
Anti-Tumor Effects of Bak-Proteoliposomes against Glioblastoma


Abstract
:1. Introduction
2. Results and Discussion
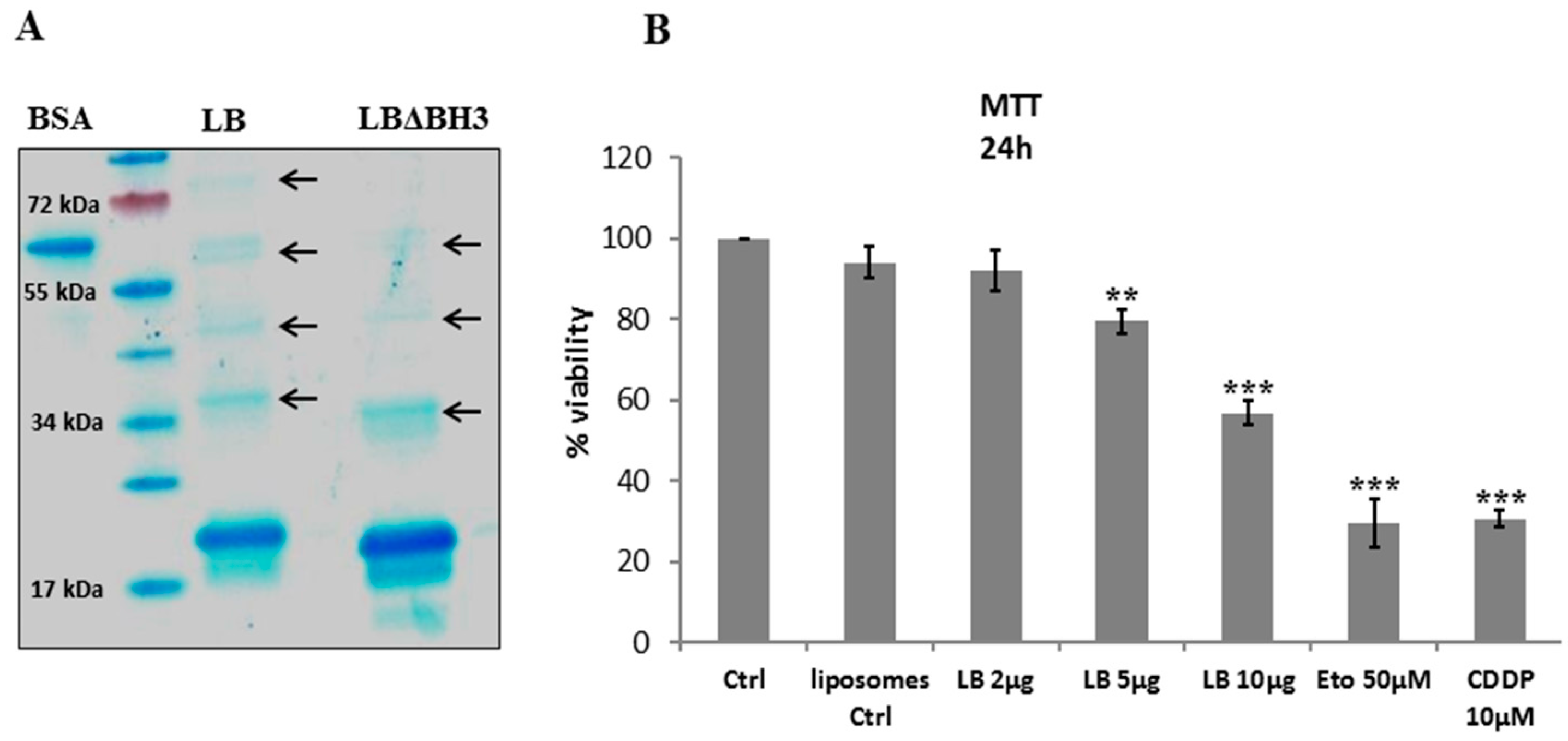
2.1. LB Production and Cell Viability

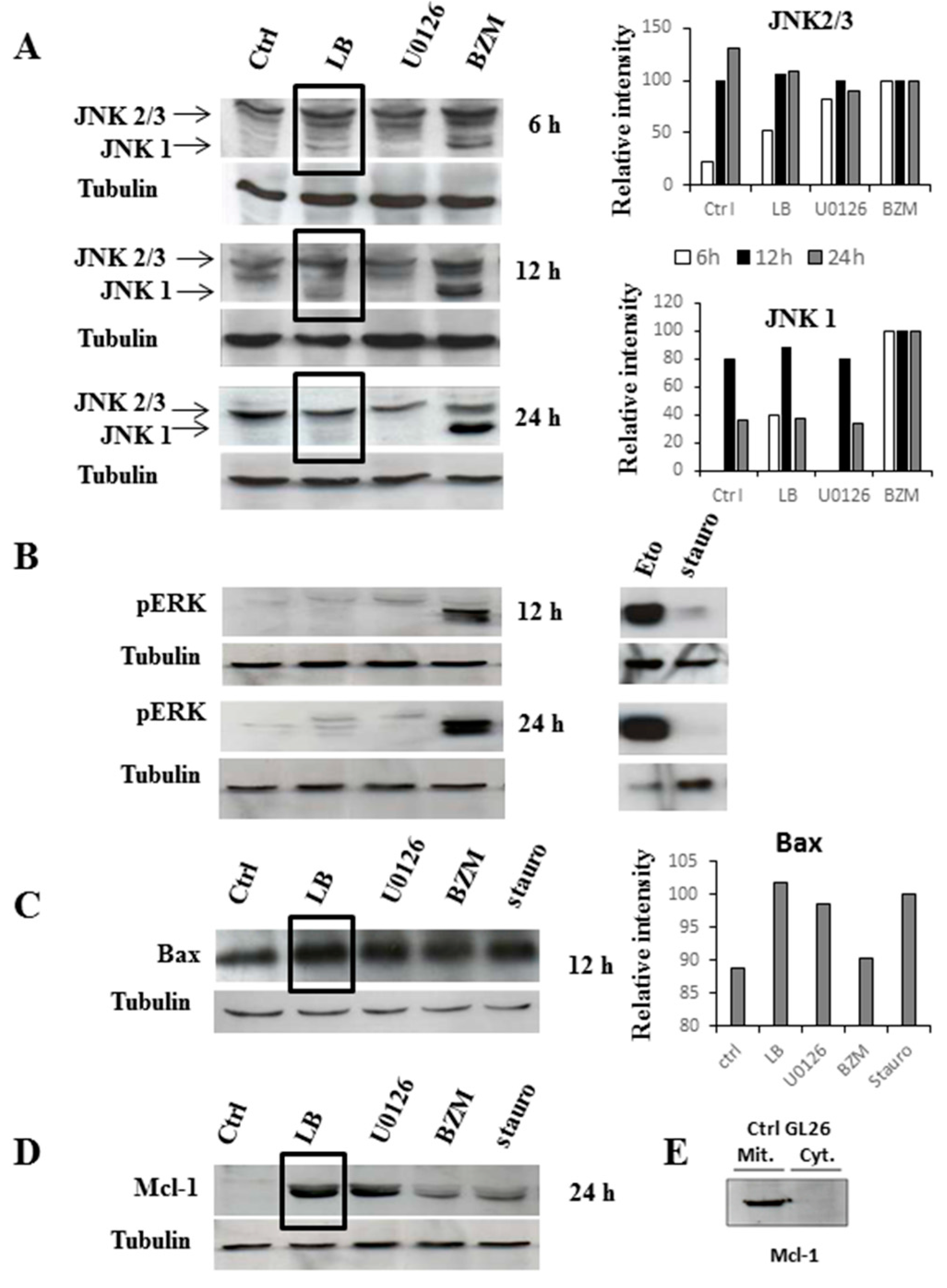
2.2. Western Blotting on GL26 Cells

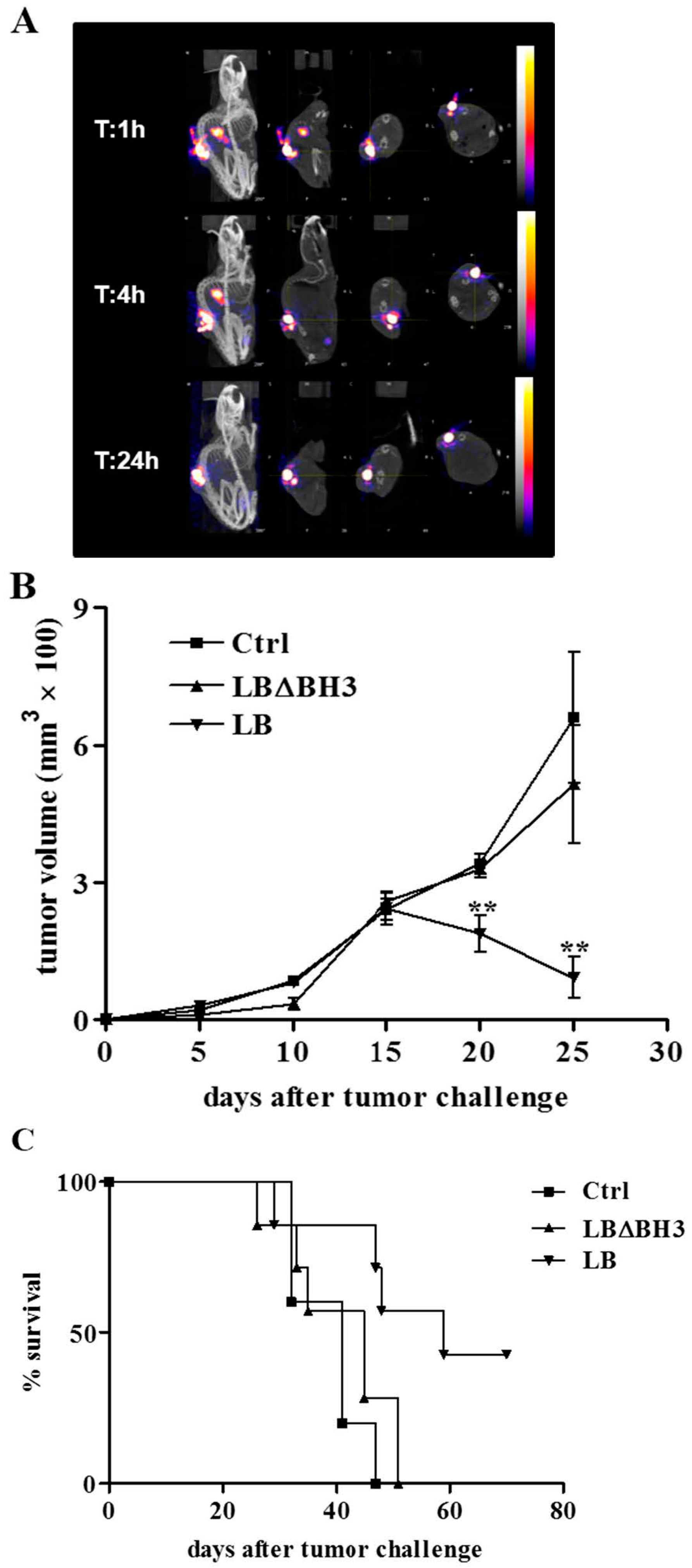
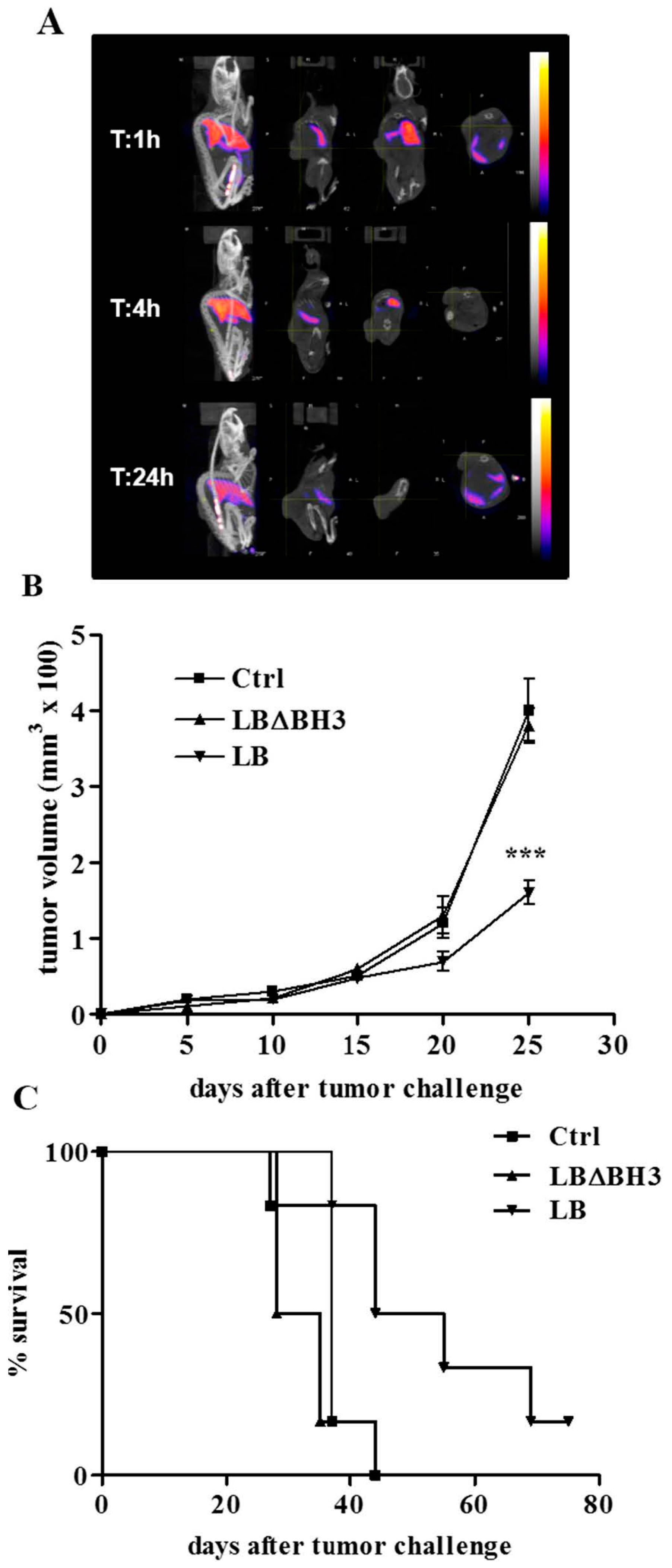
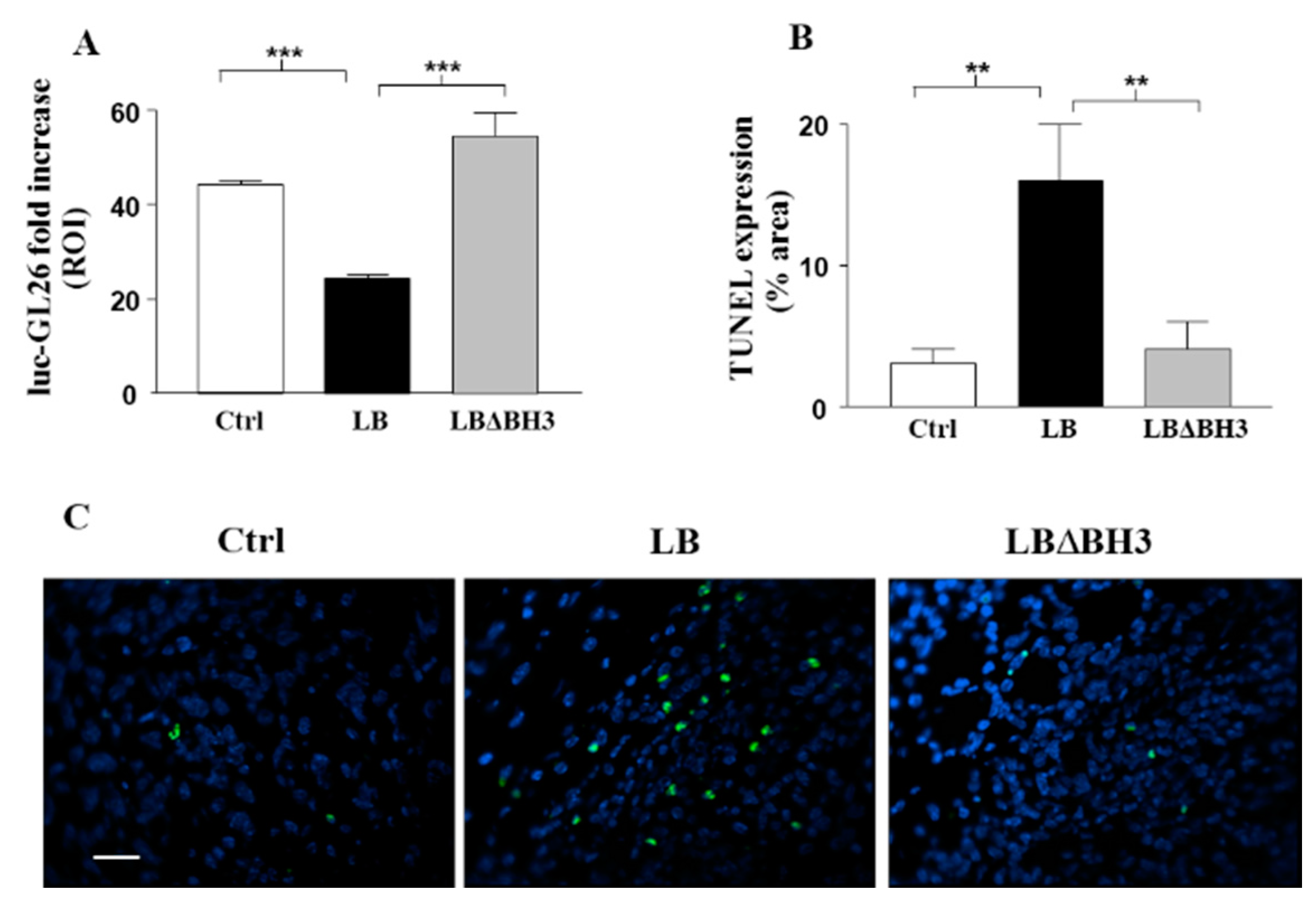
2.3. Biodistribution and Anti-Tumor Effects of LB



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Organ | Group A Intra-Tumor Injection | Group B Intravenous Injection |
|---|---|---|
| Brain | 0.005 ± 0.009 | 0.030 ± 0.006 |
| Heart | 0.004 ± 0.003 | 0.282 ± 0.057 |
| Stomach | 0.112 ± 0.007 | 0.975 ± 0.135 |
| Liver | 0.443 ± 0.630 | 75.655 ± 14.376 |
| Sal. Gland | 0.007 ± 0.005 | 0.605 ± 0.156 |
| Fat | 0.004 ± 0.003 | 0.327 ± 0.143 |
| Intestine | 0.015 ± 0.003 | 0.932 ± 0.149 |
| Muscle | 0.002 ± 0.001 | 0.103 ± 0.023 |
| Skin | 0.007 ± 0.003 | 0.685 ± 0.718 |
| Lung | 0.152 ± 0.254 | 25.655 ± 11.674 |
| Spleen | 0.342 ± 0.473 | 45.252 ± 4.626 |
| Kidney | 0.221 ± 0.104 | 6.032 ± 1.007 |
| Blood | 0.014 ± 0.005 | 0.698 ± 0.115 |
| Thyroid | 0.032 ± 0.033 | 5.838 ± 4.368 |
| Urine | 0.563 ± 0.350 | 41.202 ± 9.209 |
| Tumor | 164.817 ± 101.445 | 0.538 ± 0.219 |
2.4. Apoptosis on Isolated Tumors: TUNEL Assay

3. Experimental Section
3.1. Cell-Free Production and Purification of Proteoliposomes
3.2. Cell Line and Treatments
3.3. Animal Model
3.4. Biodistribution Analysis
3.5. In Vivo Therapeutic Studies
3.6. TUNEL Assay on Tumor Sections
3.7. Statistics
