
Glycoprotein Quality Control and Endoplasmic Reticulum Stress


{kind=link}
{kind=link}
Abstract
:1. Introduction
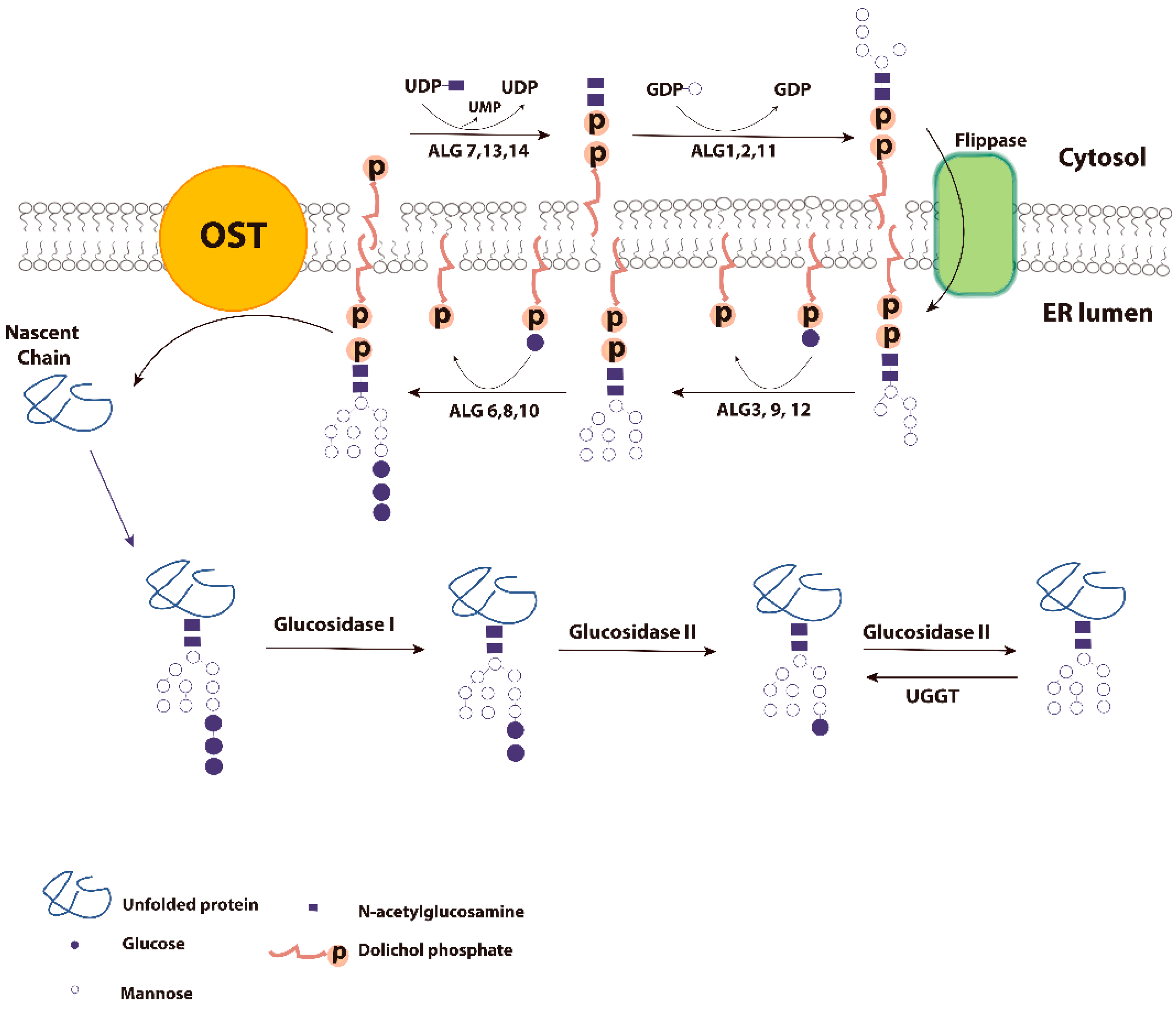
2. Glycoproteins and ER Quality Control


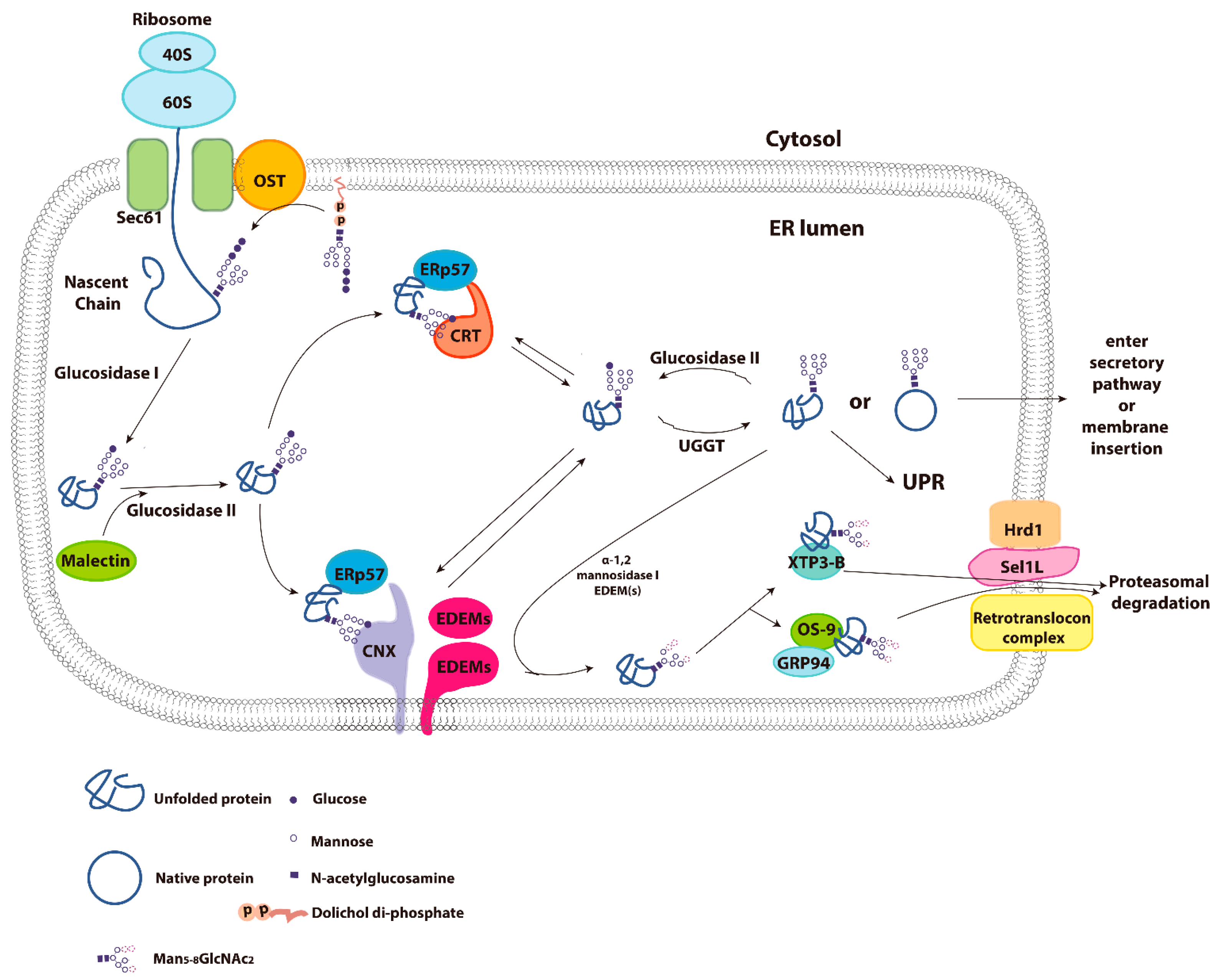
3. Calnexin/Calreticulin Cycle
4. Molecular Properties of Calnexin and Calreticulin
5. Endoplasmic Reticulum Associated Degradation (ERAD)
6. Quality Control and Endoplasmic Reticulum Stress
7. Conclusions
Acknowledgment
Authors Contributions
Conflicts of Interest
References
- Hebert, D.N.; Molinari, M. In and out of the ER: Protein folding, quality control, degradation, and related human diseases. Physiol. Rev. 2007, 87, 1377–1408. [Google Scholar] [CrossRef] [PubMed]
- Corbett, E.F.; Michalak, M. Calcium, a signaling molecule in the endoplasmic reticulum? Trends Biochem. Sci. 2000, 25, 307–311. [Google Scholar] [CrossRef]
- Mekahli, D.; Bultynck, G.; Parys, J.B.; De Smedt, H.; Missiaen, L. Endoplasmic-reticulum calcium depletion and disease. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef] [PubMed]
- Verkhratsky, A. The endoplasmic reticulum and neuronal calcium signalling. Cell Calcium 2002, 32, 393–404. [Google Scholar] [CrossRef] [PubMed]
- Rutkowski, D.T.; Kaufman, R.J. A trip to the ER: Coping with stress. Trends Cell. Biol. 2004, 14, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Agellon, L.B.; Michalak, M. Coping with endoplasmic reticulum stress in the cardiovascular system. Annu. Rev. Physiol. 2013, 75, 49–67. [Google Scholar] [CrossRef] [PubMed]
- Corbett, E.F.; Michalak, K.M.; Oikawa, K.; Johnson, S.; Campbell, I.D.; Eggleton, P.; Kay, C.; Michalak, M. The conformation of calreticulin is influenced by the endoplasmic reticulum luminal environment. J. Biol. Chem. 2000, 275, 27177–27185. [Google Scholar] [PubMed]
- Sanyal, S.; Frank, C.G.; Menon, A.K. Distinct flippases translocate glycerophospholipids and oligosaccharide diphosphate dolichols across the endoplasmic reticulum. Biochemistry 2008, 47, 7937–7946. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Bernasconi, R.; Molinari, M. ERAD substrates: Which way out? Semin. Cell Dev. Biol. 2010, 21, 526–532. [Google Scholar] [CrossRef] [PubMed]
- Zielinska, D.F.; Gnad, F.; Wisniewski, J.R.; Mann, M. Precision mapping of an in vivo N-glycoproteome reveals rigid topological and sequence constraints. Cell 2010, 141, 897–907. [Google Scholar] [CrossRef] [PubMed]
- Nilsson, T.; Warren, G. Retention and retrieval in the endoplasmic reticulum and the Golgi apparatus. Curr. Opin. Cell. Biol. 1994, 6, 517–521. [Google Scholar] [CrossRef]
- Dejgaard, K.; Theberge, J.F.; Heath-Engel, H.; Chevet, E.; Tremblay, M.L.; Thomas, D.Y. Organization of the Sec61 translocon, studied by high resolution native electrophoresis. J. Proteome Res. 2010, 9, 1763–1771. [Google Scholar] [CrossRef] [PubMed]
- Mohorko, E.; Glockshuber, R.; Aebi, M. Oligosaccharyltransferase: The central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis. 2011, 34, 869–878. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, S.; Dudek, J.; Gogala, M.; Schorr, S.; Linxweiler, J.; Lang, S.; Becker, T.; Beckmann, R.; Zimmermann, R.; Forster, F. Structure of the mammalian oligosaccharyl-transferase complex in the native ER protein translocon. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Deprez, P.; Gautschi, M.; Helenius, A. More than one glycan is needed for ER glucosidase II to allow entry of glycoproteins into the calnexin/calreticulin cycle. Mol. Cell. 2005, 19, 183–195. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N. An MBoC favorite: Malectin: A novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol. Biol. Cell. 2012, 23, 2236–2236. [Google Scholar] [CrossRef] [PubMed]
