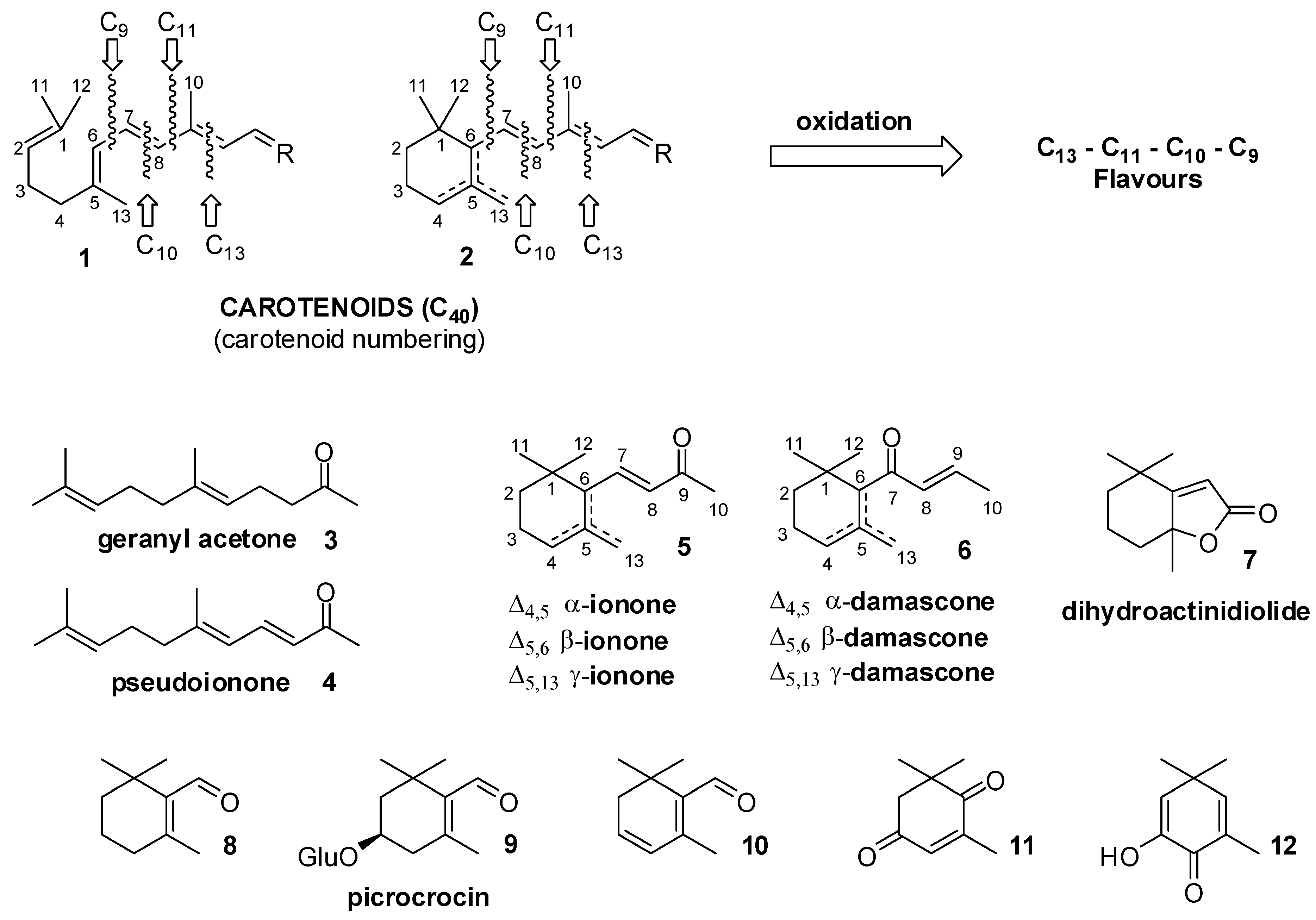
2. Preparation of “Natural” Carotenoid-Derived Flavours and Fragrances
3. New Synthetic Approaches to the Most Relevant Classes of Apocarotenoids Odorants
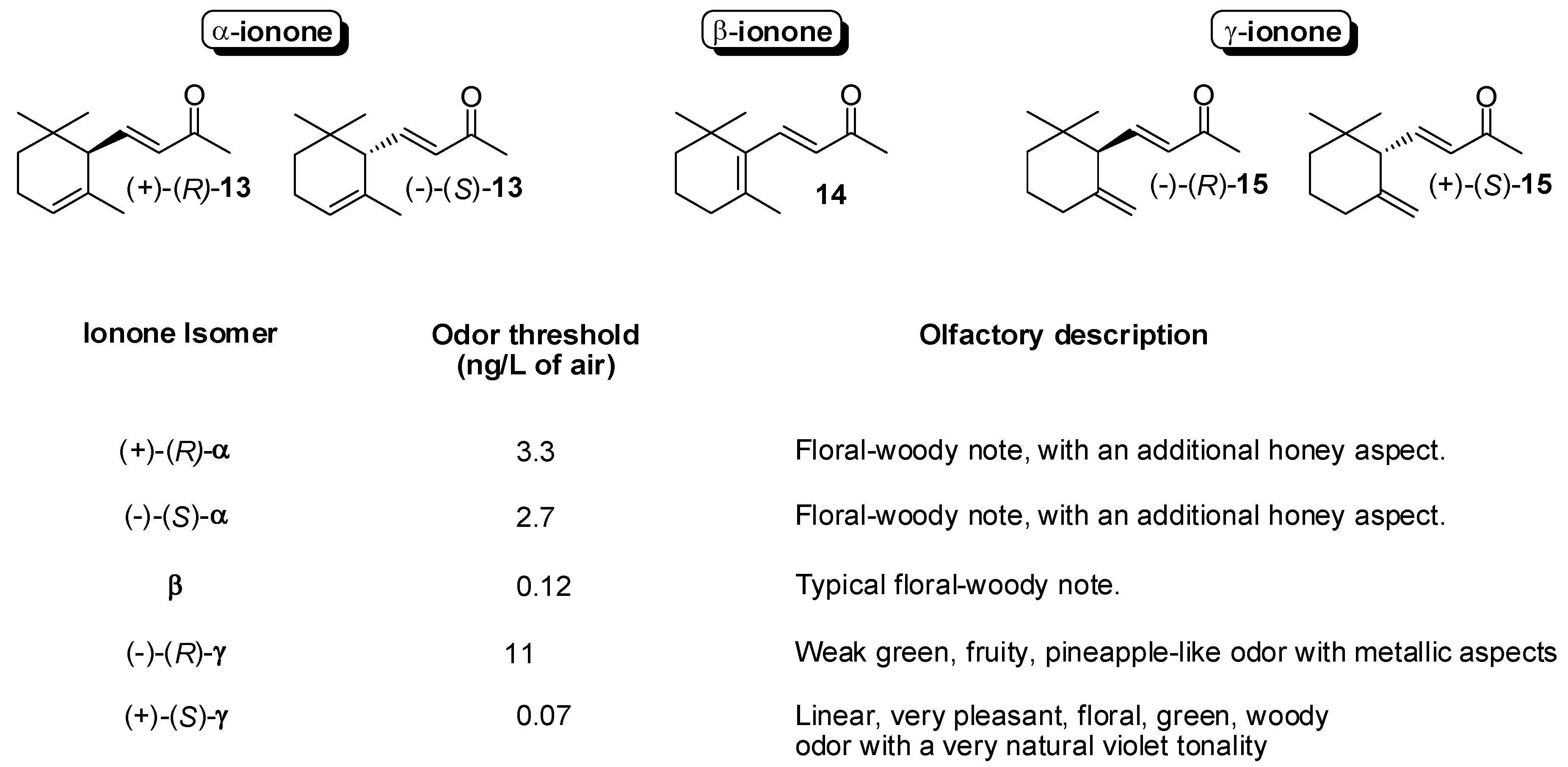
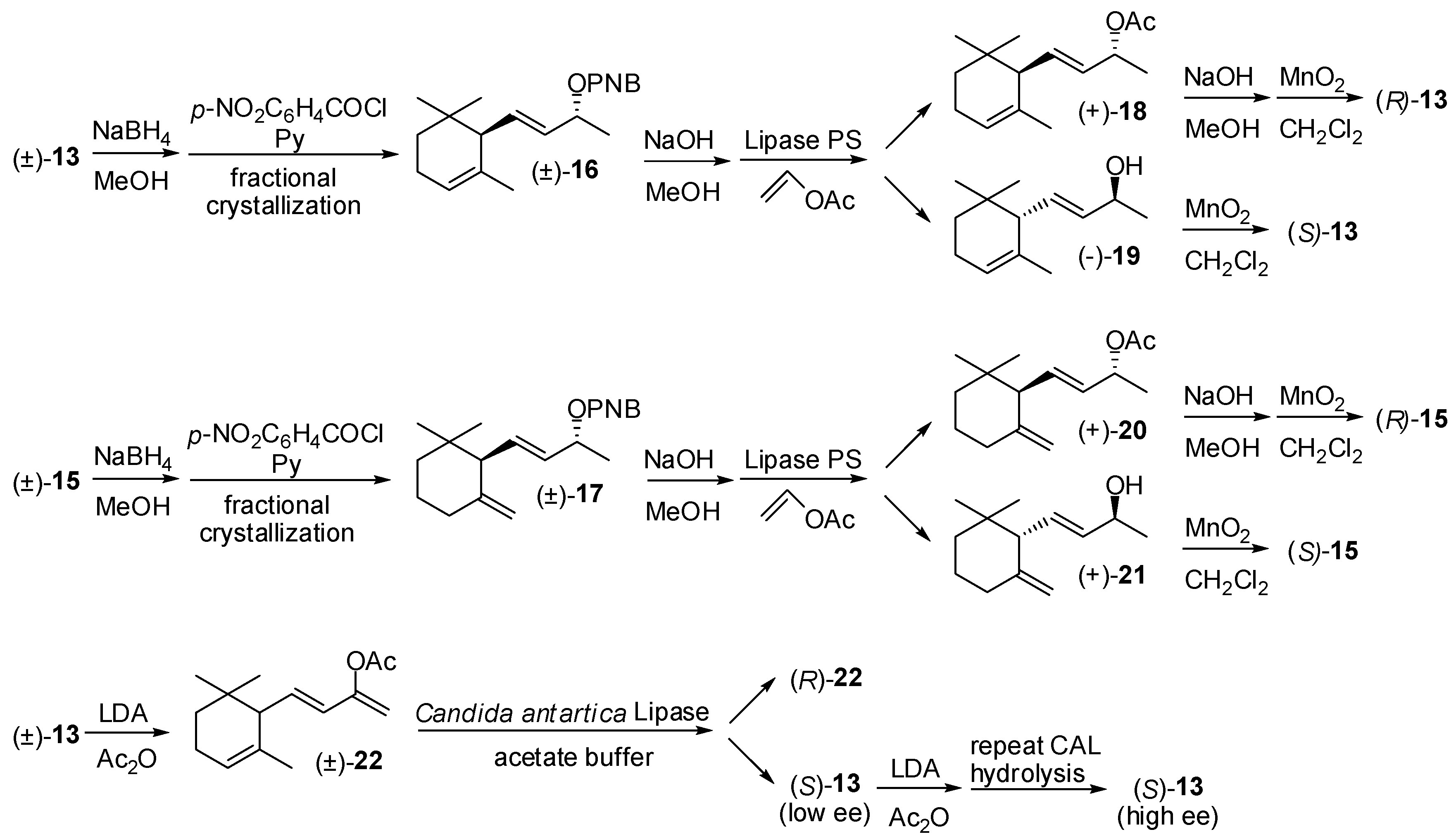
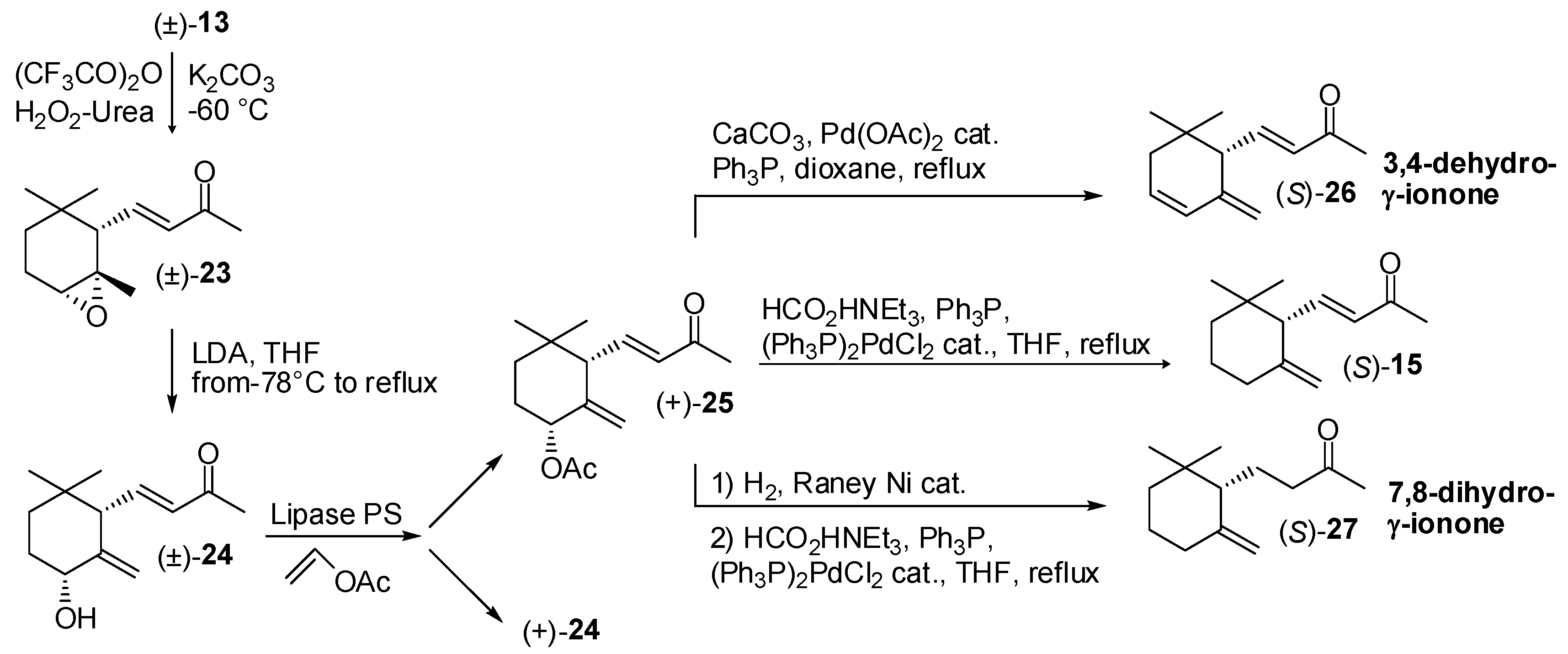
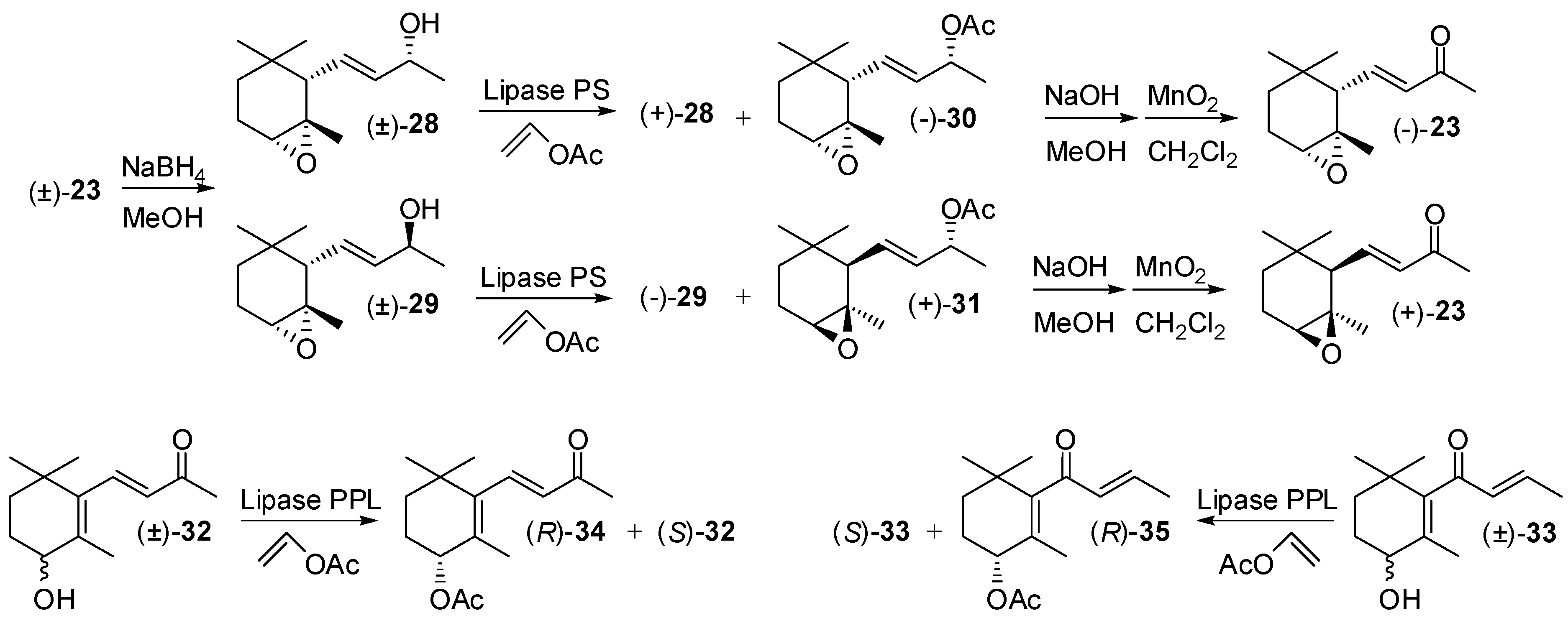
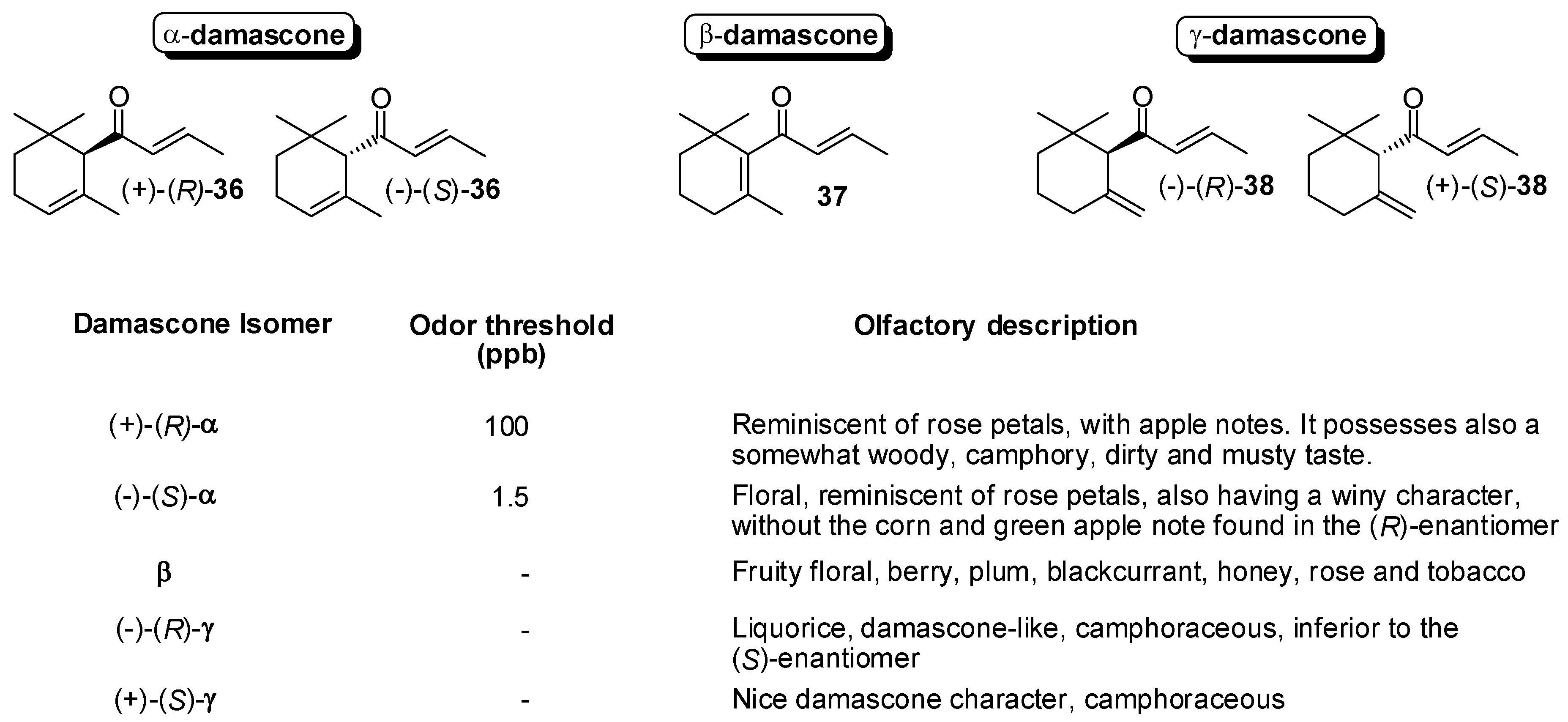
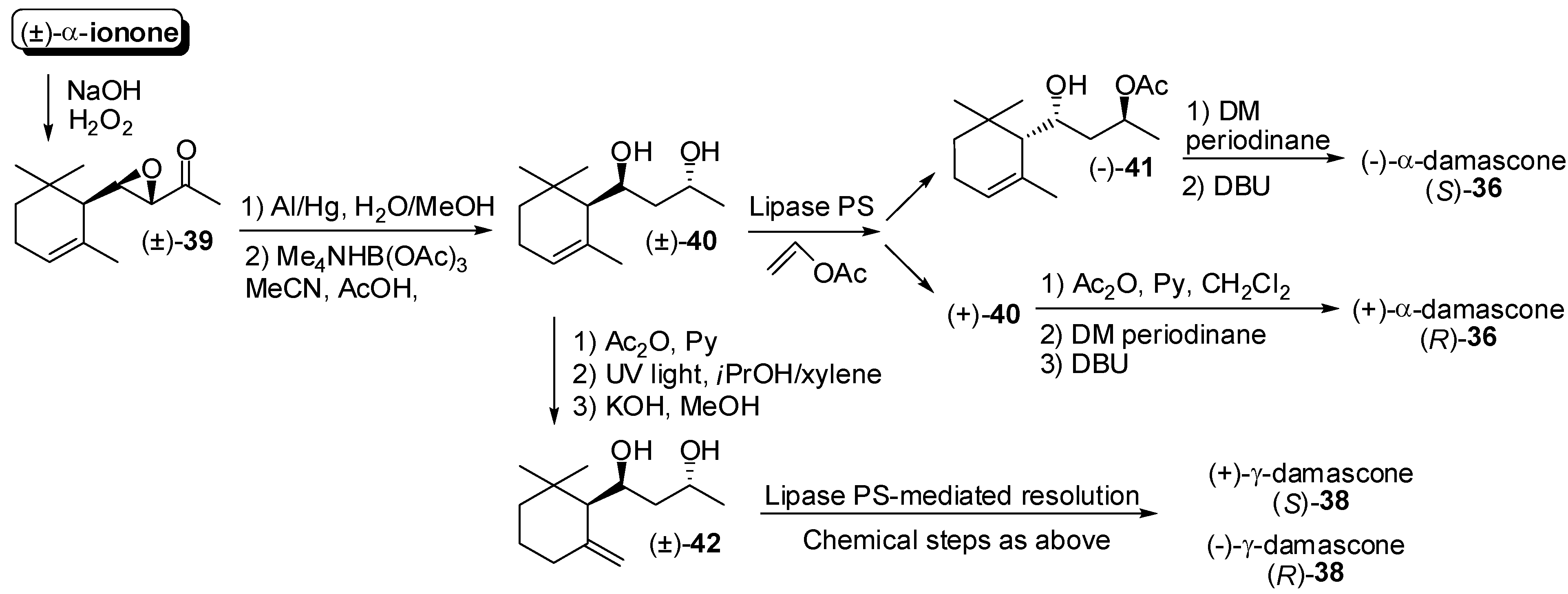
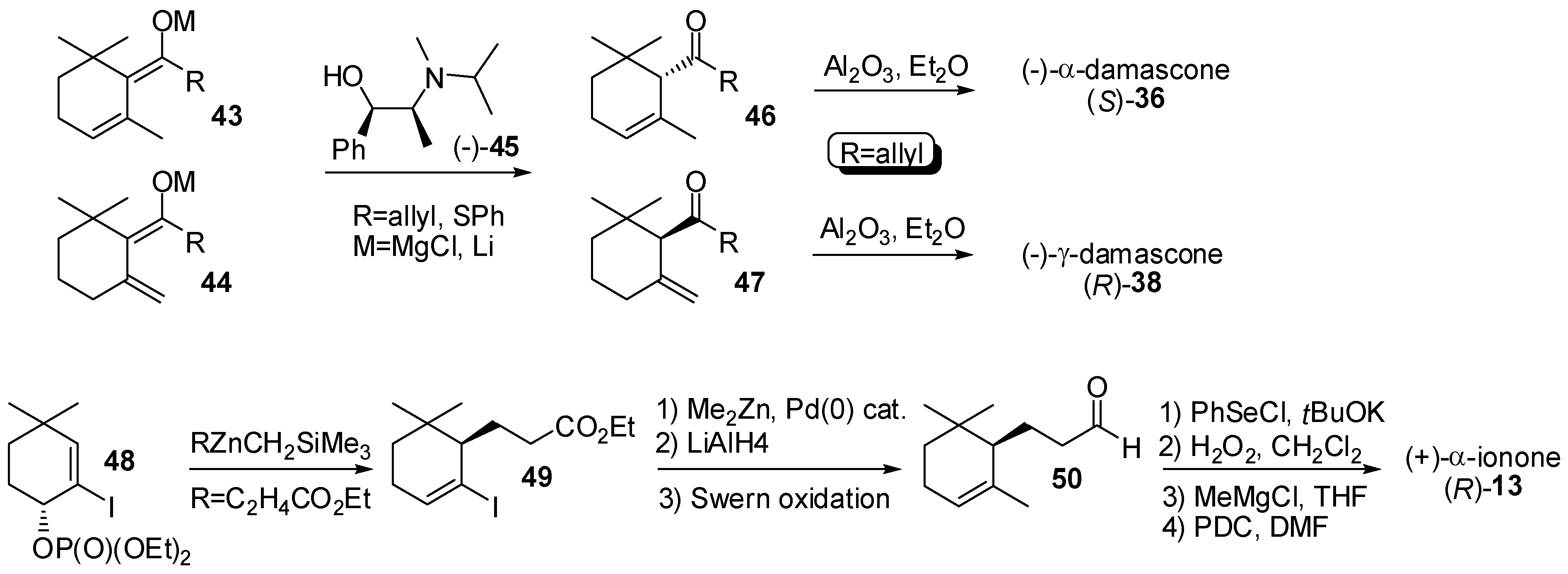
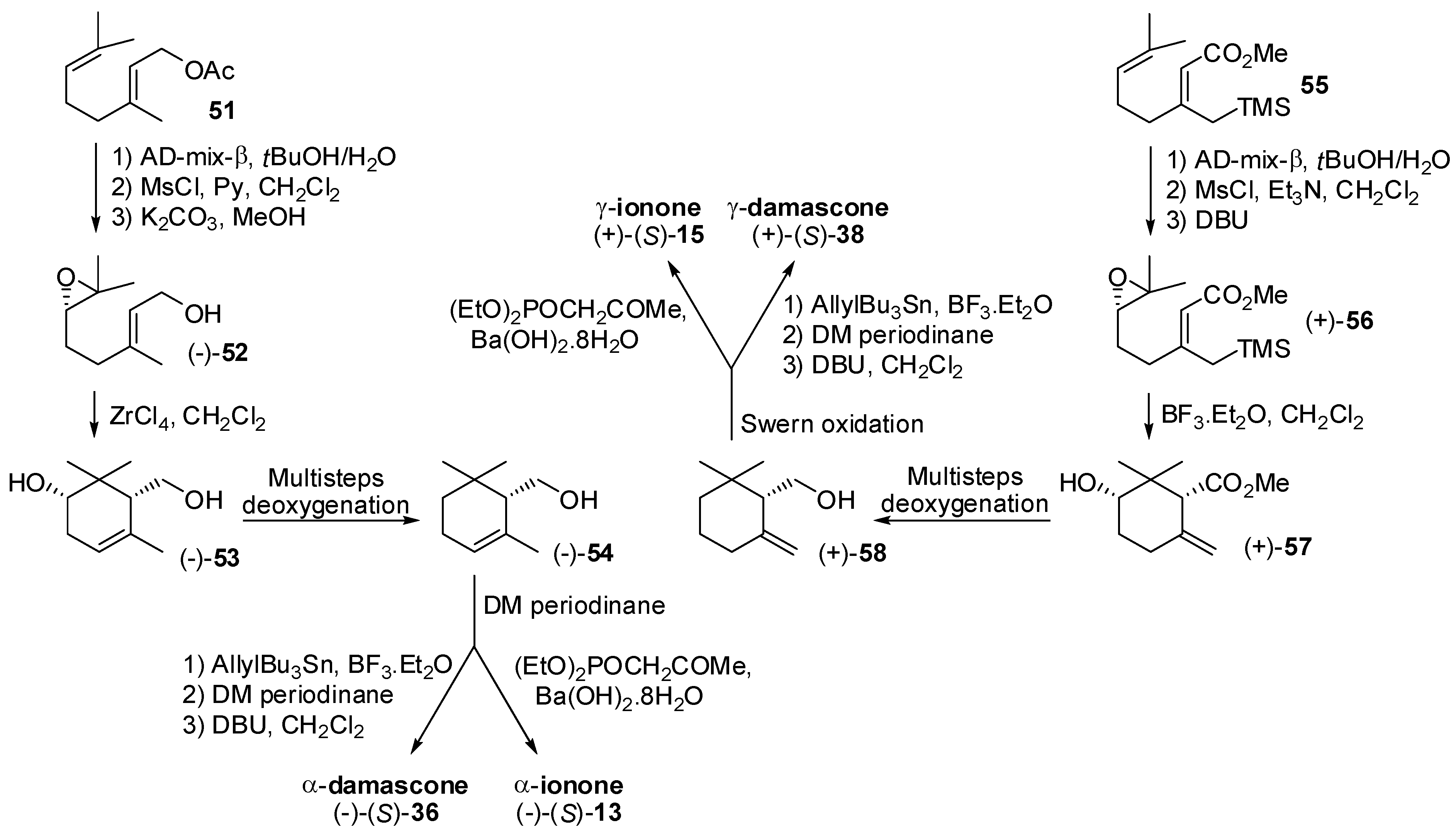
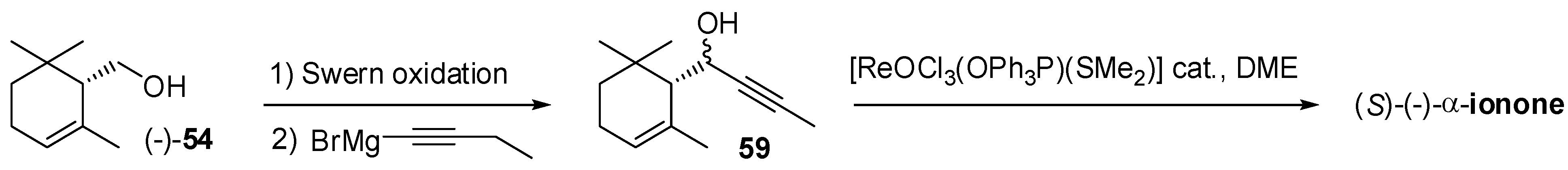
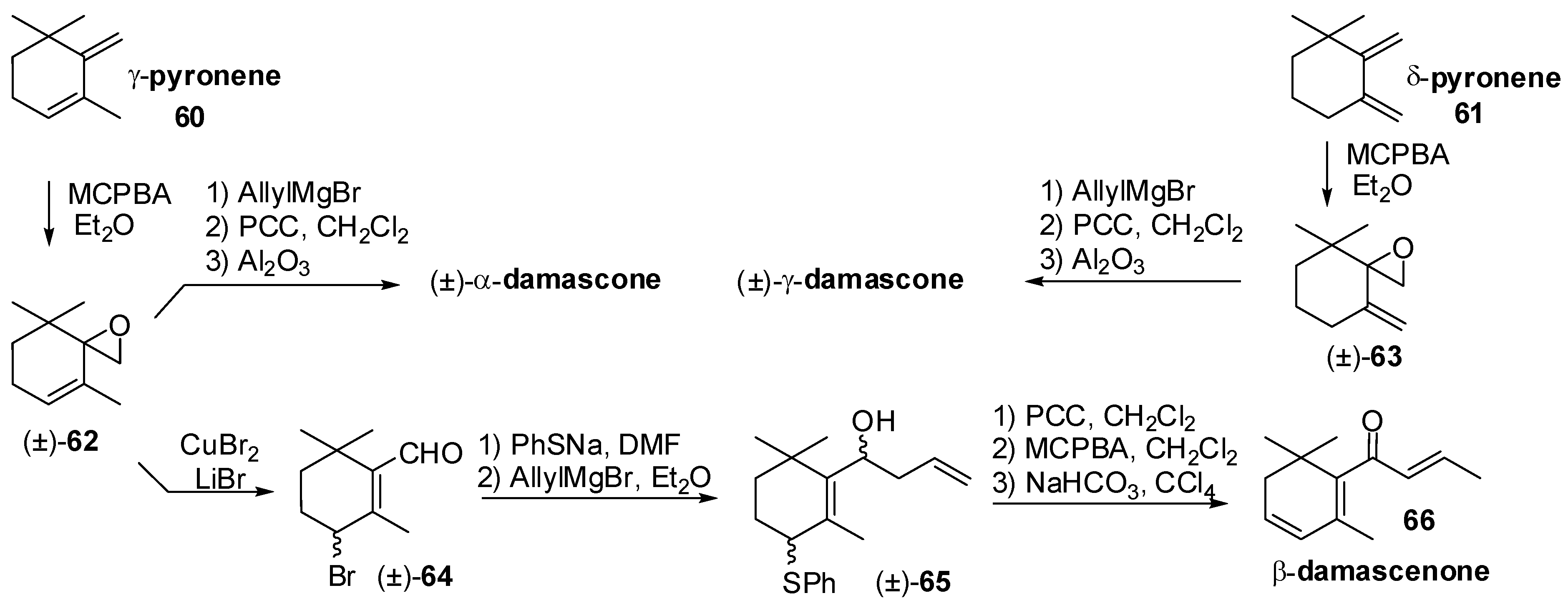
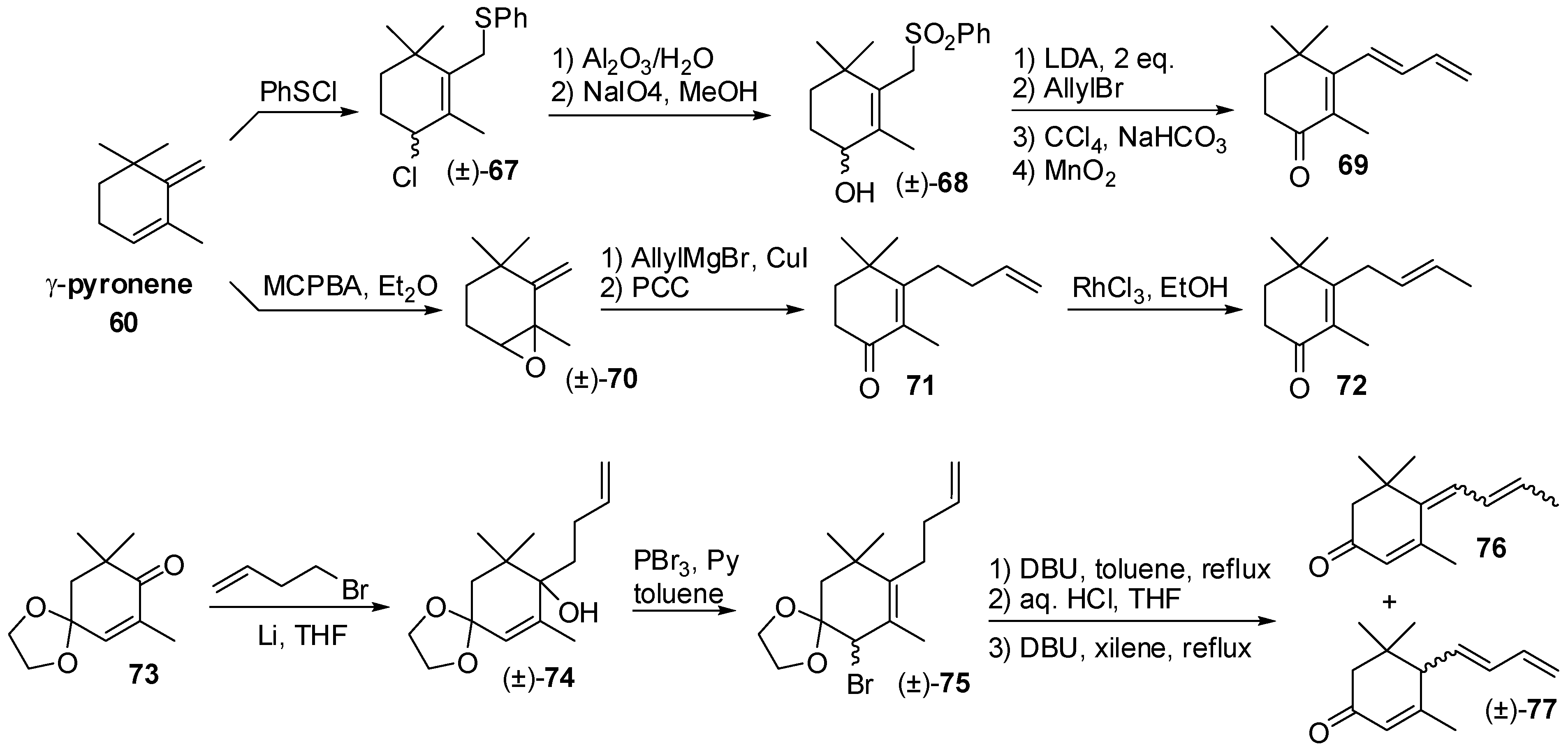