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chang, S.M.; Wen, P.; Cloughesy, T.; Greenberg, H.; Schiff, D.; Conrad, C.; Fink, K.; Robins, H.I.; de Angelis, L.; Raizer, J.; et al. Phase II study of CCI-779 in patients with recurrent glioblastoma multiforme. Investig. New Drugs 2005, 23, 357–361. [Google Scholar] [CrossRef]
- Stupp, R.; Hottinger, A.F.; van den Bent, M.J.; Dietrich, P.Y.; Brandes, A.A. Frequently asked questions in the medical management of high-grade glioma: A short guide with practical answers. Ann. Oncol. 2008, 19 (Suppl. 7), vii209–vii216. [Google Scholar] [CrossRef] [PubMed]
- Mitra, A.; Mishra, L.; Li, S. EMT, CTCs and CSCs in tumor relapse and drug-resistance. Oncotarget 2015, 6, 10697–10711. [Google Scholar] [PubMed]
- Soni, V.; Jain, A.; Khare, P.; Gulbake, A.; Jain, S.K. Potential approaches for drug delivery to the brain: Past, present, and future. Crit. Rev. Ther. Drug Carr. Syst. 2010, 27, 187–236. [Google Scholar] [CrossRef]
- Krakstad, C.; Chekenya, M. Survival signalling and apoptosis resistance in glioblastomas: Opportunities for targeted therapeutics. Mol. Cancer 2010, 9, 135. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Franco, V.I.; Henkel, J.M.; Miller, T.L.; Lipshultz, S.E. Cardiovascular effects in childhood cancer survivors treated with anthracyclines. Cardiol. Res. Pract. 2011, 2011, 134679. [Google Scholar] [CrossRef] [PubMed]
- Jiang, W.; Lionberger, R.; Yu, L.X. In vitro and in vivo characterizations of pegylated liposomal doxorubicin. Bioanalysis 2011, 3, 333–344. [Google Scholar] [CrossRef] [PubMed]
