
Synthesis of C3/C1-Substituted Tetrahydroisoquinolines


 ,
, 
 and
and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
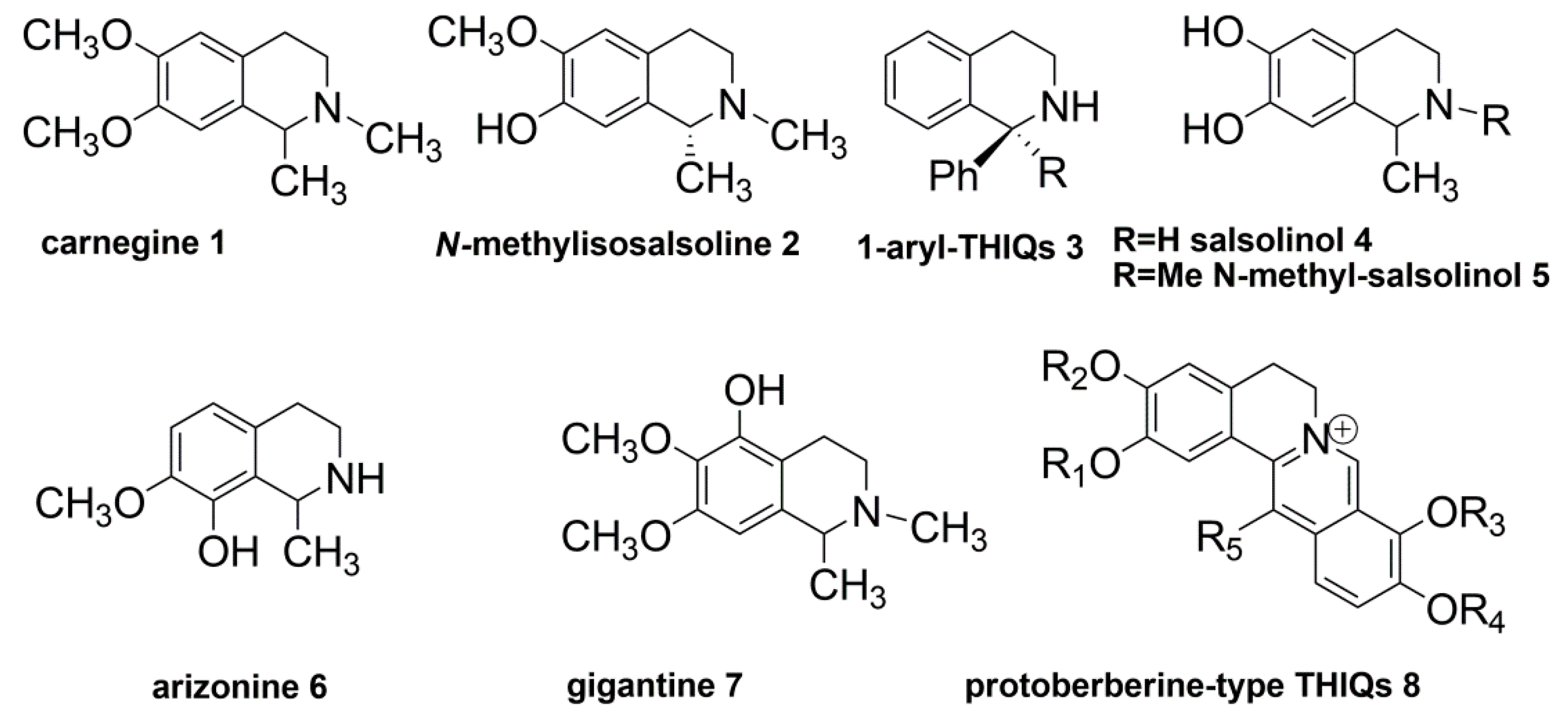
1. Introduction


2. Results and Discussion
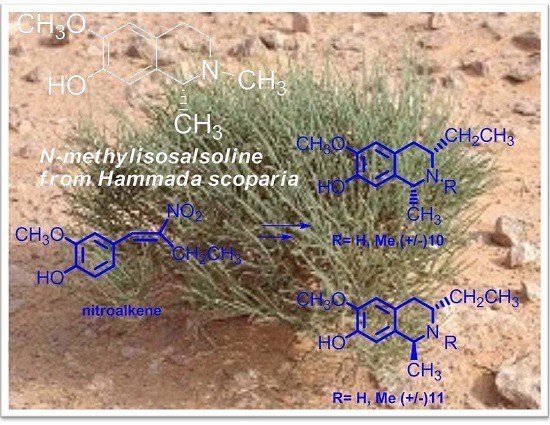
2.1. Evaluation of N-Methylisosalsoline (2) against Human and Parasitic Proteases
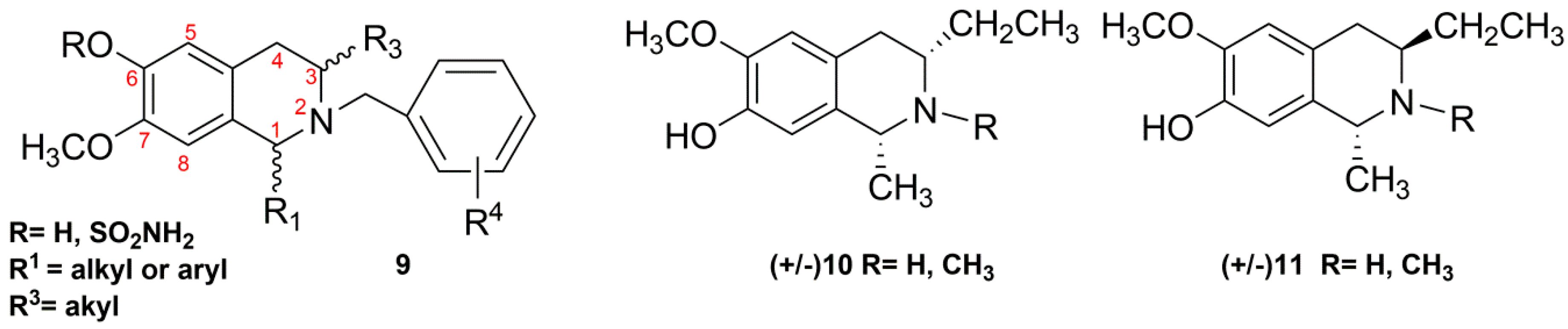
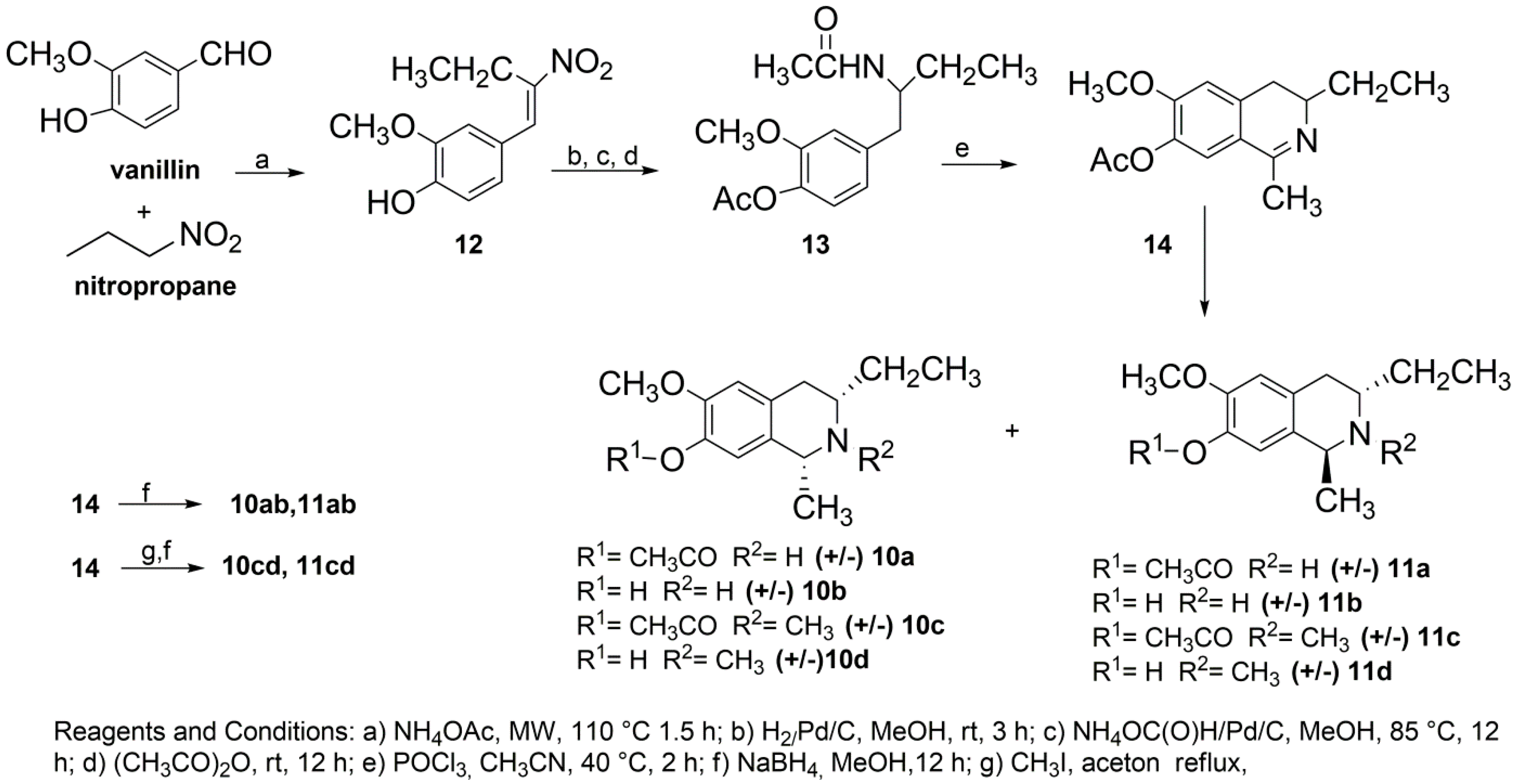
2.2. Synthesis of C3/C1-Substituted THIQs (10–11)

3. Experimental Section
3.1. General Information
3.2. Extraction of N-Methylisosalsoline 2
3.3. Evaluation of 2 against Human and Parasitic Proteases
3.3.1. Assaying the Chymotryptic Activity of the 20S Proteasome
3.3.2. Assaying the Tryptic Activity of the 20S Proteasome
3.3.3. Assaying the Post-Glutamyl Peptide Hydrolyzing Activity of the 20S Proteasome
3.3.4. Assay for Bovine Pancreatic α-Chymotrypsin Inhibition
3.3.5. Assay for Rhodesain Inhibition
3.3.6. Assay for Leishmania Mexicana Cysteine Protease CPB2.8
3.3.7. Assay for Dengue Virus NS2B/NS3 Protease
3.4. Synthesis of (E)-2-Methoxy-4-(2-nitrobut-1-enyl)phenol 12
3.5. Synthesis of 4-(2-Acetamidobutyl)-2-methoxyphenyl Acetate 13
3.6. Synthesis of 3-Ethyl-6-methoxy-1-methyl-3,4-dihydroisoquinolin-7-yl Acetate 14
3.7. Synthesis of 10a,b and 11a,b
3.8. Synthesis of 10c,d and 11c,d
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Scott, J.D.; Williams, R.M. Chemistry and biology of the tetrahydroisoquinoline antitumor antibiotics. Chem. Rev. 2002, 102, 1669–1730. [Google Scholar] [CrossRef] [PubMed]
- Xuan, B.; Li, D.; Wang, W. Protective effects of tetrahydroprotoberberines on experimental myocardial infarction in rats. Acta Pharmacol. Sin. 1992, 13, 167–171. [Google Scholar]