- Ruddock, L.W.; Molinari, M. N-glycan processing in ER quality control. J. Cell. Sci. 2006, 119, 4373–4380. [Google Scholar] [CrossRef] [PubMed]
- Shailubhai, K.; Pukazhenthi, B.S.; Saxena, E.S.; Varma, G.M.; Vijay, I.K. Glucosidase I, a transmembrane endoplasmic reticular glycoprotein with a luminal catalytic domain. J. Biol. Chem. 1991, 266, 16587–16593. [Google Scholar] [PubMed]
- Schallus, T.; Jaeckh, C.; Feher, K.; Palma, A.S.; Liu, Y.; Simpson, J.C.; Mackeen, M.; Stier, G.; Gibson, T.J.; Feizi, T.; et al. Malectin: A novel carbohydrate-binding protein of the endoplasmic reticulum and a candidate player in the early steps of protein N-glycosylation. Mol. Biol. Cell. 2008, 19, 3404–3414. [Google Scholar] [CrossRef] [PubMed]
- Pelletier, M.F.; Marcil, A.; Sevigny, G.; Jakob, C.A.; Tessier, D.C.; Chevet, E.; Menard, R.; Bergeron, J.J.; Thomas, D.Y. The heterodimeric structure of glucosidase II is required for its activity, solubility, and localization in vivo. Glycobiology 2000, 10, 815–827. [Google Scholar] [CrossRef] [PubMed]
- Ware, F.E.; Vassilakos, A.; Peterson, P.A.; Jackson, M.R.; Lehrman, M.A.; Williams, D.B. The molecular chaperone calnexin binds Glc1Man9GlcNAc2 oligosaccharide as an initial step in recognizing unfolded glycoproteins. J. Biol. Chem. 1995, 270, 4697–4704. [Google Scholar] [CrossRef] [PubMed]
- Spiro, R.G.; Zhu, Q.; Bhoyroo, V.; Söling, H.D. Definition of the lectin-like properties of the molecular chaperone, calreticulin, and demonstration of its copurification with endomannosidase from rat liver Golgi. J. Biol. Chem. 1996, 271, 11588–11594. [Google Scholar] [PubMed]
- Trombetta, E.S.; Simons, J.F.; Helenius, A. Endoplasmic reticulum glucosidase II is composed of a catalytic subunit, conserved from yeast to mammals, and a tightly bound noncatalytic HDEL-containing subunit. J. Biol. Chem. 1996, 271, 27509–27516. [Google Scholar] [CrossRef] [PubMed]
- Hu, D.; Kamiya, Y.; Totani, K.; Kamiya, D.; Kawasaki, N.; Yamaguchi, D.; Matsuo, I.; Matsumoto, N.; Ito, Y.; Kato, K.; et al. Sugar-binding activity of the MRH domain in the ERα-glucosidase II β subunit is important for efficient glucose trimming. Glycobiology 2009, 19, 1127–1135. [Google Scholar] [CrossRef] [PubMed]
- Olson, L.J.; Orsi, R.; Alculumbre, S.G.; Peterson, F.C.; Stigliano, I.D.; Parodi, A.J.; D'Alessio, C.; Dahms, N.M. Structure of the lectin mannose 6-phosphate receptor homology (MRH) domain of glucosidase II, an enzyme that regulates glycoprotein folding quality control in the endoplasmic reticulum. J. Biol. Chem. 2013, 288, 16460–16475. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kraus, A.; Prins, D.; Groenendyk, J.; Aubry, I.; Liu, W.X.; Li, H.D.; Julien, O.; Touret, N.; Sykes, B.D.; Tremblay, M.L.; Michalak, M. UBC9-dependent association between calnexin and protein tyrosine phosphatase 1B (PTP1B) at the endoplasmic reticulum. J. Biol. Chem. 2015, 290, 5725–5738. [Google Scholar] [CrossRef] [PubMed]
- Lynes, E.M.; Bui, M.; Yap, M.C.; Benson, M.D.; Schneider, B.; Ellgaard, L.; Berthiaume, L.G.; Simmen, T. Palmitoylated TMX and calnexin target to the mitochondria-associated membrane. EMBO J. 2012, 31, 457–470. [Google Scholar] [CrossRef] [PubMed]
