
Probing Water and CO2 Interactions at the Surface of Collapsed Titania Nanotubes Using IR Spectroscopy

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. X-ray Diffraction Data


2.2. Transmission Electron Microscopy

2.3. BET Surface Area

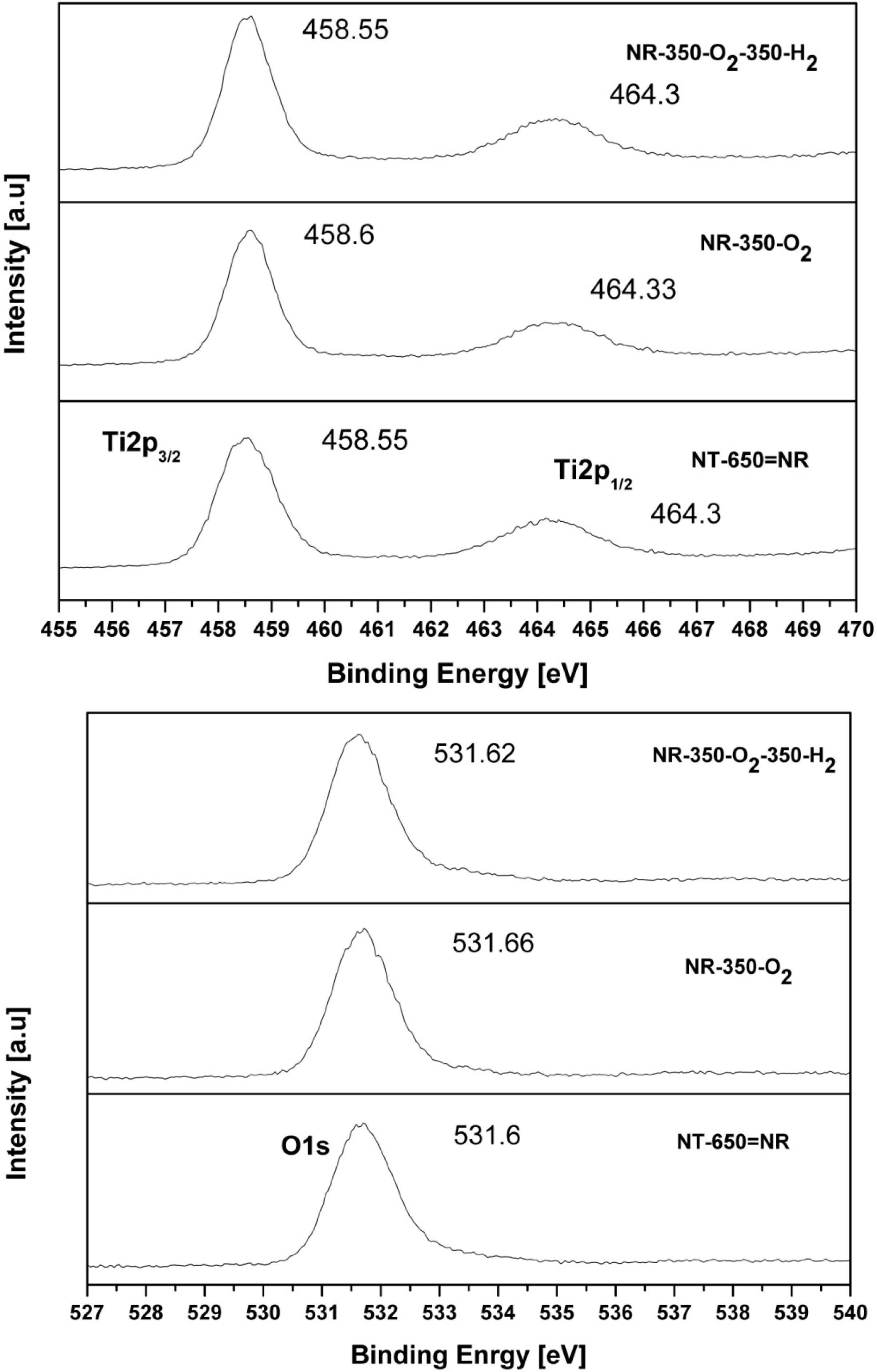
2.4. XPS

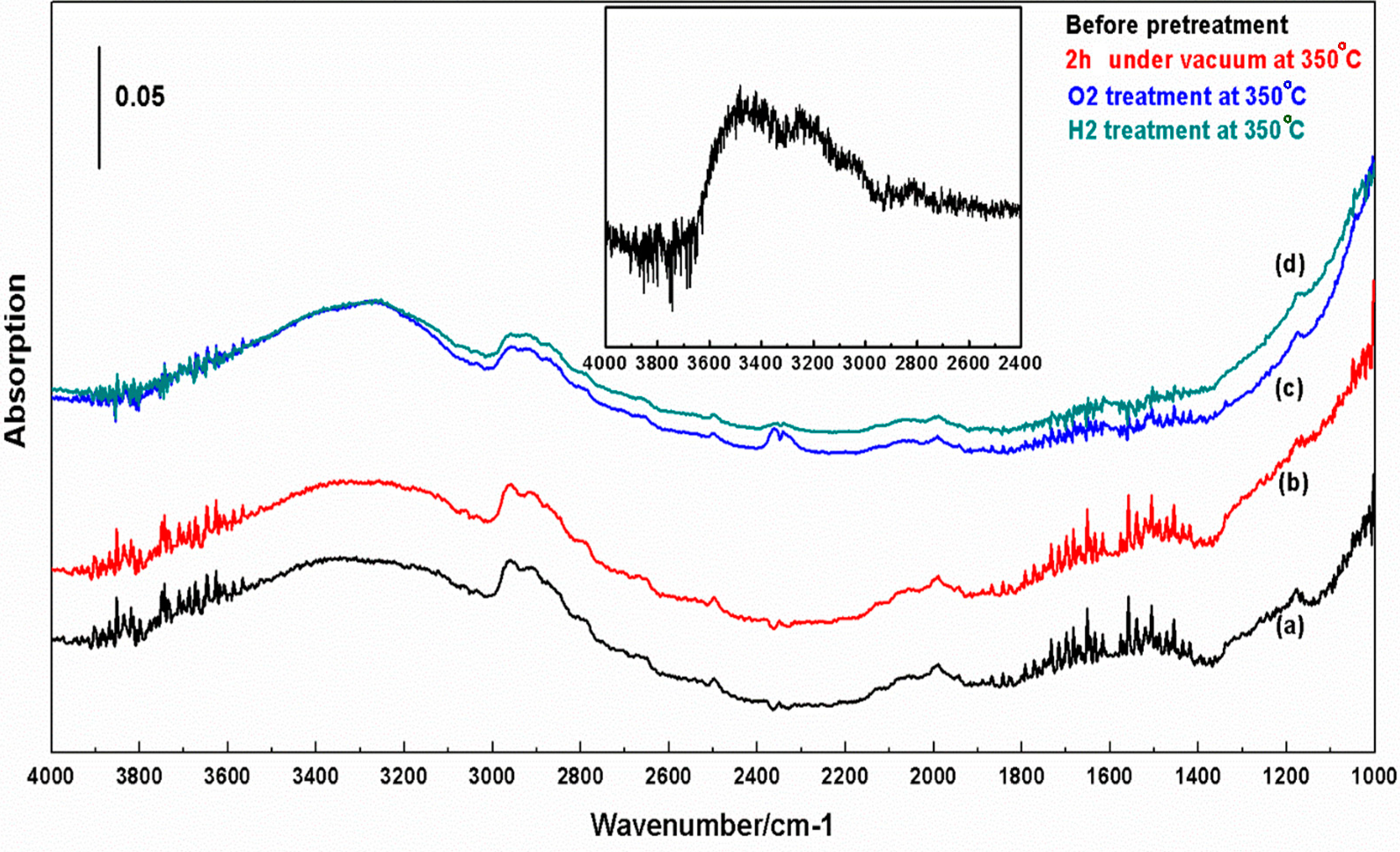
2.5. FT-IR Spectroscopy



3. Discussion
3.1. Structure of the NTs