- Chu, L.H.; Hsu, F.L.; Chueh, F.Y.; Niu, C.S.; Cheng, J.T. Antihypertensive Activity of dl-Tetrahydropalmatine, an active alkaloid isolated from the tubers of Corydalis racemosa. J. Clin. Chem. Soc. 1996, 43, 489–492. [Google Scholar]
- Lin, M.; Chueh, F.; Hsieh, M.; Chen, C. Antihypertensive effects of DL-Tetrahydropalmatine: an active principle isolated from Corydalis. Clin. Exp. Pharmacol. Physiol. 1996, 23, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Barbosa-Filho, J.M.; Piuvezam, M.R.; Moura, M.D.; Silva, M.S.; Lima, K.V.B.; da-Cunha, E.V.L.; Fechine, I.M.; Takemura, O.S. Anti-inflammatory activity of alkaloids: A twenty-century review. Rev. Bras. Farmacogn. 2006, 16, 109–139. [Google Scholar] [CrossRef]
- Nammalwar, B.; Bunce, R.A. Recent Syntheses of 1,2,3,4-Tetrahydroquinolines, 2,3-Dihydro-4(1H)-quinolinones and 4(1H)-Quinolinones using domino reactions. Molecules 2014, 19, 204–232. [Google Scholar] [CrossRef] [PubMed]
- Singh, I.P.; Mahajan, S. Berberine and its derivatives: A patent review (2009–2012). Expert Opin. Ther. Pat. 2013, 23, 215–231. [Google Scholar] [CrossRef] [PubMed]
- Wangchuk, P.; Keller, P.A.; Pyne, S.G.; Willis, A.C.; Kamchonwongpaisan, S. Antimalarial alkaloids from a Bhutanese traditional medicinal plant Corydalis dubia. J. Ethnopharmacol. 2012, 143, 310–313. [Google Scholar] [CrossRef] [PubMed]
- Wangchuk, P.; Keller, P.A.; Pyne, S.G.; Sastraruji, T.; Taweechotipatr, M.; Rattanajak, R.; Tonsomboon, A.; Kamchonwongpaisan, S. Phytochemical and biological activity studies of the bhutanese medicinal plant Corydalis crispa. Nat. Prod. Commun. 2012, 7, 575–580. [Google Scholar] [PubMed]
- Xiao, H.T.; Peng, J.; Liang, Y.; Yang, J.; Bai, X.; Hao, X.Y.; Yang, F.M.; Sun, Q.Y. Acetylcholinesterase inhibitors from Corydalis yanhusuo. Nat. Prod. Res. 2011, 25, 1418–1422. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.F.; Shen, L. Berberine: A potential multipotent natural product to combat Alzheimer’s disease. Molecules 2011, 16, 6732–6740. [Google Scholar] [CrossRef] [PubMed]