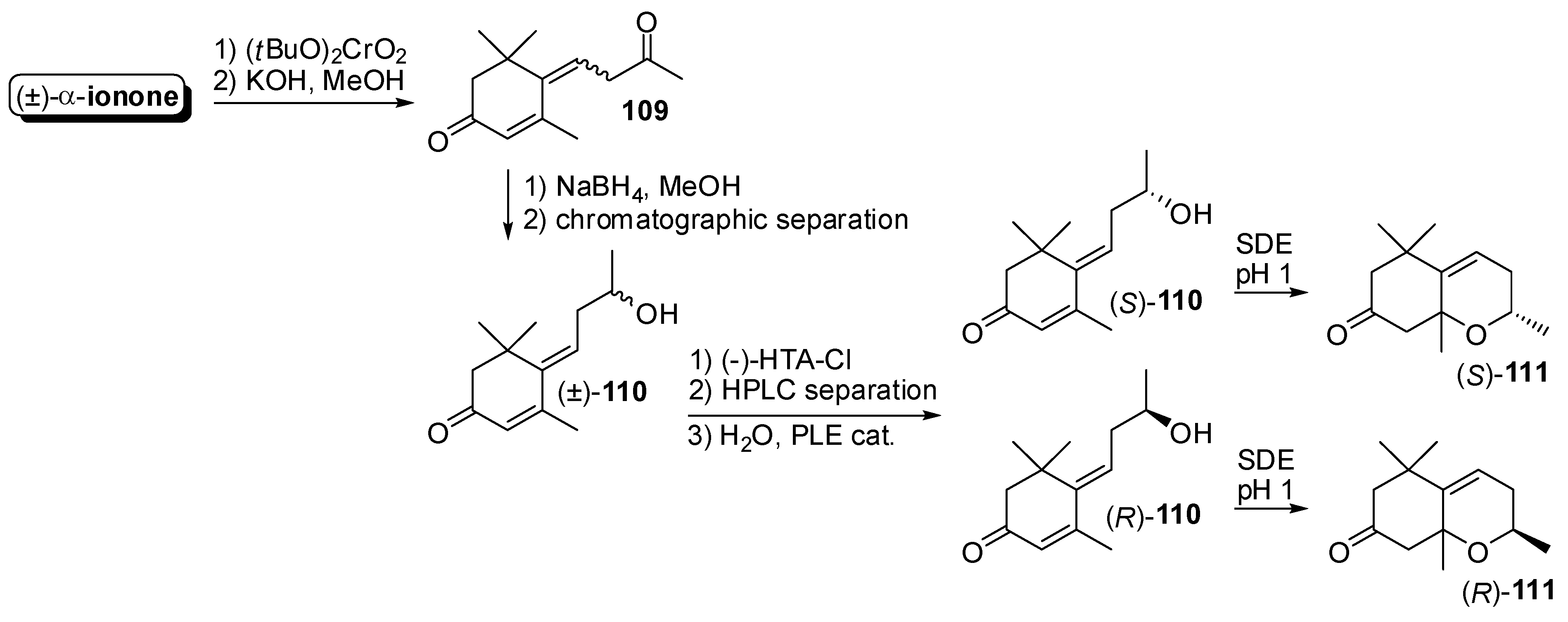
3.1. Synthesis of Ionone and Damascone Isomers










3.2. Synthesis of Other Relevant C13 Apocarotenoids





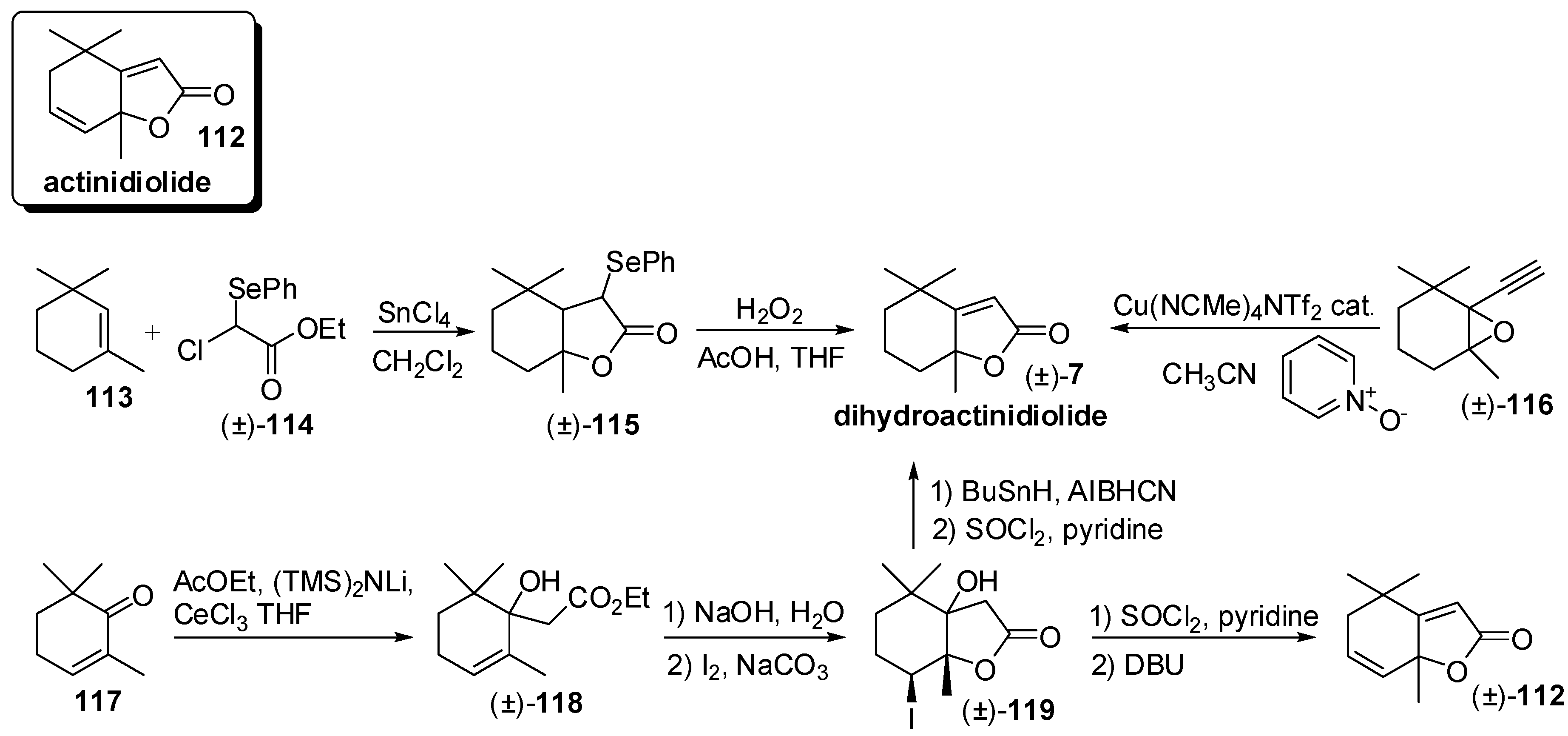
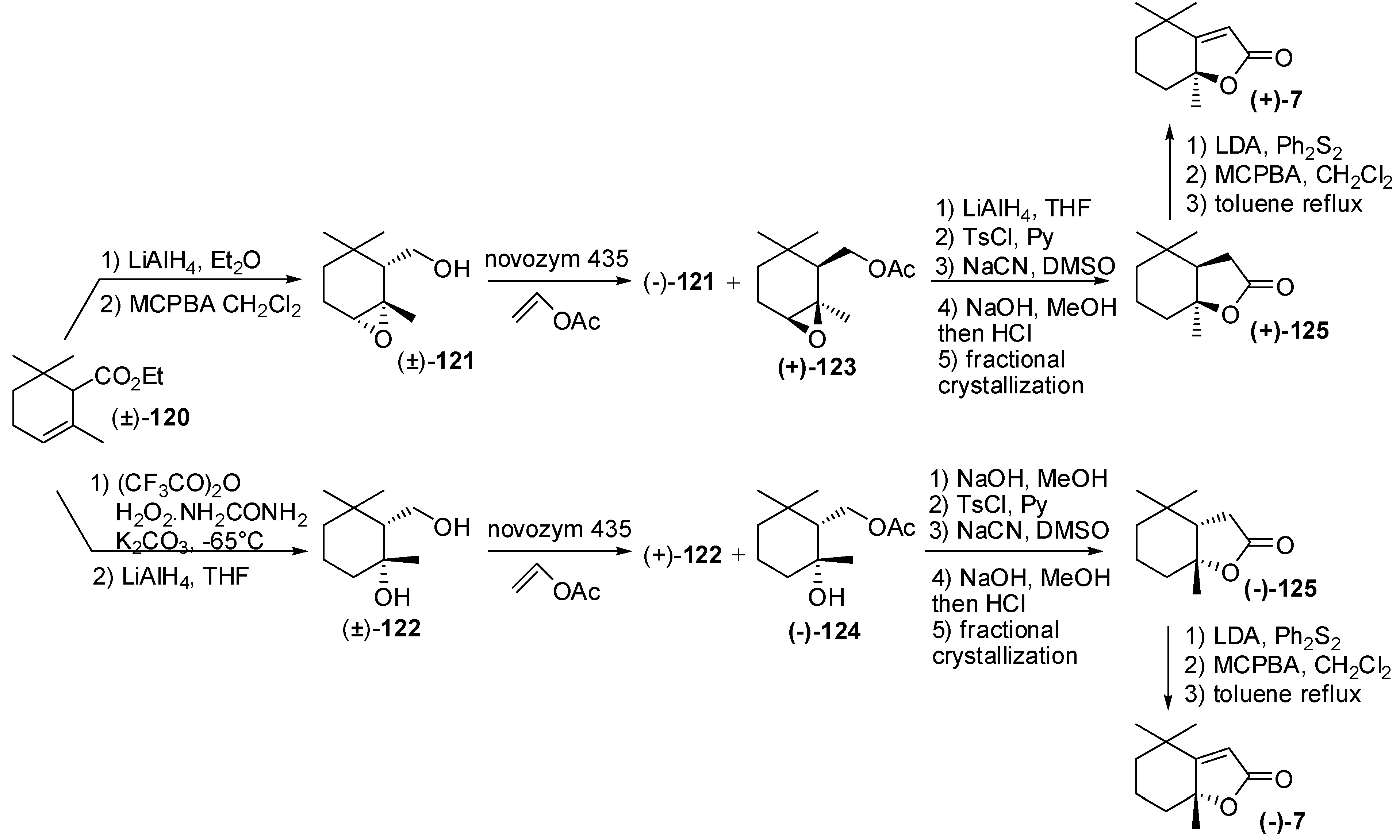
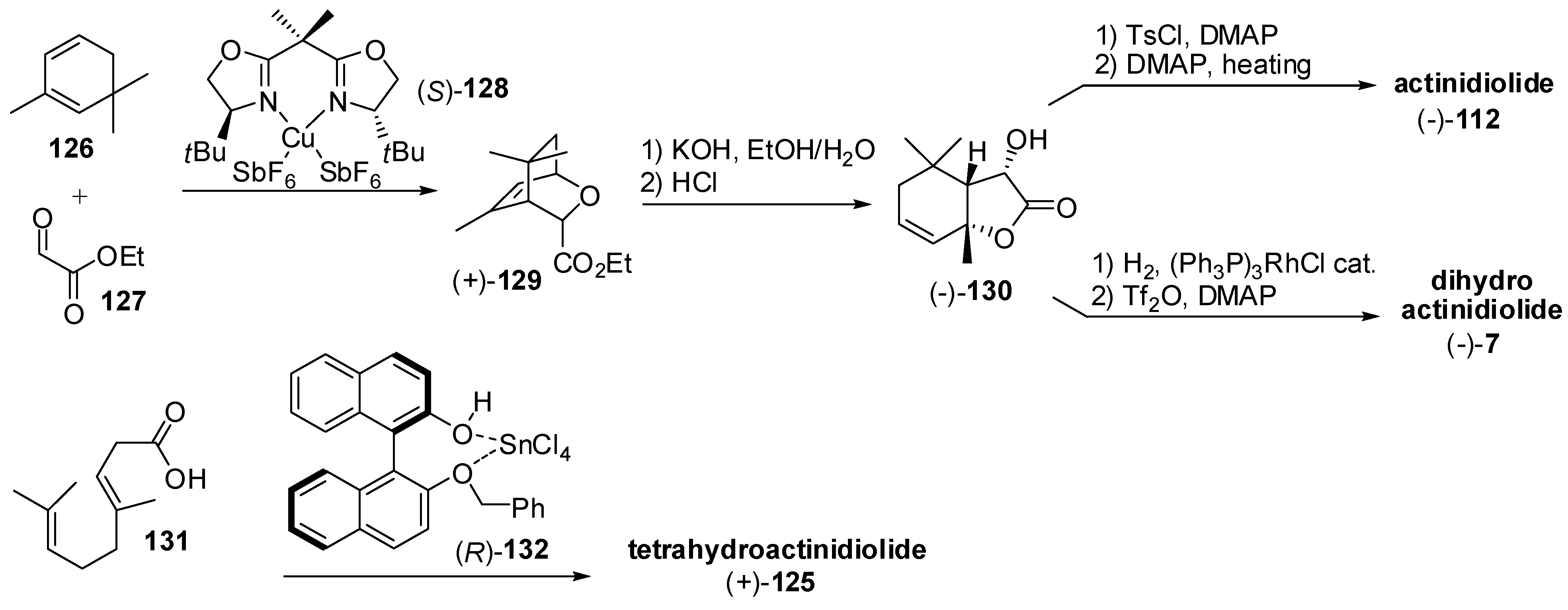
3.3. Synthesis of Relevant Apocarotenoids Having a C11 Chemical Framework



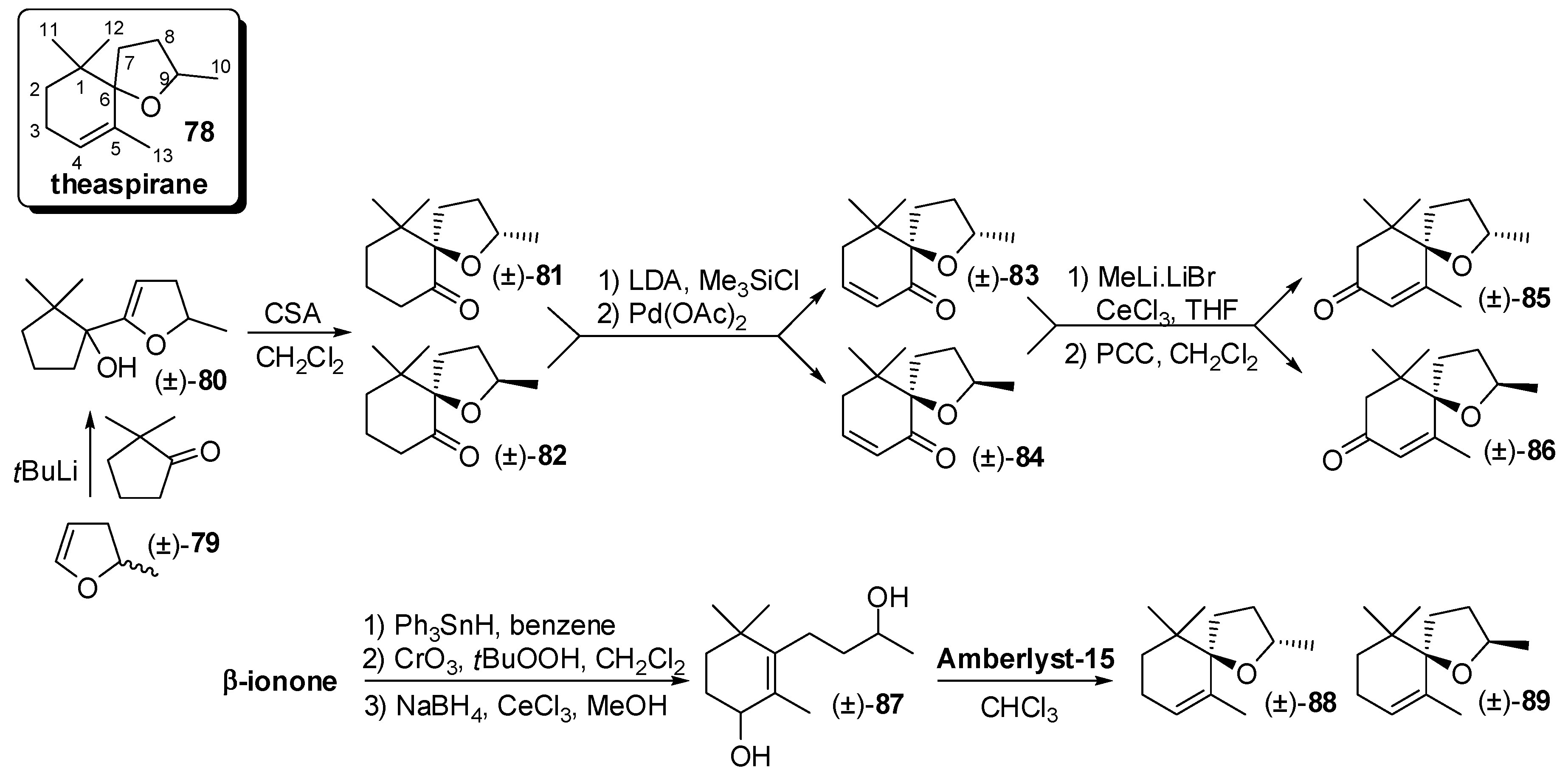
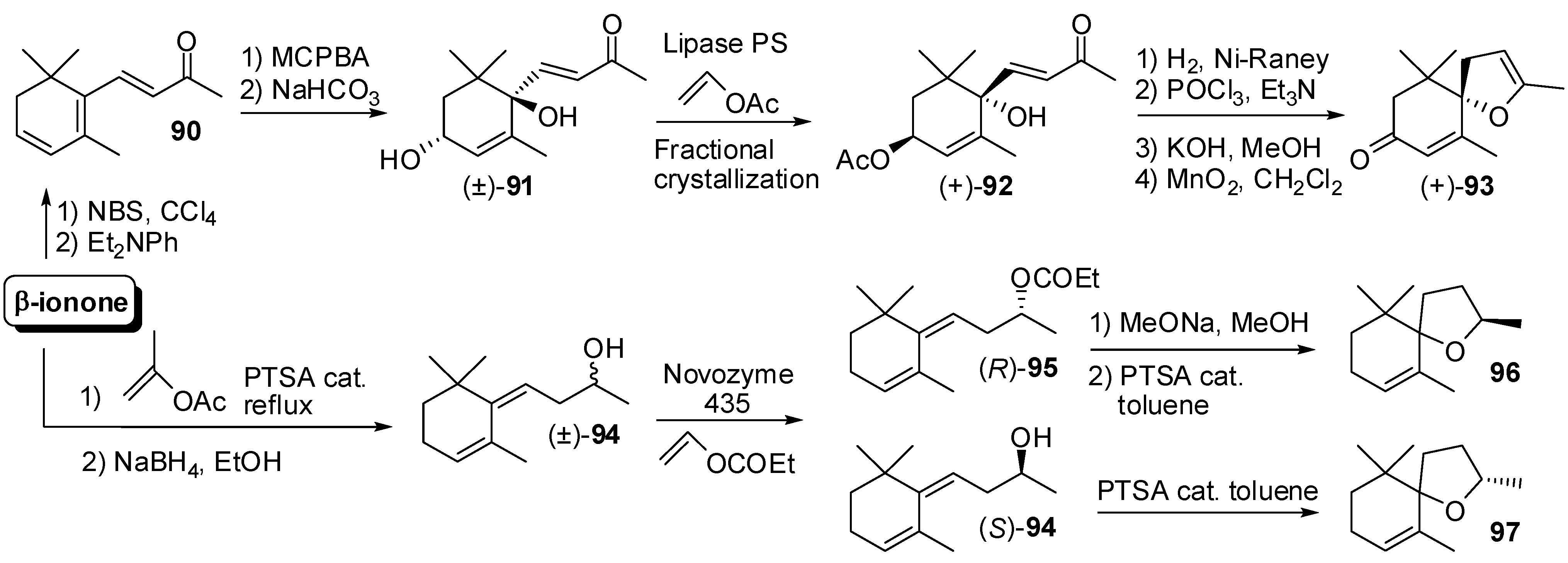
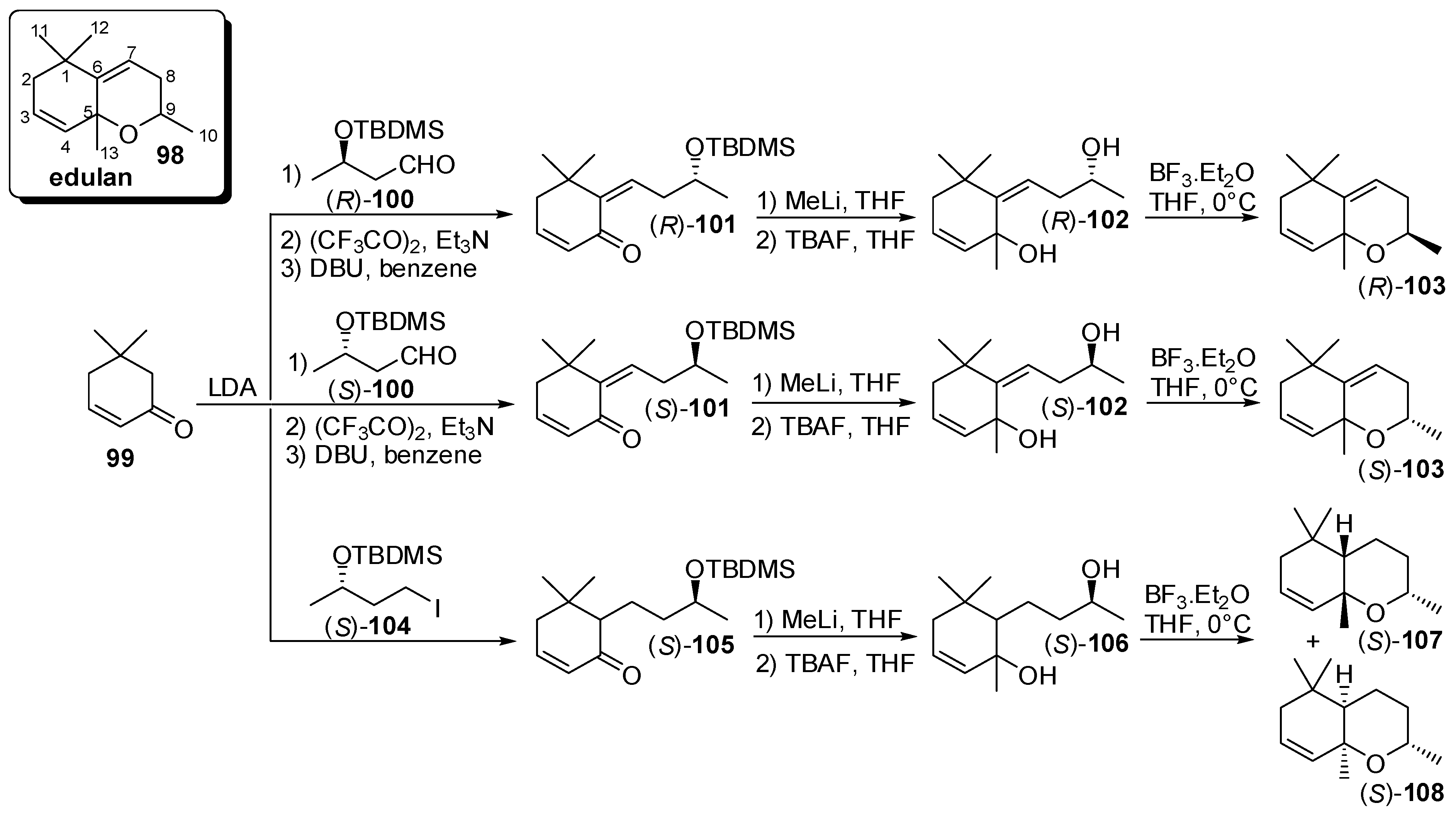
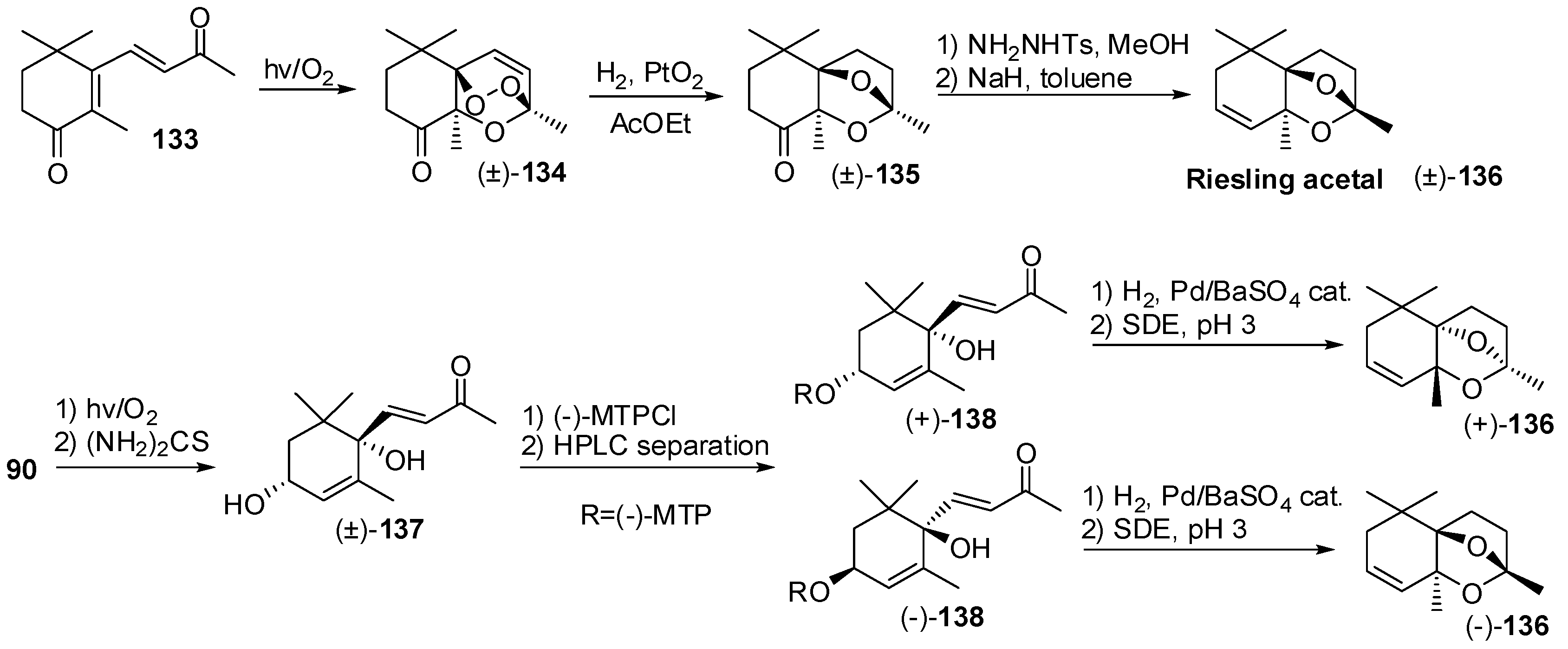
3.4. Synthesis of Odour Active Apocarotenoids Having Less Common Chemical Frameworks



4. Conclusions
Conflicts of Interest
References