- ElBayoumi, T.A.; Torchilin, V.P. Tumor-targeted nanomedicines: Enhanced antitumor efficacy in vivo of doxorubicin-loaded, long-circulating liposomes modified with cancer-specific monoclonal antibody. Clin. Cancer Res. 2009, 15, 1973–1980. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Antibody-modified liposomes for cancer chemotherapy. Expert Opin. Drug Deliv. 2008, 5, 1003–1025. [Google Scholar] [CrossRef] [PubMed]
- Torchilin, V. Tumor delivery of macromolecular drugs based on the epr effect. Adv. Drug Deliv. Rev. 2011, 63, 131–135. [Google Scholar] [CrossRef] [PubMed]
- Dubois, L.G.; Campanati, L.; Righy, C.; D’Andrea-Meira, I.; Spohr, T.C.; Porto-Carreiro, I.; Pereira, C.M.; Balca-Silva, J.; Kahn, S.A.; DosSantos, M.F.; et al. Gliomas and the vascular fragility of the blood brain barrier. Front. Cell. Neurosci 2014, 8, 418. [Google Scholar] [CrossRef] [PubMed]
- Leber, B.; Lin, J.; Andrews, D.W. Still embedded together binding to membranes regulates Bcl-2 protein interactions. Oncogene 2010, 29, 5221–5230. [Google Scholar] [CrossRef] [PubMed]
- Reed, J.C. Proapoptotic multidomain bcl-2/bax-family proteins: Mechanisms, physiological roles, and therapeutic opportunities. Cell Death Differ. 2006, 13, 1378–1386. [Google Scholar] [CrossRef] [PubMed]
- Spirin, A.S. High-throughput cell-free systems for synthesis of functionally active proteins. Trends Biotechnol. 2004, 22, 538–545. [Google Scholar] [CrossRef] [PubMed]
- Swartz, J.R.; Jewett, M.C.; Woodrow, K.A. Cell-free protein synthesis with prokaryotic combined transcription-translation. Methods Mol. Biol. 2004, 267, 169–182. [Google Scholar] [PubMed]