- Naoi, M.; Maruyama, W.; Nagy, G.M. Dopamine-derived salsolinol derivatives as endogenous monoamine oxidase inhibitors: Occurrence, metabolism and function in human brains. NeuroToxicol. 2004, 25, 193–204. [Google Scholar] [CrossRef]
- Tiwari, R.K.; Singh, D.; Singh, J.; Chhillar, A.K.; Chandra, R.; Verma, A.K. Synthesis, antibacterial activity and QSAR studies of 1,2-Disubstituted-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinolines. Eur. J. Med. Chem. 2006, 41, 40–49. [Google Scholar] [CrossRef] [PubMed]
- Ludwig, M.; Hoesl, C.E.; Hӧfner, G.; Wanner, K.T. Affinity of 1-aryl-1,2,3,4-tetrahydroisoquinoline derivatives to the ion channel binding site of the NMDA receptor complex. Eur. J. Med. Chem. 2006, 41, 1003–1010. [Google Scholar] [CrossRef] [PubMed]
- Gitto, R.; Caruso, R.; Pagano, B.; de Luca, L.; Citraro, R.; Russo, E.; De Sarro, G.; Chimirri, A. Novel potent anticonvulsant agent containing a tetrahydroisoquinoline skeleton. J. Med. Chem. 2006, 49, 5618–5622. [Google Scholar] [CrossRef] [PubMed]
- Gao, M.; Kong, D.Y.; Clearfield, A.; Zeng, Q.H. Synthesis of Carbon-11 and Fluorine-18 labeled N-Acetyl-1-aryl-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline derivatives as new potential PET AMPA receptor ligands. Bioorg. Med. Chem. Lett. 2006, 16, 2229–2233. [Google Scholar] [CrossRef] [PubMed]
- Gitto, R.; Ficarra, R.; Stancanelli, R.; Guardo, M.; de Luca, L.; Barreca, M.L.; Pagano, B.; Rotondo, A.; Bruno, G.; Russo, E.; et al. Synthesis, resolution, stereochemistry, and molecular modeling of (R)-and (S)-2-acetyl-1-(4ʹ-chlorophenyl)-6,7-dimethoxy-1,2,3,4-tetrahydroisoquinoline AMPAR antagonists. Bioorg. Med. Chem. 2007, 15, 5417–5423. [Google Scholar] [CrossRef] [PubMed]