- Lynes, E.M.; Raturi, A.; Shenkman, M.; Sandoval, C.O.; Yap, M.C.; Wu, J.; Janowicz, A.; Myhill, N.; Benson, M.D.; Campbell, R.E.; et al. Palmitoylation is the switch that assigns calnexin to quality control or ER Ca2+ signaling. J. Cell Sci. 2013, 126, 3893–3903. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, A.K.; Abrami, L.; Lemmin, T.; Blaskovic, S.; Kunz, B.; Kihara, A.; Dal Peraro, M.; van der Goot, F.G. Palmitoylated calnexin is a key component of the ribosome-translocon complex. EMBO J. 2012, 31, 1823–1835. [Google Scholar] [CrossRef] [PubMed]
- Lakkaraju, A.K.; van der Goot, F.G. Calnexin Controls the STAT3-Mediated Transcriptional Response to EGF. Mol. Cell 2013, 51, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Chevet, E.; Smirle, J.; Cameron, P.H.; Thomas, D.Y.; Bergeron, J.J. Calnexin phosphorylation: Linking cytoplasmic signalling to endoplasmic reticulum lumenal functions. Semin. Cell Dev. Biol. 2010, 21, 486–490. [Google Scholar] [CrossRef] [PubMed]
- Michalak, M.; Groenendyk, J.; Szabo, E.; Gold, L.I.; Opas, M. Calreticulin, a multi-process calcium-buffering chaperone of the endoplasmic reticulum. Biochem. J. 2009, 417, 651–666. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Foellmer, B.; Helenius, A. Glucose trimming and reglucosylation determine glycoprotein association with calnexin in the endoplasmic reticulum. Cell 1995, 81, 425–433. [Google Scholar] [CrossRef]
- Trombetta, E.S.; Parodi, A.J. Quality Control and Protein Folding in the Secretory Pathway. Annu. Rev. Cell Dev. Biol. 2003, 649–676. [Google Scholar] [CrossRef] [PubMed]
- Hebert, D.N.; Garman, S.C.; Molinari, M. The glycan code of the endoplasmic reticulum: Asparagine-linked carbohydrates as protein maturation and quality-control tags. Trends Cell Biol. 2005, 15, 364–370. [Google Scholar] [CrossRef] [PubMed]
- Zhu, T.; Satoh, T.; Kato, K. Structural insight into substrate recognition by the endoplasmic reticulum folding-sensor enzyme: Crystal structure of third thioredoxin-like domain of UDP-glucose:glycoprotein glucosyltransferase. Sci. Rep. 2014, 4. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.; Parodi, A.J. The molecular basis for the recognition of misfolded glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. EMBO J. 1995, 14, 4196–4203. [Google Scholar] [PubMed]
- Fink, A.L. Chaperone-mediated protein folding. Physiol. Rev. 1999, 79, 425–449. [Google Scholar] [PubMed]
- Braakman, I.; Hebert, D.N. Protein folding in the endoplasmic reticulum. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Williams, D.B. Beyond lectins: The calnexin/calreticulin chaperone system of the endoplasmic reticulum. J. Cell Sci. 2006, 119, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Schrag, J.D.; Procopio, D.O.; Cygler, M.; Thomas, D.Y.; Bergeron, J.J. Lectin control of protein folding and sorting in the secretory pathway. Trends Biochem. Sci. 2003, 28, 49–57. [Google Scholar] [CrossRef]
- Schrag, J.D.; Bergeron, J.J.; Li, Y.; Borisova, S.; Hahn, M.; Thomas, D.Y.; Cygler, M. The Structure of calnexin, an ER chaperone involved in quality control of protein folding. Mol. Cell 2001, 8, 633–644. [Google Scholar] [CrossRef]
- Pollock, S.; Kozlov, G.; Pelletier, M.F.; Trempe, J.F.; Jansen, G.; Sitnikov, D.; Bergeron, J.J.; Gehring, K.; Ekiel, I.; Thomas, D.Y. Specific interaction of ERp57 and calnexin determined by NMR spectroscopy and an ER two-hybrid system. EMBO J. 2004, 23, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Ramsamooj, P.; Notario, V.; Dritschilo, A. Enhanced expression of calreticulin in the nucleus of radioresistant squamous carcinoma cells in response to ionizing radiation. Cancer Res. 1995, 55, 3016–3021. [Google Scholar] [PubMed]