- Liguori, L.; Marques, B.; Villegas-Mendez, A.; Rothe, R.; Lenormand, J.L. Liposomes-mediated delivery of pro-apoptotic therapeutic membrane proteins. J. Control. Release 2008, 126, 217–227. [Google Scholar] [CrossRef] [PubMed]
- Louis, D.N.; Ohgaki, H.; Wiestler, O.D.; Cavenee, W.K.; Burger, P.C.; Jouvet, A.; Scheithauer, B.W.; Kleihues, P. The 2007 who classification of tumours of the central nervous system. Acta Neuropathol. 2007, 114, 97–109. [Google Scholar] [CrossRef] [PubMed]
- Van Meir, E.G.; Hadjipanayis, C.G.; Norden, A.D.; Shu, H.K.; Wen, P.Y.; Olson, J.J. Exciting new advances in neuro-oncology: The avenue to a cure for malignant glioma. CA Cancer J. Clin. 2010, 60, 166–193. [Google Scholar] [CrossRef] [PubMed]
- Indran, I.R.; Tufo, G.; Pervaiz, S.; Brenner, C. Recent advances in apoptosis, mitochondria and drug resistance in cancer cells. Biochim. Biophys. Acta 2011, 1807, 735–745. [Google Scholar] [CrossRef] [PubMed]
- Liguori, L.; Lenormand, J.L. Production of recombinant proteoliposomes for therapeutic uses. Methods Enzymol. 2009, 465, 209–223. [Google Scholar] [PubMed]
- Csibi, A.; Fendt, S.M.; Li, C.; Poulogiannis, G.; Choo, A.Y.; Chapski, D.J.; Jeong, S.M.; Dempsey, J.M.; Parkhitko, A.; Morrison, T.; et al. The mTORC1 pathway stimulates glutamine metabolism and cell proliferation by repressing SIRT4. Cell 2013, 153, 840–854. [Google Scholar] [CrossRef] [PubMed]
- Allen, C.; Saigal, K.; Nottingham, L.; Arun, P.; Chen, Z.; van Waes, C. Bortezomib-induced apoptosis with limited clinical response is accompanied by inhibition of canonical but not alternative nuclear factor-κB subunits in head and neck cancer. Clin. Cancer Res. 2008, 14, 4175–4185. [Google Scholar] [CrossRef] [PubMed]