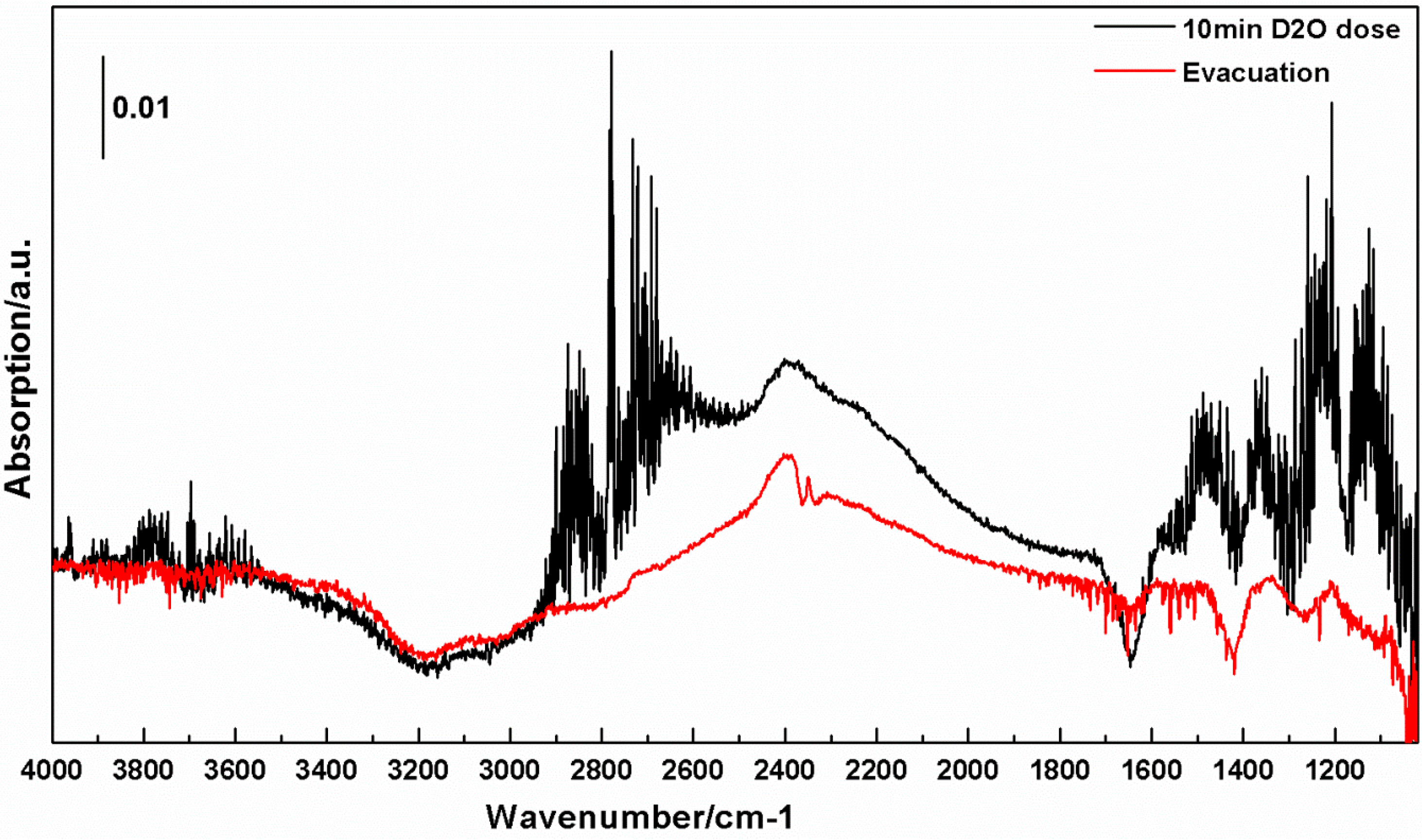
3.2. Interactions of H2O and D2O with c-TiNT
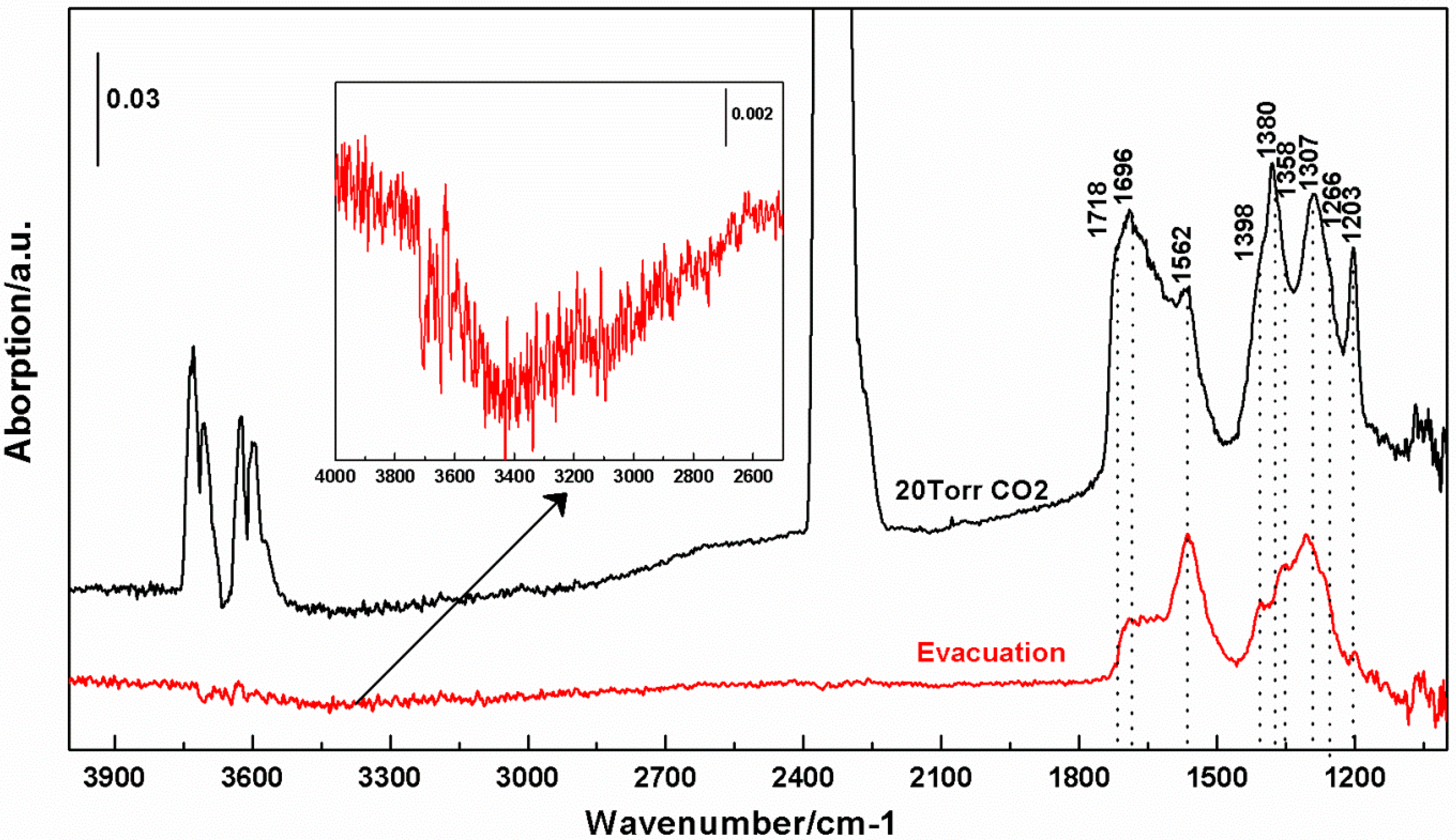
3.3. Interactions of CO2 with cTiNT
- Bidentate carbonate (in cm−1): 1690, 1562, 1380, 1358, 1307
- Monodentate carbonate (in cm−1): 1266
- Bicarbonate (in cm−1): 1404, 1398, 1203
- The 1290 cm−1 absorption is likely a convolution of peaks including a bidentate carbonate reported at 1278 cm−1.