- Shan, H.; Wei, J.; Zhang, M.; Lin, L.; Yan, R.; Zhu, Y.; Zhang, R. Calreticulin is localized at mitochondria of rat cardiomyocytes and affected by furazolidone. Mol. Cell Biochem. 2014, 397, 125–130. [Google Scholar] [CrossRef] [PubMed]
- Afshar, N.; Black, B.E.; Paschal, B.M. Retrotranslocation of the chaperone calreticulin from the endoplasmic reticulum lumen to the cytosol. Mol. Cell. Biol. 2005, 25, 8844–8853. [Google Scholar]
- Orr, A.W.; Elzie, C.A.; Kucik, D.F.; Murphy-Ullrich, J.E. Thrombospondin signaling through the calreticulin/LDL receptor-related protein co-complex stimulates random and directed cell migration. J. Cell Sci. 2003, 116, 2917–2927. [Google Scholar] [CrossRef] [PubMed]
- Gardai, S.J.; McPhillips, K.A.; Frasch, S.C.; Janssen, W.J.; Starefeldt, A.; Murphy-Ullrich, J.E.; Bratton, D.L.; Oldenborg, P.A.; Michalak, M.; Henson, P.M. Cell-surface calreticulin initiates clearance of viable or apoptotic cells through trans-activation of LRP on the phagocyte. Cell 2005, 123, 321–334. [Google Scholar] [CrossRef] [PubMed]
- Gold, L.I.; Eggleton, P.; Sweetwyne, M.T.; Van Duyn, L.B.; Greives, M.R.; Naylor, S.M.; Michalak, M.; Murphy-Ullrich, J.E. Calreticulin: Non-endoplasmic reticulum functions in physiology and disease. FASEB J. 2010, 24, 665–683. [Google Scholar] [CrossRef] [PubMed]
- Frickel, E.M.; Riek, R.; Jelesarov, I.; Helenius, A.; Wuthrich, K.; Ellgaard, L. TROSY-NMR reveals interaction between ERp57 and the tip of the calreticulin P-domain. Proc. Natl. Acad. Sci. USA 2002, 99, 1954–1959. [Google Scholar] [CrossRef] [PubMed]
- Leach, M.R.; Cohen-Doyle, M.F.; Thomas, D.Y.; Williams, D.B. Localization of the Lectin, ERp57 Binding, and Polypeptide Binding Sites of Calnexin and Calreticulin. J. Biol. Chem. 2002, 277, 29686–29697. [Google Scholar] [CrossRef] [PubMed]
- Kapoor, M.; Ellgaard, L.; Gopalakrishnapai, J.; Schirra, C.; Gemma, E.; Oscarson, S.; Helenius, A.; Surolia, A. Mutational analysis provides molecular insight into the carbohydrate-binding region of calreticulin: Pivotal roles of tyrosine-109 and aspartate-135 in carbohydrate recognition. Biochemistry 2004, 43, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Thomson, S.P.; Williams, D.B. Delineation of the lectin site of the molecular chaperone calreticulin. Cell Stress Chaperones 2005, 10, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Kozlov, G.; Bastos-Aristizabal, S.; Maattanen, P.; Rosenauer, A.; Zheng, F.; Killikelly, A.; Trempe, J.F.; Thomas, D.Y.; Gehring, K. Structural basis of cyclophilin B binding by the calnexin/calreticulin P-domain. J. Biol. Chem. 2010, 285, 35551–35557. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; Tremblay, L.O.; You, Z.; Herscovics, A.; Wada, I.; Nagata, K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded Null Hong Kong α1-antitrypsin by human ER mannosidase I. J. Biol. Chem. 2003, 278, 26287–26294. [Google Scholar] [CrossRef] [PubMed]
- Ninagawa, S.; Okada, T.; Sumitomo, Y.; Kamiya, Y.; Kato, K.; Horimoto, S.; Ishikawa, T.; Takeda, S.; Sakuma, T.; Yamamoto, T.; et al. EDEM2 initiates mammalian glycoprotein ERAD by catalyzing the first mannose trimming step. J. Cell Biol. 2014, 206, 347–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Olzmann, J.A.; Kopito, R.R.; Christianson, J.C. The mammalian endoplasmic reticulum-associated degradation system. Cold Spring Harb. Perspect. Biol. 2013, 5. [Google Scholar] [CrossRef] [PubMed]