- Karpinich, N.O.; Tafani, M.; Rothman, R.J.; Russo, M.A.; Farber, J.L. The course of etoposide-induced apoptosis from damage to DNA and p53 activation to mitochondrial release of cytochromec. J. Biol. Chem. 2002, 277, 16547–16552. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.D.; Gillespie, S.K.; Hersey, P. Staurosporine induces apoptosis of melanoma by both caspase-dependent and -independent apoptotic pathways. Mol. Cancer Ther. 2004, 3, 187–197. [Google Scholar] [PubMed]
- Moll, U.M.; Wolff, S.; Speidel, D.; Deppert, W. Transcription-independent pro-apoptotic functions of p53. Curr. Opin. Cell Biol. 2005, 17, 631–636. [Google Scholar] [CrossRef] [PubMed]
- Tianhu, Z.; Shiguang, Z.; Xinghan, L. Bmf is upregulated by PS-341-mediated cell death of glioma cells through JNK phosphorylation. Mol. Biol. Rep. 2010, 37, 1211–1219. [Google Scholar] [CrossRef] [PubMed]
- Li, J.Y.; Wang, H.; May, S.; Song, X.; Fueyo, J.; Fuller, G.N. Constitutive activation of C-Jun N-terminal kinase correlates with histologic grade and EGFR expression in diffuse gliomas. J. Neurooncol. 2008, 88, 11–17. [Google Scholar] [CrossRef] [PubMed]
- Powis, G.; Ihle, N.T.; Yung, W.K. Inhibiting PI-3-K for glioma therapy. Cell Cycle 2009, 8, 335–337. [Google Scholar] [CrossRef] [PubMed]