- Cheng, P.; Huang, N.; Jiang, Z.Y.; Zhang, Q.; Zheng, Y.T.; Chen, J.J.; Zhang, X.M.; Ma, Y.B. 1-Aryl-tetrahydroisoquinoline analogs as active anti-HIV agents in vitro. Bioorg. Med. Chem. Lett. 2008, 18, 2475–2478. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.X.; Xie, H.Y.; Li, Z.G.; Gao, J.R. Quantitative structure-activity relationship studies on 1-aryl-tetrahydroisoquinoline analogs as active anti-HIV agents. Bioorg. Med. Chem. Lett. 2008, 18, 5381–5386. [Google Scholar] [CrossRef] [PubMed]
- Micale, N.; Ettari, R.; Schirmeister, T.; Evers, A.; Gelhaus, C.; Leippe, M.; Zappalà, M.; Grasso, S. Novel 2H-isoquinolin-3-ones as antiplasmodial falcipain-2 inhibitors. Bioorg. Med. Chem. 2009, 17, 6505–6511. [Google Scholar] [CrossRef] [PubMed]
- Ivanov, I.; Nikolova, S.; Aladjov, D.; Stefanova, I.; Zagorchev, P. Synthesis and contractile activity of substituted 1,2,3,4-Tetrahydroisoquinolines. Molecules 2011, 16, 7019–7042. [Google Scholar] [CrossRef] [PubMed]
- Galán, A.; Moreno, L.; Párraga, J.; Serrano, Á.; Jesús Sanz, M.; Cortes, D.; Cabedo, N. Novel isoquinoline derivatives as antimicrobial agents. Bioorg. Med. Chem. 2013, 21, 3221–3230. [Google Scholar] [CrossRef] [PubMed]
- Iranshahy, M.; Quinn, R.J.; Iranshahi, M. Biologically active isoquinoline alkaloids with drug-like properties from the genus Corydalis. RSC Adv. 2014, 4, 15900–15913. [Google Scholar] [CrossRef]
- Mezghani-Jarraya, R.; Hammami, H.; Ayadi, A.; Damak, M. Molluscicidal activity of Hammada scoparia (Pomel) Iljin leaf extracts and the principal alkaloids isolated from them against Galba truncatula. Mem. Inst. Mem. I. 2009, 104, 1035–1038. [Google Scholar] [CrossRef]