- Aebi, M.; Bernasconi, R.; Clerc, S.; Molinari, M. N-glycan structures: Recognition and processing in the ER. Trends Biochem. Sci. 2010, 35, 74–82. [Google Scholar] [CrossRef] [PubMed]
- Leitman, J.; Ron, E.; Ogen-Shtern, N.; Lederkremer, G.Z. Compartmentalization of endoplasmic reticulum quality control and ER-associated degradation factors. DNA Cell Biol. 2013, 32, 2–7. [Google Scholar] [CrossRef] [PubMed]
- Benyair, R.; Ogen-Shtern, N.; Lederkremer, G.Z. Glycan regulation of ER-associated degradation through compartmentalization. Semin. Cell Dev. Biol. 2015, 41, 99–109. [Google Scholar] [CrossRef] [PubMed]
- Avezov, E.; Frenkel, Z.; Ehrlich, M.; Herscovics, A.; Lederkremer, G.Z. Endoplasmic reticulum (ER) mannosidase I is compartmentalized and required for N-glycan trimming to Man5–6GlcNAc2 in glycoprotein ER-associated degradation. Mol. Biol. Cell 2008, 19, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, N.; You, Z.; Tremblay, L.O.; Nagata, K.; Herscovics, A. Stimulation of ERAD of misfolded null Hong Kong α1-antitrypsin by Golgi α1,2-mannosidases. Biochem. Biophys. Res. Commun. 2007, 362, 626–632. [Google Scholar] [CrossRef] [PubMed]
- Benyair, R.; Ogen-Shtern, N.; Mazkereth, N.; Shai, B.; Ehrlich, M.; Lederkremer, G.Z. Mammalian ER mannosidase I resides in quality control vesicles, where it encounters its glycoprotein substrates. Mol. Biol. Cell 2015, 26, 172–184. [Google Scholar] [CrossRef] [PubMed]
- Cali, T.; Galli, C.; Olivari, S.; Molinari, M. Segregation and rapid turnover of EDEM1 by an autophagy-like mechanism modulates standard ERAD and folding activities. Biochem. Biophys. Res. Commun. 2008, 371, 405–410. [Google Scholar] [CrossRef] [PubMed]
- Zuber, C.; Cormier, J.H.; Guhl, B.; Santimaria, R.; Hebert, D.N.; Roth, J. EDEM1 reveals a quality control vesicular transport pathway out of the endoplasmic reticulum not involving the COPII exit sites. Proc. Natl. Acad. Sci. USA 2007, 104, 4407–4412. [Google Scholar] [CrossRef] [PubMed]
- Frenkel, Z.; Gregory, W.; Kornfeld, S.; Lederkremer, G.Z. Endoplasmic reticulum-associated degradation of mammalian glycoproteins involves sugar chain trimming to Man6–5GlcNAc2. J. Biol. Chem. 2003, 278, 34119–34124. [Google Scholar] [CrossRef] [PubMed]
- Tannous, A.; Pisoni, G.B.; Hebert, D.N.; Molinari, M. N-linked sugar-regulated protein folding and quality control in the ER. Semin. Cell Dev. Biol. 2015, 41, 79–89. [Google Scholar] [CrossRef] [PubMed]
- Leitman, J.; Shenkman, M.; Gofman, Y.; Shtern, N.O.; Ben-Tal, N.; Hendershot, L.M.; Lederkremer, G.Z. Herp coordinates compartmentalization and recruitment of HRD1 and misfolded proteins for ERAD. Mol. Biol. Cell 2014, 25, 1050–1060. [Google Scholar] [CrossRef] [PubMed]
- Sousa, M.C.; Ferrero-Garcia, M.A.; Parodi, A.J. Recognition of the oligosaccharide and protein moieties of glycoproteins by the UDP-Glc:glycoprotein glucosyltransferase. Biochemistry 1992, 31, 97–105. [Google Scholar] [CrossRef] [PubMed]
- Stigliano, I.D.; Alculumbre, S.G.; Labriola, C.A.; Parodi, A.J.; D'Alessio, C. Glucosidase II and N-glycan mannose content regulate the half-lives of monoglucosylated species in vivo. Mol. Biol. Cell 2011, 22, 1810–1823. [Google Scholar] [CrossRef] [PubMed]