- Ihrlund, L.S.; Hernlund, E.; Viktorsson, K.; Panaretakis, T.; Barna, G.; Sabapathy, K.; Linder, S.; Shoshan, M.C. Two distinct steps of bak regulation during apoptotic stress signaling: Different roles of MEKK1 and JNK1. Exp. Cell Res. 2006, 312, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Stefanelli, C.; Tantini, B.; Fattori, M.; Stanic, I.; Pignatti, C.; Clo, C.; Guarnieri, C.; Caldarera, C.M.; Mackintosh, C.A.; Pegg, A.E.; et al. Caspase activation in etoposide-treated fibroblasts is correlated to ERK phosphorylation and both events are blocked by polyamine depletion. FEBS Lett. 2002, 527, 223–228. [Google Scholar] [CrossRef]
- Shankar, S.; Srivastava, R.K. Bax and Bak genes are essential for maximum apoptotic response by curcumin, a polyphenolic compound and cancer chemopreventive agent derived from turmeric, Curcuma longa. Carcinogenesis 2007, 28, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Gelinas, C.; White, E. BH3-only proteins in control: Specificity regulates MCL-1 and BAK-mediated apoptosis. Genes Dev. 2005, 19, 1263–1268. [Google Scholar] [CrossRef] [PubMed]
- Quinn, B.A.; Dash, R.; Azab, B.; Sarkar, S.; Das, S.K.; Kumar, S.; Oyesanya, R.A.; Dasgupta, S.; Dent, P.; Grant, S.; et al. Targeting MCL-1 for the therapy of cancer. Expert Opin. Investig. Drugs 2011, 20, 1397–1411. [Google Scholar] [CrossRef] [PubMed]
- Navon, A.; Ciechanover, A. The 26 S proteasome: From basic mechanisms to drug targeting. J. Biol. Chem. 2009, 284, 33713–33718. [Google Scholar] [CrossRef] [PubMed]
- Newman, M.S.; Colbern, G.T.; Working, P.K.; Engbers, C.; Amantea, M.A. Comparative pharmacokinetics, tissue distribution, and therapeutic effectiveness of cisplatin encapsulated in long-circulating, pegylated liposomes (SPI-077) in tumor-bearing mice. Cancer Chemother. Pharmacol. 1999, 43, 1–7. [Google Scholar] [CrossRef] [PubMed]