4. Experimental Section
4.1. Synthesis of TiNT, cTiNT, and in Situ Pretreatments
4.2. Characterization of the TiNTs
4.3. In Situ FT-IR Spectroscopy
5. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Kasuga, T.; Hiramatsu, M.; Hoson, A.; Sekino, T.; Niihara, K. Titania Nanotubes Prepared by Chemical Processing. Adv. Mater. 1999, 11, 1307–1311. [Google Scholar] [CrossRef]
- Kasuga, T.; Hiramatsu, M.; Hoson, A.; Sekino, T.; Niihara, K. Formation of titanium oxide nanotube. Langmuir 1998, 14, 3160–3163. [Google Scholar] [CrossRef]
- Du, G.H.; Chen, Q.; Che, R.C.; Yuan, Z.Y.; Peng, L.M. Preparation and structure analysis of titanium oxide nanotubes. Appl. Phys. Lett. 2001, 79, 3702–3704. [Google Scholar] [CrossRef]
- Chen, Q.; Zhou, W.Z.; Du, G.H.; Peng, L.M. Trititanate nanotubes made via a single alkali treatment. Adv. Mater. 2002, 14, 1208–1211. [Google Scholar] [CrossRef]
- Chen, Q.; Du, G.H.; Zhang, S.; Peng, L.M. The structure of trititanate nanotubes. Acta Crystallogr. Sect. B Struct. Sci. 2002, 58, 587–593. [Google Scholar] [CrossRef]
- Nakahira, A.; Kato, W.; Tamai, M.; Isshiki, T.; Nishio, K.; Aritani, H. Synthesis of nanotube from a layered H2Ti4O9·H2O in a hydrothermal treatment using various titania sources. J. Mater. Sci. 2004, 39, 4239–4245. [Google Scholar] [CrossRef]
- Poudel, B.; Wang, W.Z.; Dames, C.; Huang, J.Y.; Kunwar, S.; Wang, D.Z.; Banerjee, D.; Chen, G.; Ren, Z.F. Formation of crystallized titania nanotubes and their transformation into nanowires. Nanotechnology 2005, 16, 1935–1940. [Google Scholar] [CrossRef]
- Tsai, C.C.; Teng, H.S. Regulation of the physical characteristics of Titania nanotube aggregates synthesized from hydrothermal treatment. Chem. Mater. 2004, 16, 4352–4358. [Google Scholar] [CrossRef]
- Yoshida, R.; Suzuki, Y.; Yoshikawa, S. Effects of synthetic conditions and heat-treatment on the structure of partially ion-exchanged titanate nanotubes. Mater. Chem. Phys. 2005, 91, 409–416. [Google Scholar] [CrossRef]
- Vijayan, B.; Dimitrijevic, N.M.; Rajh, T.; Gray, K. Effect of Calcination Temperature on the Photocatalytic Reduction and Oxidation Processes of Hydrothermally Synthesized Titania Nanotubes. J. Phys. Chem. C 2010, 114, 12994–13002. [Google Scholar] [CrossRef]
- Suzuki, Y.; Yoshikawa, S. Synthesis and thermal analyses of TiO2-derived nanotubes prepared by the hydrothermal method. J. Mater. Res. 2004, 19, 982–985. [Google Scholar] [CrossRef]
- Bhattacharyya, K.; Danon, A.; Vijayan, B.K.; Gray, K.A.; Stair, P.C.; Weitz, E. Role of the Surface Lewis Acid and Base Sites in the Adsorption of CO2 on Titania Nanotubes and Platinized Titania Nanotubes: An in Situ FT-IR Study. J. Phys. Chem. C 2013, 117, 12661–12678. [Google Scholar] [CrossRef]
- Zhang, J.; Li, M.J.; Feng, Z.C.; Chen, J.; Li, C. UV Raman spectroscopic study on TiO2. I. Phase transformation at the surface and in the bulk. J. Phys. Chem. B 2006, 110, 927–935. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.Z.; Banfield, J.F. Thermodynamic analysis of phase stability of nanocrystalline titania. J. Mater. Chem. 1998, 8, 2073–2076. [Google Scholar] [CrossRef]
- Muscat, J.; Swamy, V.; Harrison, N.M. First-principles calculations of the phase stability of TiO2. Phys. Rev. B 2002, 65, 22412–22415. [Google Scholar] [CrossRef]
- Kumar, K.N.P. Growth of Rutile Crystallites during the Initial-Stage of Anatase-to-Rutile Transformation in Pure Titania and in Titania-Alumina Nanocomposites. Scr. Metall. Mater. 1995, 32, 873–877. [Google Scholar] [CrossRef]