- Christianson, J.C.; Shaler, T.A.; Tyler, R.E.; Kopito, R.R. OS-9 and GRP94 deliver mutant α1-antitrypsin to the Hrd1-SEL1L ubiquitin ligase complex for ERAD. Nat. Cell Biol. 2008, 10, 272–282. [Google Scholar] [CrossRef] [PubMed]
- Kikkert, M.; Doolman, R.; Dai, M.; Avner, R.; Hassink, G.; van Voorden, S.; Thanedar, S.; Roitelman, J.; Chau, V.; Wiertz, E. Human HRD1 is an E3 ubiquitin ligase involved in degradation of proteins from the endoplasmic reticulum. J. Biol. Chem. 2004, 279, 3525–3534. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. Multiprotein complexes that link dislocation, ubiquitination, and extraction of misfolded proteins from the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14296–14301. [Google Scholar] [CrossRef] [PubMed]
- Schulze, A.; Standera, S.; Buerger, E.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Koning, F.; Kloetzel, P.M.; Seeger, M. The ubiquitin-domain protein HERP forms a complex with components of the endoplasmic reticulum associated degradation pathway. J. Mol. Biol. 2005, 354, 1021–1027. [Google Scholar] [CrossRef] [PubMed]
- Tsai, B.; Ye, Y.; Rapoport, T.A. Retro-translocation of proteins from the endoplasmic reticulum into the cytosol. Nat. Rev. Mol. Cell Biol. 2002, 3, 246–255. [Google Scholar] [CrossRef] [PubMed]
- Bernardi, K.M.; Forster, M.L.; Lencer, W.I.; Tsai, B. Derlin-1 facilitates the retro-translocation of cholera toxin. Mol. Biol. Cell 2008, 19, 877–884. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Kikkert, M.; van Voorden, S.; Wiertz, E.; Rapoport, T.A. Inaugural Article: Recruitment of the p97 ATPase and ubiquitin ligases to the site of retrotranslocation at the endoplasmic reticulum membrane. Proc. Natl. Acad. Sci. USA 2005, 102, 14132–14138. [Google Scholar] [CrossRef] [PubMed]
- Oda, Y.; Okada, T.; Yoshida, H.; Kaufman, R.J.; Nagata, K.; Mori, K. Derlin-2 and Derlin-3 are regulated by the mammalian unfolded protein response and are required for ER-associated degradation. J. Cell Biol. 2006, 172, 383–393. [Google Scholar] [CrossRef] [PubMed]
- Katiyar, S.; Joshi, S.; Lennarz, W.J. The retrotranslocation protein Derlin-1 binds peptide: N-glycanase to the endoplasmic reticulum. Mol. Biol. Cell 2005, 16, 4584–4594. [Google Scholar] [CrossRef] [PubMed]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Lilley, B.N.; Ploegh, H.L. A membrane protein required for dislocation of misfolded proteins from the ER. Nature 2004, 429, 834–840. [Google Scholar] [CrossRef] [PubMed]
- Shen, X.; Zhang, K.; Kaufman, R.J. The unfolded protein response--a stress signaling pathway of the endoplasmic reticulum. J. Chem. Neuroanat. 2004, 28, 79–92. [Google Scholar] [CrossRef] [PubMed]
- Travers, K.J.; Patil, C.K.; Wodicka, L.; Lockhart, D.J.; Weissman, J.S.; Walter, P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 2000, 101, 249–258. [Google Scholar] [CrossRef]
- Schroder, M. Endoplasmic reticulum stress responses. Cell Mol. Life Sci. 2008, 65, 862–894. [Google Scholar] [CrossRef] [PubMed]
- Oyadomari, S.; Araki, E.; Mori, M. Endoplasmic reticulum stress-mediated apoptosis in pancreatic β-cells. Apoptosis 2002, 7, 335–345. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.Y.; Wong, H.N.; Schauerte, J.A.; Kaufman, R.J. The protein kinase/endoribonuclease IRE1α that signals the unfolded protein response has a luminal N-terminal ligand-independent dimerization domain. J. Biol. Chem. 2002, 277, 18346–18356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.; Kaufman, R.J. Protein folding in the endoplasmic reticulum and the unfolded protein response. Handb. Exp. Pharmacol. 2006, 69–91. [Google Scholar]