- Walter, M.H.; Strack, D. Carotenoids and their cleavage products: Biosynthesis and functions. Nat. Prod. Rep. 2011, 28, 663–692. [Google Scholar] [CrossRef] [PubMed]
- Moise, A.R.; Al-Babili, S.; Wurtzel, E.T. Mechanistic aspects of carotenoid biosynthesis. Chem. Rev. 2014, 114, 164–193. [Google Scholar] [CrossRef] [PubMed]
- Khoo, H.E.; Prasad, K.N.; Kong, K.W.; Jiang, Y.; Ismail, A. Carotenoids and their isomers: Color pigments in fruits and vegetables. Molecules 2011, 16, 1710–1738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walter, M.; Floss, D.; Strack, D. Apocarotenoids: Hormones, mycorrhizal metabolites and aroma volatiles. Planta 2010, 232, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Cazzonelli, C.I. Carotenoids in nature: Insights from plants and beyond. Funct. Plant Biol. 2011, 38, 833–847. [Google Scholar] [CrossRef]
- Sui, X.; Kiser, P.D.; Lintig, J.V.; Palczewski, K. Structural basis of carotenoid cleavage: From bacteria to mammals. Arch. Biochem. Biophys. 2013, 539, 203–213. [Google Scholar] [CrossRef] [PubMed]
- Harrison, P.J.; Bugg, T.D.H. Enzymology of the carotenoid cleavage dioxygenases: Reaction mechanisms, inhibition and biochemical roles. Arch. Biochem. Biophys. 2014, 544, 105–111. [Google Scholar] [CrossRef] [PubMed]