- Ovenstone, J.; Yanagisawa, K. Effect of hydrothermal treatment of amorphous titania on the phase change from anatase to rutile during calcination. Chem. Mater. 1999, 11, 2770–2774. [Google Scholar] [CrossRef]
- Regonini, D.; Jaroenworaluck, A.; Stevens, R.; Bowen, C.R. Effect of heat treatment on the properties and structure of TiO2 nanotubes: Phase composition and chemical composition. Surf. Interface Anal. 2010, 42, 139–144. [Google Scholar] [CrossRef]
- Schulte, K.L.; DeSario, P.A.; Gray, K.A. Effect of crystal phase composition on the reductive and oxidative abilities of TiO2 nanotubes under UV and visible light. Appl. Catal. B Environ. 2010, 97, 354–360. [Google Scholar] [CrossRef]
- Albu, S.P.; Ghicov, A.; Aldabergenova, S.; Drechsel, P.; LeClere, D.; Thompson, G.E.; Macak, J.M.; Schmuki, P. Formation of Double-Walled TiO2 Nanotubes and Robust Anatase Membranes. Adv. Mater. 2008, 20, 4135–4139. [Google Scholar]
- Bhattacharyya, K.; Varma, S.; Tripathi, A.K.; Bharadwaj, S.R.; Tyagi, A.K. Effect of Vanadia Doping and Its Oxidation State on the Photocatalytic Activity of TiO2 for Gas-Phase Oxidation of Ethene. J. Phys. Chem. C 2008, 112, 19102–19112. [Google Scholar] [CrossRef]
- Sodergren, S.; Siegbahn, H.; Rensmo, H.; Lindstrom, H.; Hagfeldt, A.; Lindquist, S.E. Lithium intercalation in nanoporous anatase TiO2 studied with XPS. J. Phys. Chem. B 1997, 101, 3087–3090. [Google Scholar] [CrossRef]
- Li, J.; Zeng, H.C. Preparation of monodisperse Au/TiO2 nanocatalysts via self-assembly. Chem. Mater. 2006, 18, 4270–4277. [Google Scholar] [CrossRef]
- Finnie, K.S.; Cassidy, D.J.; Bartlett, J.R.; Woolfrey, J.L. IR Spectroscopy of Surface Water and Hydroxyl Species on Nanocrystalline TiO2 Films. Langmuir 2001, 17, 816–820. [Google Scholar] [CrossRef]
- Herzberg, G. Infrared and Raman Spectra; Van Nostrand Rheinhold Company Inc: New York, NY, USA, 1945. [Google Scholar]
- Busca, G.; Lorenzelli, V. Infrared Spectroscopic Identification os Species arising from reactive adsorption of carbon oxides on metal-oxide surfaces. Mater. Chem. 1982, 7, 89–126. [Google Scholar] [CrossRef]
- Li, J.; Tang, S.; Lu, L.; Zeng, H.C. Preparation of nanocomposites of metals, metal oxides, and carbon nanotubes via self-assembly. J. Am. Chem. Soc. 2007, 129, 9401–9409. [Google Scholar] [CrossRef] [PubMed]
- Suda, Y.; Morimoto, T. Molecularly Adsorbed H2O on the Bare Surface of TiO2 (Rutile). Langmuir 1987, 3, 786–788. [Google Scholar] [CrossRef]
- Nakamura, R.; Ueda, K.; Sato, S. In Situ Observation of the Photoenhanced Adsorption of Water on TiO2 Films by Surface-Enhanced IR Absorption Spectroscopy. Langmuir 2001, 17, 2298–2300. [Google Scholar] [CrossRef]
- Tsuchiya, H.; Macak, J.M.; Müller, L.; Kunze, J.; Müller, F.; Greil, P.; Virtanen, S.; Schmuki, P. Hydroxyapatite growth on anodic TiO2 nanotubes. J. Biomed. Mater. Res. Part A 2006, 77A, 534–541. [Google Scholar] [CrossRef] [PubMed]
- Wu, W.; Bhattacharyya, K.; Gray, K.; Weitz, E. Photoinduced Reactions of Surface-Bound Species on Titania Nanotubes and Platinized Titania Nanotubes: An in Situ FTIR Study. J. Phys. Chem. C 2013, 117, 20643–20655. [Google Scholar] [CrossRef]
- Berglund, B.; Lindgren, J.; Tegenfeldt, J. O-H and O-D Stretching Vibrations in Isotopically Dilue HDO Molecules in Some Solid Hydrates. J. Mol. Struct. 1978, 43, 169–177. [Google Scholar] [CrossRef]