- Back, S.H.; Schroder, M.; Lee, K.; Zhang, K.; Kaufman, R.J. ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods 2005, 35, 395–416. [Google Scholar] [CrossRef] [PubMed]
- Harding, H.P.; Zhang, Y.; Ron, D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 1999, 397, 271–274. [Google Scholar] [PubMed]
- Ye, J.; Rawson, R.B.; Komuro, R.; Chen, X.; Dave, U.P.; Prywes, R.; Brown, M.S.; Goldstein, J.L. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol. Cell 2000, 6, 1355–1364. [Google Scholar] [CrossRef]
- Mesaeli, N.; Nakamura, K.; Zvaritch, E.; Dickie, P.; Dziak, E.; Krause, K.H.; Opas, M.; MacLennan, D.H.; Michalak, M. Calreticulin is essential for cardiac development. J. Cell Biol. 1999, 144, 857–868. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.; Mao, C.; Lee, B.; Lee, A.S. GRP78/BiP is required for cell proliferation and protecting the inner cell mass from apoptosis during early mouse embryonic development. Mol. Cell. Biol. 2006, 26, 5688–5697. [Google Scholar] [CrossRef] [PubMed]
- Wanderling, S.; Simen, B.B.; Ostrovsky, O.; Ahmed, N.T.; Vogen, S.M.; Gidalevitz, T.; Argon, Y. GRP94 is essential for mesoderm induction and muscle development because it regulates insulin-like growth factor secretion. Mol. Biol. Cell 2007, 18, 3764–3775. [Google Scholar] [CrossRef] [PubMed]
- Coe, H.; Jung, J.; Groenendyk, J.; Prins, D.; Michalak, M. ERp57 modulates STAT3 signaling from the lumen of the endoplasmic reticulum. J. Biol. Chem. 2010, 285, 6725–6738. [Google Scholar] [CrossRef] [PubMed]
- Kraus, A.; Groenendyk, J.; Bedard, K.; Baldwin, T.A.; Krause, K.H.; Dubois-Dauphin, M.; Dyck, J.; Rosenbaum, E.E.; Korngut, L.; Colley, N.J.; et al. Calnexin deficiency leads to dysmyelination. J. Biol. Chem. 2010, 285, 18928–18938. [Google Scholar] [CrossRef] [PubMed]
- Thuerauf, D.J.; Marcinko, M.; Belmont, P.J.; Glembotski, C.C. Effects of the isoform-specific characteristics of ATF6α and ATF6β on endoplasmic reticulum stress response gene expression and cell viability. J. Biol. Chem. 2007, 282, 22865–22878. [Google Scholar] [CrossRef] [PubMed]
- Doroudgar, S.; Thuerauf, D.J.; Marcinko, M.C.; Belmont, P.J.; Glembotski, C.C. Ischemia activates the ATF6 branch of the endoplasmic reticulum stress response. J. Biol. Chem. 2009, 284, 29735–29745. [Google Scholar] [CrossRef] [PubMed]
- Okada, K.; Minamino, T.; Tsukamoto, Y.; Liao, Y.; Tsukamoto, O.; Takashima, S.; Hirata, A.; Fujita, M.; Nagamachi, Y.; Nakatani, T.; et al. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: Possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation 2004, 110, 705–712. [Google Scholar] [CrossRef] [PubMed]
- Martindale, J.J.; Fernandez, R.; Thuerauf, D.; Whittaker, R.; Gude, N.; Sussman, M.A.; Glembotski, C.C. Endoplasmic reticulum stress gene induction and protection from ischemia/reperfusion injury in the hearts of transgenic mice with a tamoxifen-regulated form of ATF6. Circ. Res. 2006, 98, 1186–1193. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.; Valdes, P.; Hetz, C. An ERcentric view of Parkinson’s disease. Trends Mol. Med. 2013, 19, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Mercado, G.; Castillo, V.; Vidal, R.; Hetz, C. ER proteostasis disturbances in Parkinson’s disease: Novel insights. Front. Aging Neurosci. 2015, 7, 39. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Castillo, K.; Armisen, R.; Stutzin, A.; Soto, C.; Hetz, C. Prion protein misfolding affects calcium homeostasis and sensitizes cells to endoplasmic reticulum stress. PLoS ONE 2010, 5, e15658. [Google Scholar] [CrossRef] [PubMed]