- Britton, M.; Lucas, M.M.; Downey, S.L.; Screen, M.; Pletnev, A.A.; Verdoes, M.; Tokhunts, R.A.; Amir, O.; Goddard, A.L.; Pelphrey, P.M.; et al. Selective inhibitor of proteasome’s caspase-like sites sensitizes cells to specific inhibition of chymotrypsin-like sites. Chem. Biol. 2009, 16, 1278–1289. [Google Scholar] [CrossRef] [PubMed]
- Screen, M.; Britton, M.; Downey, S.L.; Verdoes, M.; Voges, M.J.; Blom, A.E.; Geurink, P.P.; Risseeuw, M.D.; Florea, B.I.; van der Linden, W.A.; et al. Nature of pharmacophore influences active site specificity of proteasome inhibitors. J. Biol. Chem. 2010, 285, 40125–40134. [Google Scholar] [CrossRef] [PubMed]
- Mirabella, A.C.; Pletnev, A.A.; Downey, S.L.; Florea, B.I.; Shabaneh, T.B.; Britton, M.; Verdoes, M.; Filippov, D.V.; Overkleeft, H.S; Kisselev, A.F. Specific cell-permeable inhibitor of proteasome trypsin-like sites selectively sensitizes myeloma cells to bortezomib and carfilzomib. Chem. Biol. 2011, 18, 608–618. [Google Scholar] [CrossRef] [PubMed]
- Parlati, F.; Lee, S.J.; Aujay, M.; Suzuk, E.; Levitsky, K.; Lorens, J.B.; Micklem, D.R.; Ruurs, P.; Sylvain, C.; Lu, Y.; et al. Carfilzomib can induce tumor cell death through selective inhibition of the chymotrypsin-like activity of the proteasome. Blood 2009, 114, 3439–3447. [Google Scholar] [CrossRef] [PubMed]
- Bonaccorso, C.; Micale, N.; Ettari, R.; Grasso, S.; Zappalà, M. Glutamate binding-site ligands of NMDA receptors. Curr. Med. Chem. 2011, 18, 5483–5550. [Google Scholar] [CrossRef] [PubMed]
- Bracca, A.B.J.; Kaufman, T.S. Synthetic Approaches to Carnegine, a Simple Tetrahydroisoquinoline Alkaloid. Tetrahedron 2004, 60, 10575–10610. [Google Scholar] [CrossRef]
- Awuah, E.; Capretta, A. Strategies and synthetic methods directed toward the preparation of libraries of substituted isoquinolines. J. Org. Chem. 2010, 75, 5627–5634. [Google Scholar] [CrossRef] [PubMed]