- Nakamoto, K.; Margoshes, M.; Rundle, R.E. Stretching Frequencies as a Function of Distances in Hydrogen Bonds. J. Am. Chem. Soc. 1955, 77, 6480–6486. [Google Scholar] [CrossRef]
- Ferrari, A.M.; Lessio, M.; Szieberth, D.; Maschil, L. On the Stability of Dititanate Nanotubes: A Density Functional Theory Study. J. Phys. Chem. C 2010, 114, 21219–21225. [Google Scholar] [CrossRef]
- Izawa, H.; Kikkawa, B.; Koizumi, M. Ion Exchange and Dehydration of Layered Titanates: Na2Ti3O7 and K2Ti4O9. J. Phys. Chem. 1982, 86, 5023–5026. [Google Scholar] [CrossRef]
- Banwell, C.N. Fundamentals of Molecular Spectroscopy; McGraw Hill Book Company Ltd: London, UK, 1972. [Google Scholar]
- Miller, K.L.; Faconer, J.L.; Medin, J.W. Effect of water on the adsorbed structure of formic acid on TiO2 anatase (101). J. Catal. 2011, 278, 321–328. [Google Scholar] [CrossRef]
- Nanayakkara, C.E.; Dillon, J.K.; Grassian, V.H. Surface Adsorption and Photochemistry of Gas-Phase Formic Acid on TiO2 Nanoparticles: The Role of Adsorbed Water in Surface Coordination, Adsorption Kinetics, and Rate of Photoproduct Formation. J. Phys. Chem. C 2014, 118, 25487–25495. [Google Scholar] [CrossRef]
- Kim, S.Y.; van Duin, A.C.T.; Kubicki, J.D. Molecular dynamics simulations of the interactions between TiO2 nanoparticles and water with Na+ and Cl−, methanol, and formic acid using a reactive force field. J. Mater. Res. 2012, 28, 513–520. [Google Scholar] [CrossRef]
- Baiju, K.V.; Shukla, S.; Biju, S.; Reddy, M.L.P.; Warrier, K.G.K. Hydrothermal processing of dye-adsorbing one-dimensional hydrogen titanate. Mater. Lett. 2009, 63, 923–926. [Google Scholar] [CrossRef]
- Chung, J.S.; Miranda, R.; Bennett, C. Study of Methanol and Water Chemisorbed on Molybdenum Oxide. J. Chem. Soc. Faraday Trans. 1985, 81, 19–36. [Google Scholar] [CrossRef]
- Hind, A.A.; Grassian, V.H. FT-IR Study of Water Adsorption on Aluminum Oxide Surfaces. Langmuir 2003, 19, 341–347. [Google Scholar]
- Dimitrijevic, N.D.; Vijayan, B.K.; Poluektov, O.G.; Rajh, R.; Gray, K.A.; He, H.; Zapol, P. Role of Water and Carbonates in Photocatalytic Transformation of CO2 to CH4 on Titania. J. Am. Chem. Soc. 2011, 133, 3964–3971. [Google Scholar] [CrossRef] [PubMed]
- Samples Availability: The methods by which material samples were synthesized are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bhattacharyya, K.; Wu, W.; Weitz, E.; Vijayan, B.K.; Gray, K.A. Probing Water and CO2 Interactions at the Surface of Collapsed Titania Nanotubes Using IR Spectroscopy. Molecules 2015, 20, 15469-15487. https://doi.org/10.3390/molecules200915469
Bhattacharyya K, Wu W, Weitz E, Vijayan BK, Gray KA. Probing Water and CO2 Interactions at the Surface of Collapsed Titania Nanotubes Using IR Spectroscopy. Molecules. 2015; 20(9):15469-15487. https://doi.org/10.3390/molecules200915469
Chicago/Turabian StyleBhattacharyya, Kaustava, Weiqiang Wu, Eric Weitz, Baiju K. Vijayan, and Kimberly A. Gray. 2015. "Probing Water and CO2 Interactions at the Surface of Collapsed Titania Nanotubes Using IR Spectroscopy" Molecules 20, no. 9: 15469-15487. https://doi.org/10.3390/molecules200915469
APA StyleBhattacharyya, K., Wu, W., Weitz, E., Vijayan, B. K., & Gray, K. A. (2015). Probing Water and CO2 Interactions at the Surface of Collapsed Titania Nanotubes Using IR Spectroscopy. Molecules, 20(9), 15469-15487. https://doi.org/10.3390/molecules200915469