- McCaskill, J.; Singhania, R.; Burgess, M.; Allavena, R.; Wu, S.; Blumenthal, A.; McMillan, N.A. Efficient biodistribution and gene silencing in the lung epithelium via intravenous liposomal delivery of sirna. Mol. Ther. Nucleic Acids 2013, 2, e96. [Google Scholar] [CrossRef] [PubMed]
- Drummond, D.C.; Noble, C.O.; Hayes, M.E.; Park, J.W.; Kirpotin, D.B. Pharmacokinetics and in vivo drug release rates in liposomal nanocarrier development. J. Pharm. Sci. 2008, 97, 4696–4740. [Google Scholar] [CrossRef] [PubMed]
- Van Tellingen, O.; Yetkin-Arik, B.; de Gooijer, M.C.; Wesseling, P.; Wurdinger, T.; de Vries, H.E. Overcoming the blood-brain tumor barrier for effective glioblastoma treatment. Drug Resist. Updat. 2015, 19, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Dams, E.T.; Laverman, P.; Oyen, W.J.; Storm, G.; Scherphof, G.L.; van Der Meer, J.W.; Corstens, F.H.; Boerman, O.C. Accelerated blood clearance and altered biodistribution of repeated injections of sterically stabilized liposomes. J. Pharmacol. Exp. Ther. 2000, 292, 1071–1079. [Google Scholar] [PubMed]
- Loi, M.; di Paolo, D.; Soster, M.; Brignole, C.; Bartolini, A.; Emionite, L.; Sun, J.; Becherini, P.; Curnis, F.; Petretto, A.; et al. Novel phage display-derived neuroblastoma-targeting peptides potentiate the effect of drug nanocarriers in preclinical settings. J. Control. Release 2013, 170, 233–241. [Google Scholar] [CrossRef] [PubMed]