- Leese, M.P.; Jourdan, F.L.; Major, M.R.; Dohle, W.; Thomas, M.P.; Hamel, E.; Ferrandis, E.; Mahon, M. F.; Newman, S.P.; Purohit, A.; Potter, B.V.L. Synthesis, antitubulin and antiproliferative SAR of steroidomimetic dihydroisoquinolinones. Chem. Med. Chem. 2014, 9, 798–812. [Google Scholar] [CrossRef] [PubMed]
- Dohle, W.; Leese, M.P.; Jourdan, F.L.; Major, M.R.; Bai, R.; Hamel, E.; Ferrandis, E.; Kasprzyk, F.G.; Fiore, A.; Newman, S.P.; Purohit, A.; Potter, B.V.L. Synthesis, antitubulin, and antiproliferative SAR of C3/C1-substituted tetrahydroisoquinolines. Chem. Med. Chem. 2014, 9, 350–370. [Google Scholar] [CrossRef] [PubMed]
- Thomas, M.P.; Potter, B.V.L. Discovery and development of the aryl O-sulfamate pharmacophore for oncology and women’s health. J. Med. Chem. 2015. [CrossRef]
- Ram, S.; Ehrenkaufer, R.E. A general procedure for mild and rapid reduction of aliphatic and aromatic nitro compounds using ammonium formate as a catalytic hydrogen transfer agent. Tetrahedron Lett. 1984, 25, 3415–3418. [Google Scholar] [CrossRef]
- Caffrey, C.R; Hansell, E.; Lucas, K.D.; Brinen, L.S.; Hernandez, A.A.; Cheng, J.; Gwaltney, S.L., II; Roush, W.R.; Stierhof, Y.-D.; Bogyo, M.; et al. Active site mapping, biochemical properties and subcellular localization of rhodesain, the major cysteine protease of Trypanosoma brucei rhodesiense. Mol. Biochem. Parasit. 2001, 118, 61–73. [Google Scholar] [CrossRef]
- Sanderson, S.J.; Pollock, K.G.; Hilley, J.D.; Meldal, M.; Hilaire, P.S.; Juliano, M.A.; Juliano, L.; Mottram, J.C.; Coombs, G.H. Expression and characterization of a recombinant cysteine proteinase of Leishmania Mexicana. Biochem. J. 2000, 347, 383–388. [Google Scholar] [CrossRef] [PubMed]