- Burton, G.W.; Daroszewski, J.; Nickerson, J.G.; Johnston, J.B.; Mogg, T.J.; Nikiforov, G.B. β-Carotene autoxidation: Oxygen copolymerization, non-vitamin A products, and immunological activity. Can. J. Chem. 2014, 92, 305–316. [Google Scholar] [CrossRef]
- Carail, M.; Caris-Veyrat, C. Carotenoid oxidation products: From villain to saviour? Pure Appl. Chem. 2006, 78, 1493–1503. [Google Scholar] [CrossRef]
- Mendes-Pinto, M.M. Carotenoid breakdown products the—norisoprenoids—in wine aroma. Arch. Biochem. Biophys. 2009, 483, 236–245. [Google Scholar] [CrossRef] [PubMed]
- Surburg, H.; Panten, J. Common fragrance and flavor materials: Preparation, Properties and Uses, 5th ed.; WILEY-VCH: Weinheim, Germany, 2006. [Google Scholar]
- D’Auria, M.; Mauriello, G.; Rana, G.L. Volatile organic compounds from saffron. Flavour Frag. J. 2004, 19, 17–23. [Google Scholar] [CrossRef]
- Silva, I.; Coimbra, M.A.; Barros, A.S.; Marriott, P.J.; Rocha, S.M. Can volatile organic compounds be markers of sea salt? Food Chem. 2015, 169, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Fuganti, C.; Brenna, E. Biocatalytic preparation of natural flavours and fragrances. Trends Biotechnol. 2005, 23, 193–198. [Google Scholar] [CrossRef] [PubMed]
- US Code of Federal Regulations; Food and Drug Administration: Washington, WA, USA, 1985.