- Loi, M.; Marchio, S.; Becherini, P.; di Paolo, D.; Soster, M.; Curnis, F.; Brignole, C.; Pagnan, G.; Perri, P.; Caffa, I.; et al. Combined targeting of perivascular and endothelial tumor cells enhances anti-tumor efficacy of liposomal chemotherapy in neuroblastoma. J. Control. Release 2010, 145, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.Q.; Lv, Q.; Li, L.M.; Tang, X.J.; Li, F.Z.; Hu, Y.L.; Han, M. Glioma targeting and blood-brain barrier penetration by dual-targeting doxorubincin liposomes. Biomaterials 2013, 34, 5628–5639. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds are available from the authors.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Liguori, L.; Pastorino, F.; Rousset, X.; Alfano, S.; Cortes, S.; Emionite, L.; Daga, A.; Ponzoni, M.; Lenormand, J.-L. Anti-Tumor Effects of Bak-Proteoliposomes against Glioblastoma. Molecules 2015, 20, 15893-15909. https://doi.org/10.3390/molecules200915893
Liguori L, Pastorino F, Rousset X, Alfano S, Cortes S, Emionite L, Daga A, Ponzoni M, Lenormand J-L. Anti-Tumor Effects of Bak-Proteoliposomes against Glioblastoma. Molecules. 2015; 20(9):15893-15909. https://doi.org/10.3390/molecules200915893
Chicago/Turabian StyleLiguori, Lavinia, Fabio Pastorino, Xavier Rousset, Silvia Alfano, Sandra Cortes, Laura Emionite, Antonio Daga, Mirco Ponzoni, and Jean-Luc Lenormand. 2015. "Anti-Tumor Effects of Bak-Proteoliposomes against Glioblastoma" Molecules 20, no. 9: 15893-15909. https://doi.org/10.3390/molecules200915893
APA StyleLiguori, L., Pastorino, F., Rousset, X., Alfano, S., Cortes, S., Emionite, L., Daga, A., Ponzoni, M., & Lenormand, J. -L. (2015). Anti-Tumor Effects of Bak-Proteoliposomes against Glioblastoma. Molecules, 20(9), 15893-15909. https://doi.org/10.3390/molecules200915893