- Moreno, J.A.; Radford, H.; Peretti, D.; Steinert, J.R.; Verity, N.; Martin, M.G.; Halliday, M.; Morgan, J.; Dinsdale, D.; Ortori, C.A.; et al. Sustained translational repression by eIF2α-P mediates prion neurodegeneration. Nature 2012, 485, 507–511. [Google Scholar] [CrossRef] [PubMed]
- Greenman, C.; Stephens, P.; Smith, R.; Dalgliesh, G.L.; Hunter, C.; Bignell, G.; Davies, H.; Teague, J.; Butler, A.; Stevens, C.; et al. Patterns of somatic mutation in human cancer genomes. Nature 2007, 446, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Parsons, D.W.; Jones, S.; Zhang, X.; Lin, J.C.; Leary, R.J.; Angenendt, P.; Mankoo, P.; Carter, H.; Siu, I.M.; Gallia, G.L.; et al. An integrated genomic analysis of human glioblastoma multiforme. Science 2008, 321, 1807–1812. [Google Scholar] [CrossRef] [PubMed]
- Guichard, C.; Amaddeo, G.; Imbeaud, S.; Ladeiro, Y.; Pelletier, L.; Maad, I.B.; Calderaro, J.; Bioulac-Sage, P.; Letexier, M.; Degos, F.; et al. Integrated analysis of somatic mutations and focal copy-number changes identifies key genes and pathways in hepatocellular carcinoma. Nat. Genet. 2012, 44, 694–698. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Iliopoulos, D.; Zhang, Q.; Tang, Q.; Greenblatt, M.B.; Hatziapostolou, M.; Lim, E.; Tam, W.L.; Ni, M.; Chen, Y.; et al. XBP1 promotes triple-negative breast cancer by controlling the HIF1α pathway. Nature 2014, 508, 103–107. [Google Scholar] [CrossRef] [PubMed]
- Groenendyk, J.; Sreenivasaiah, P.K.; Kim do, H.; Agellon, L.B.; Michalak, M. Biology of endoplasmic reticulum stress in the heart. Circ. Res. 2010, 107, 1185–1197. [Google Scholar] [CrossRef] [PubMed]
- Lenna, S.; Trojanowska, M. The role of endoplasmic reticulum stress and the unfolded protein response in fibrosis. Curr. Opin. Rheumatol. 2012, 24, 663–668. [Google Scholar] [CrossRef] [PubMed]
- Torres, M.; Encina, G.; Soto, C.; Hetz, C. Abnormal calcium homeostasis and protein folding stress at the ER: A common factor in familial and infectious prion disorders. Commun. Integr. Biol. 2011, 4, 258–261. [Google Scholar] [CrossRef] [PubMed]
- Scheuner, D.; Kaufman, R.J. The unfolded protein response: A pathway that links insulin demand with β-cell failure and diabetes. Endocr. Rev. 2008, 29, 317–333. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, K.; Yoshida, H.; Kokame, K.; Kaufman, R.J.; Mori, K. Differential contributions of ATF6 and XBP1 to the activation of endoplasmic reticulum stress-responsive cis-acting elements ERSE, UPRE and ERSE-II. J. Biochem. 2004, 136, 343–350. [Google Scholar] [CrossRef] [PubMed]
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Q.; Groenendyk, J.; Michalak, M. Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules 2015, 20, 13689-13704. https://doi.org/10.3390/molecules200813689
Wang Q, Groenendyk J, Michalak M. Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules. 2015; 20(8):13689-13704. https://doi.org/10.3390/molecules200813689
Chicago/Turabian StyleWang, Qian, Jody Groenendyk, and Marek Michalak. 2015. "Glycoprotein Quality Control and Endoplasmic Reticulum Stress" Molecules 20, no. 8: 13689-13704. https://doi.org/10.3390/molecules200813689
APA StyleWang, Q., Groenendyk, J., & Michalak, M. (2015). Glycoprotein Quality Control and Endoplasmic Reticulum Stress. Molecules, 20(8), 13689-13704. https://doi.org/10.3390/molecules200813689