- The European Commission. Regulation (EU) No 872/2012. 1 October 2012. [Google Scholar]
- Schwartz, S.H.; Qin, X.Q.; Zeevaart, J.A.D. Characterization of a novel carotenoid cleavage dioxygenase from plants. J. Biol. Chem. 2001, 276, 25208–25211. [Google Scholar] [CrossRef] [PubMed]
- Simkin, A.J.; Underwood, B.A.; Auldridge, M.; Loucas, H.M.; Shibuya, K.; Schmelz, E.; Clark, D.G.; Klee, H.J. Circadian regulation of the PhCCD1 carotenoid cleavage dioxygenase controls emission of β-ionone, a fragrance volatile of petunia flowers. Plant Physiol. 2004, 136, 3504–3514. [Google Scholar] [CrossRef] [PubMed]
- Ibdah, M.; Azulay, Y.; Portnoy, V.; Wasserman, B.; Bar, E.; Meir, A.; Burger, Y.; Hirschberg, J.; Schaffer, A.A.; Katzir, N.; et al. Functional characterization of CmCCD1, a carotenoid cleavage dioxygenase from melon. Phytochemistry 2006, 67, 1579–1589. [Google Scholar] [CrossRef] [PubMed]
- Vogel, J.T.; Tan, B.C.; Mc Carty, D.R.; Klee, H.J. The carotenoid cleavage dioxygenase 1 enzyme has broad substrate specificity, cleaving multiple carotenoids at two different bond positions. J. Biol. Chem. 2008, 283, 11364–11373. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.C.; Horváth, G.; Molnár, P.; Turcsi, E.; Deli, J.; Schrader, J.; Sandmann, G.; Schmidt, H.; Schwab, W. Substrate promiscuity of RdCCD1, a carotenoid cleavage oxygenase from Rosa damascena. Phytochemistry 2009, 70, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Baldermann, S.; Kato, M.; Kurosawa, M.; Kurobayashi, Y.; Fujita, A.; Fleischmann, P.; Watanabe, N. Functional characterization of a carotenoid cleavage dioxygenase 1 and its relation to the carotenoid accumulation and volatile emission during the floral development of Osmanthus fragrans Lour. J. Exp. Bot. 2010, 61, 2967–2977. [Google Scholar] [CrossRef] [PubMed]
- Yahyaa, M.; Bar, E.; Dubey, N.K.; Meir, A.; Davidovich-Rikanati, R.; Hirschberg, J.; Aly, R.; Tholl, D.; Simon, P.W.; Tadmor, Y.; et al. Formation of norisoprenoid flavor compounds in carrot (Daucus carota L.) roots: Characterization of a cyclic-specific carotenoid cleavage dioxygenase 1 gene. J. Agric. Food Chem. 2013, 61, 12244–12252. [Google Scholar] [CrossRef] [PubMed]
- Lashbrooke, J.; Young, P.; Dockrall, S.; Vasanth, K.; Vivier, M. Functional characterisation of three members of the Vitis vinifera L. Carotenoid cleavage dioxygenase gene family. BMC Plant Biol. 2013, 13. [Google Scholar] [CrossRef] [PubMed]
- Ilg, A.; Bruno, M.; Beyer, P.; Al-Babili, S. Tomato carotenoid cleavage dioxygenases 1A and 1B: Relaxed double bond specificity leads to a plenitude of dialdehydes, mono-apocarotenoids and isoprenoid volatiles. FEBS Open Biol. 2014, 4, 584–593. [Google Scholar] [CrossRef] [PubMed]
- Frusciante, S.; Diretto, G.; Bruno, M.; Ferrante, P.; Pietrella, M.; Prado-Cabrero, A.; Rubio-Moraga, A.; Beyer, P.; Gomez-Gomez, L.; Al-Babili, S.; et al. Novel carotenoid cleavage dioxygenase catalyzes the first dedicated step in saffron crocin biosynthesis. Proc. Natl. Acad. Sci. USA 2014, 111, 12246–12251. [Google Scholar] [CrossRef] [PubMed]
- Beekwilder, J.; van Rossum, H.M.; Koopman, F.; Sonntag, F.; Buchhaupt, M.; Schrader, J.; Hall, R.D.; Bosch, D.; Pronk, J.T.; van Maris, A.J.A.; et al. Polycistronic expression of a β-carotene biosynthetic pathway in Saccharomyces cerevisiae coupled to β-ionone production. J. Biotechnol. 2014, 192, 383–392. [Google Scholar] [CrossRef] [PubMed]
- Zorn, H.; Langhoff, S.; Scheibner, M.; Berger, R.G. Cleavage of β,β-carotene to flavor compounds by fungi. Appl. Microbiol. Biotechnol. 2003, 62, 331–336. [Google Scholar] [CrossRef] [PubMed]
- Zelena, K.; Hardebusch, B.; Hülsdau, B.; Berger, R.G.; Zorn, H. Generation of norisoprenoid flavors from carotenoids by fungal peroxidases. J. Agric. Food Chem. 2009, 57, 9951–9955. [Google Scholar] [CrossRef] [PubMed]
- Nacke, C.; Hüttmann, S.; Etschmann, M.W.; Schrader, J. Enzymatic production and in situ separation of natural β-ionone from β-carotene. J. Ind. Microbiol. Biotechnol. 2012, 39, 1771–1778. [Google Scholar] [CrossRef] [PubMed]
- Brenna, E.; Fuganti, C.; Serra, S. Enantioselective perception of chiral odorants. Tetrahedron Asymmetry 2003, 14, 1–42. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Serra, S.; Kraft, P. Optically active ionones and derivatives: Preparation and olfactory properties. Eur. J. Org. Chem. 2002, 967–978. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C.; Brenna, E. Two easy photochemical methods for the conversion of commercial ionone alpha into regioisomerically enriched γ-ionone and γ-dihydroionone. Flavour Frag. J. 2007, 22, 505–511. [Google Scholar] [CrossRef]
- Sobotka, H.; Bloch, E.; Cahnmann, H.; Feldbau, E.; Rosen, E. Studies on ionone. II. Optical resolution of dl-α-ionone. J. Am. Chem. Soc. 1943, 65, 2061–2062. [Google Scholar] [CrossRef]
- Brenna, E.; Fuganti, C.; Gatti, F.G.; Serra, S. Biocatalytic methods for the synthesis of enantioenriched odor active compounds. Chem. Rev. 2011, 111, 4036–4072. [Google Scholar] [CrossRef] [PubMed]
- Brenna, E.; Fuganti, C.; Grasselli, P.; Redaelli, M.; Serra, S. Enzyme-mediated synthesis of (R)- and (S)-α-ionone. J. Chem. Soc. Perkin Trans. 1 1998, 4129–4134. [Google Scholar] [CrossRef]
- Fuganti, C.; Serra, S.; Zenoni, A. Synthesis and olfactory evaluation of (+)- and (−)-γ-ionone. Helv. Chim. Acta 2000, 83, 2761–2768. [Google Scholar] [CrossRef]
- Yamamoto, T.; Yagi, K.; Ishida, K. Method for Producing Optically Active Alpha-Ionone. US Patent 2009/0216039 A1, 27 August 2009. [Google Scholar]
- Serra, S.; Fuganti, C.; Brenna, E. Synthesis, olfactory evaluation, and determination of the absolute configuration of the 3,4-didehydroionone stereoisomers. Helv. Chim. Acta 2006, 89, 1110–1122. [Google Scholar] [CrossRef]
- Serra, S. An expedient preparation of enantio-enriched ambergris odorants starting from commercial ionone alpha. Flavour Frag. J. 2013, 28, 46–52. [Google Scholar] [CrossRef]
- Serra, S.; Lissoni, V. First enantioselective synthesis of marine diterpene ambliol-A. Eur. J. Org. Chem. 2015, 2226–2234. [Google Scholar] [CrossRef]
- Serra, S.; Cominetti, A.A.; Lissoni, V. A general synthetic approach to hydroquinone meroterpenoids: Stereoselective synthesis of (+)-(S)-metachromin V and alliodorol. Nat. Prod. Commun. 2014, 9, 303–308. [Google Scholar] [PubMed]
- Serra, S.; Cominetti, A.A.; Lissoni, V. Use of (S)-trans-γ-monocyclofarnesol as a useful chiral building block for the stereoselective synthesis of diterpenic natural products. Nat. Prod. Commun. 2014, 9, 329–335. [Google Scholar] [PubMed]
- Serra, S. A practical, enantiospecific synthesis of (S)-trans-γ-monocyclofarnesol. Nat. Prod. Commun. 2012, 7, 1569–1572. [Google Scholar] [PubMed]
- Aleu, J.; Brenna, E.; Fuganti, C.; Serra, S. Lipase-mediated synthesis of the enantiomeric forms of 4,5-epoxy-4,5-dihydro-α-ionone and 5,6-epoxy-5,6-dihydro-β-ionone. A new direct access to enantiopure (R)- and (S)-α-ionone. J. Chem. Soc. Perkin Trans. 1 1999, 271–278. [Google Scholar] [CrossRef]
- More, G.P.; Bhat, S.V. Facile lipase catalysed syntheses of (S)-(+)-4-hydroxy-β-ionone and (S)-(+)-4-hydroxy-β-damascone: Chiral flavorants and synthons. Tetrahedron Lett. 2013, 54, 4148–4149. [Google Scholar] [CrossRef]
- Serra, S.; Fuganti, C. Synthesis of the enantiomeric forms of α- and γ-damascone starting from commercial racemic α-ionone. Tetrahedron Asymmetry 2006, 17, 1573–1580. [Google Scholar] [CrossRef]