- Vicik, R.; Busemann, M.; Gelhaus, C.; Stiefl, N.; Scheiber, J.; Schmitz, W.; Schulz, F.; Mladenovic, M.; Engels, B.; Leippe, M.; et al. Aziridide-based inhibitors of Cathepsin, L: Synthesis, inhibition activity, and docking studies. ChemMedChem 2006, 1, 1126–1141. [Google Scholar] [CrossRef] [PubMed]
- Wu, H.; Bock, S.; Snitko, M.; Berger, T.; Weidner, T.; Holloway, S.; Kanitz, M.; Diederich, W.E.; Steuber, H.; Hofmann, C.W.D; et al. Novel Dengue Virus NS2B/NS3 Protease Inhibitors. Antimicrob Agents Ch. 2015, 59, 1100–1109. [Google Scholar]
- Jarraya, R.M.; Bouaziz, A.; Hamdi, B.; Salah, A.; Damak, M. N-Methylisosalsoline from Hammada scoparia. Acta Cryst. 2008, E64, o1714. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 2, 10 and 11 are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mihoubi, M.; Micale, N.; Scala, A.; Jarraya, R.M.; Bouaziz, A.; Schirmeister, T.; Risitano, F.; Piperno, A.; Grassi, G. Synthesis of C3/C1-Substituted Tetrahydroisoquinolines. Molecules 2015, 20, 14902-14914. https://doi.org/10.3390/molecules200814902
Mihoubi M, Micale N, Scala A, Jarraya RM, Bouaziz A, Schirmeister T, Risitano F, Piperno A, Grassi G. Synthesis of C3/C1-Substituted Tetrahydroisoquinolines. Molecules. 2015; 20(8):14902-14914. https://doi.org/10.3390/molecules200814902
Chicago/Turabian StyleMihoubi, Mohamed, Nicola Micale, Angela Scala, Raoudha Mezghani Jarraya, Amira Bouaziz, Tanja Schirmeister, Francesco Risitano, Anna Piperno, and Giovanni Grassi. 2015. "Synthesis of C3/C1-Substituted Tetrahydroisoquinolines" Molecules 20, no. 8: 14902-14914. https://doi.org/10.3390/molecules200814902
APA StyleMihoubi, M., Micale, N., Scala, A., Jarraya, R. M., Bouaziz, A., Schirmeister, T., Risitano, F., Piperno, A., & Grassi, G. (2015). Synthesis of C3/C1-Substituted Tetrahydroisoquinolines. Molecules, 20(8), 14902-14914. https://doi.org/10.3390/molecules200814902