- Fehr, C.; Galindo, J. Synthesis of (R)-(+)- and (S)-(−)-α-damascone by tandem grignard reaction-enantioselective protonation: Evidence for the intermediacy of a chiral complex. J. Am. Chem. Soc. 1988, 110, 6909–6911. [Google Scholar] [CrossRef]
- Fehr, C.; Galindo, J. Catalytic enantioselective protonation of enolates. Angew. Chem. Int. Edit. 1994, 33, 1888–1889. [Google Scholar] [CrossRef]
- Fehr, C.; Galindo, J. Syntheses of the enantiomers of γ-cyclogeranic acid, γ-cyclocitral, and γ-damascone: Enantioselective protonation of enolates. Helv. Chim. Acta 1995, 78, 539–552. [Google Scholar] [CrossRef]
- Fehr, C.; Guntern, O. Efficient synthesis of enantiomerically pure α-ionone from (R)- and (S)-α-damascone. Helv. Chim. Acta 1992, 75, 1023–1028. [Google Scholar] [CrossRef]
- Soorukram, D.; Knochel, P. Enantioselective synthesis of α-ionone derivatives using an anti SN2′ substitution of functionalized zinc organometallics. Org. Lett. 2004, 6, 2409–2411. [Google Scholar] [CrossRef] [PubMed]
- Bovolenta, M.; Castronovo, F.; Vadalà, A.; Zanoni, G.; Vidari, G. A simple and efficient highly enantioselective synthesis of α-ionone and α-damascone. J. Org. Chem. 2004, 69, 8959–8962. [Google Scholar] [CrossRef] [PubMed]
- Beszant, S.; Giannini, E.; Zanoni, G.; Vidari, G. Enantioselective synthesis of both enantiomers of γ-ionone, γ-damascone, karahana lactone and karahana ether. Tetrahedron Asymmetry 2002, 13, 1245–1255. [Google Scholar] [CrossRef]
- Stefanoni, M.; Luparia, M.; Porta, A.; Zanoni, G.; Vidari, G. A simple and versatile Re-catalyzed Meyer—Schuster rearrangement of propargylic alcohols to α,β-unsaturated carbonyl compounds. Chem. Eur. J. 2009, 15, 3940–3944. [Google Scholar] [CrossRef] [PubMed]
- Boulin, B.; Taran, M.; Miguel, B.A.S.; Delmond, B. New preparation methods for α-damascone, γ-damascone, and β-damascenone using pyronenes. Synth. Commun. 2007, 37, 2579–2591. [Google Scholar] [CrossRef]
- Boulin, B.; Arreguy-San Miguel, B.; Delmond, B. Nouvelles voies d'accès aux theaspiranes, mégastigma-5,7,9-trién-4-one et mégastigma-5,8-dién-4-one à partir du γ-pyronène. Tetrahedron 2000, 56, 3927–3932. [Google Scholar] [CrossRef]
- Ito, N.; Etoh, T.; Hagiwara, H.; Kato, M. Novel synthesis of degradation products of carotenoids, megastigmatrienone analogues and blumenol-A. J. Chem. Soc. Perkin Trans. 1 1997, 1571–1580. [Google Scholar] [CrossRef]
- Paquette, L.A.; Lanter, J.C.; Wang, H.-L. Concise synthesis of cis- and trans-theaspirones via oxonium ion-initiated pinacol ring expansion. J. Org. Chem. 1996, 61, 1119–1121. [Google Scholar] [CrossRef]
- Young, J.J.; Jung, L.J.; Cheng, K.M. Amberlyst-15-catalyzed intramolecular SN2′ oxaspirocyclization of secondary allylic alcohols. Application to the total synthesis of spirocyclic ethers theaspirane and theaspirone. Tetrahedron Lett. 2000, 41, 3415–3418. [Google Scholar] [CrossRef]
- Serra, S.; Barakat, A.; Fuganti, C. Chemoenzymatic resolution of cis- and trans-3,6-dihydroxy-α-ionone. Synthesis of the enantiomeric forms of dehydrovomifoliol and 8,9-dehydrotheaspirone. Tetrahedron Asymmetry 2007, 18, 2573–2580. [Google Scholar] [CrossRef]
- Harada, M.; Matsuda, H.; Ishida, K.; Yamazaki, Y. Process for Producing Optically Active Theaspirane. US Patent 2009/0270516 A1, 29 October 2009. [Google Scholar]
- Honda, T.; Yamauchi, A.; Tsubuki, M.; Matsumoto, T. Chiral syntheses of edulans I, II and dihydroedulans I, II and absolute configurations of edulans I, II. Tetrahedron: Asymmetry 1997, 8, 837–839. [Google Scholar] [CrossRef]
- Schmidt, G.; Neugebauer, W.; Winterhalter, P.; Schreier, P. Synthesis and enantiodifferentiation of isomeric 2,3,5,6,8,8a-hexahydro-2,5,5,8a-tetramethyl-7H-1-benzopyran-7-ones (3,4-dihydro-3-oxoedulans). J. Agr. Food Chem. 1995, 43, 1898–1902. [Google Scholar] [CrossRef]
- Dabdoub, M.J.; Silveira, C.C.; Lenardão, E.J.; Guerrero, P.G.; Viana, L.H.; Kawasoko, C.Y.; Baroni, A.C.M. Total synthesis of (±)-dihydroactinidiolide using selenium-stabilized carbenium ion. Tetrahedron Lett. 2009, 50, 5569–5571. [Google Scholar] [CrossRef]
- Gronnier, C.; Kramer, S.; Odabachian, Y.; Gagosz, F. Cu(I)-catalyzed oxidative cyclization of alkynyl oxiranes and oxetanes. J. Am. Chem. Soc. 2011, 134, 828–831. [Google Scholar] [CrossRef] [PubMed]
- Eidman, K.F.; MacDougall, B.S. Synthesis of loliolide, actinidiolide, dihydroactinidiolide, and aeginetolide via cerium enolate chemistry. J. Org. Chem. 2006, 71, 9513–9516. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Piccioni, O. A new chemo-enzymatic approach to the stereoselective synthesis of the flavors tetrahydroactinidiolide and dihydroactinidiolide. Tetrahedron Asymmetry 2015, 26, 584–592. [Google Scholar] [CrossRef]
- Yao, S.; Johannsen, M.; Hazell, R.G.; Jørgensen, K.A. Total synthesis of (R)-dihydroactinidiolide and (R)-actinidiolide using asymmetric catalytic hetero-Diels-Alder methodology. J. Org. Chem. 1998, 63, 118–121. [Google Scholar] [CrossRef] [PubMed]
- Upar, K.B.; Mishra, S.J.; Nalawade, S.P.; Singh, S.A.; Khandare, R.P.; Bhat, S.V. Efficient enantioselective synthesis of (+)-sclareolide and (+)-tetrahydroactinidiolide: Chiral LBA-induced biomimetic cyclization. Tetrahedron Asymmetry 2009, 20, 1637–1640. [Google Scholar] [CrossRef]
- Riveira, M.N.J.; La-Venia, A.; Mischne, M.P. New strategy for the construction of epoxy-bridged tetrahydropyran frameworks from trioxane precursors: Application to a concise synthesis of a Riesling acetal. J. Org. Chem. 2008, 73, 8678–8681. [Google Scholar] [CrossRef] [PubMed]
- Dollmann, B.; Full, G.; Schreier, P.; Winterhalter, P.; Güntert, M.; Sommer, H. Synthesis and enantiodifferentiation of riesling acetals. Phytochem. Anal. 1995, 6, 106–111. [Google Scholar] [CrossRef]
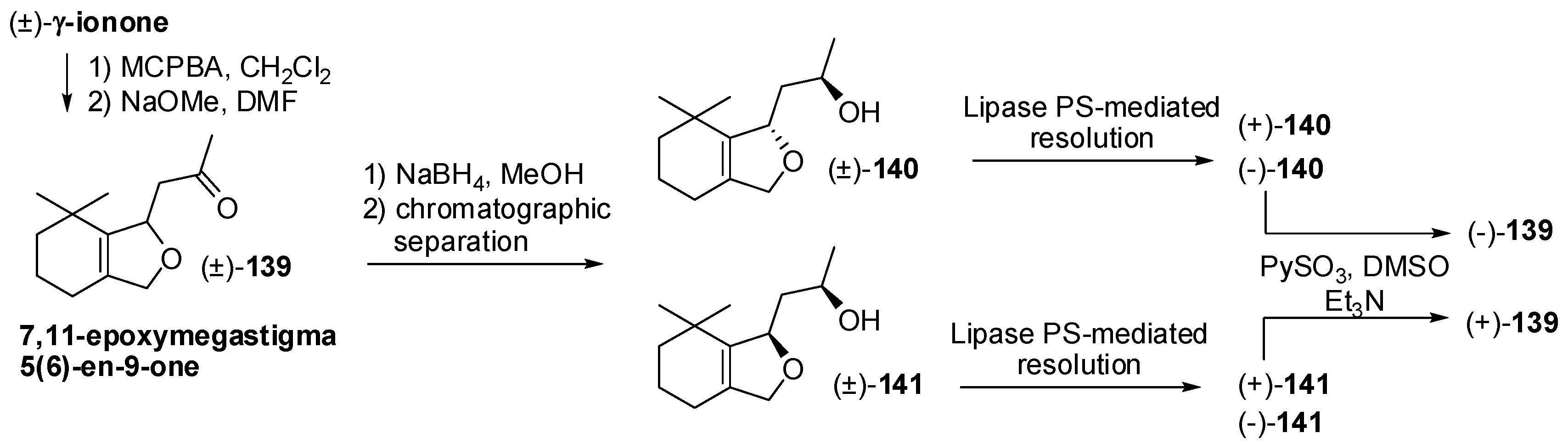
- Brenna, E.; Fuganti, C.; Serra, S. Synthesis and olfactory evaluation of the enantiomerically enriched forms of 7,11-epoxymegastigma-5(6)-en-9-one and 7,11-epoxymegastigma-5(6)-en-9-ols isomers, identified in Passiflora edulis. Tetrahedron Asymmetry 2005, 16, 1699–1704. [Google Scholar] [CrossRef]
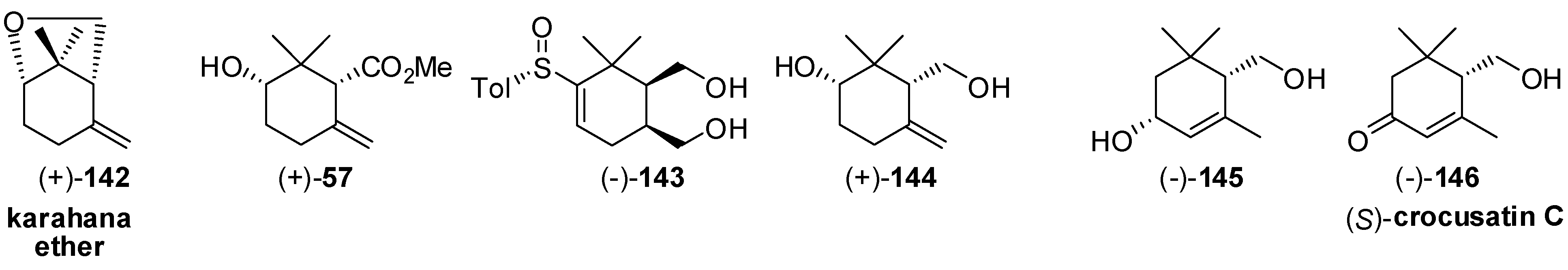
- Gosselin, P.; Bonfand, E.; Maignan, C. Application of the stereoselective Diels-Alder reaction of enantiopure 2-sulfinyl dienes: Synthesis of (−)-(1S,5R)-karahana ether. J. Org. Chem. 1996, 61, 9049–9052. [Google Scholar] [CrossRef] [PubMed]
- Serra, S.; Gatti, F.G.; Fuganti, C. Lipase-mediated resolution of the hydroxy-cyclogeraniol isomers: Application to the synthesis of the enantiomers of karahana lactone, karahana ether, crocusatin C and γ-cyclogeraniol. Tetrahedron Asymmetry 2009, 20, 1319–1329. [Google Scholar] [CrossRef]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Serra, S. Recent Advances in the Synthesis of Carotenoid-Derived Flavours and Fragrances. Molecules 2015, 20, 12817-12840. https://doi.org/10.3390/molecules200712817
Serra S. Recent Advances in the Synthesis of Carotenoid-Derived Flavours and Fragrances. Molecules. 2015; 20(7):12817-12840. https://doi.org/10.3390/molecules200712817
Chicago/Turabian StyleSerra, Stefano. 2015. "Recent Advances in the Synthesis of Carotenoid-Derived Flavours and Fragrances" Molecules 20, no. 7: 12817-12840. https://doi.org/10.3390/molecules200712817
APA StyleSerra, S. (2015). Recent Advances in the Synthesis of Carotenoid-Derived Flavours and Fragrances. Molecules, 20(7), 12817-12840. https://doi.org/10.3390/molecules200712817