
Quantification of CH-π Interactions Using Calix[4]pyrrole Receptors as Model Systems



Abstract
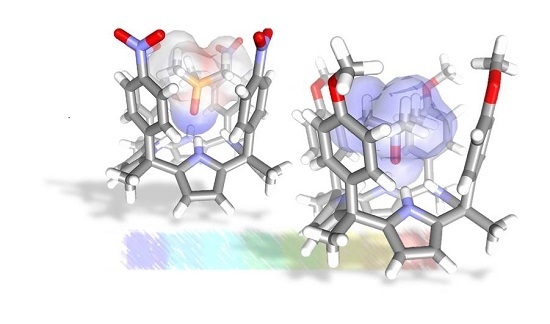
:
1. Introduction

2. Results and Discussion
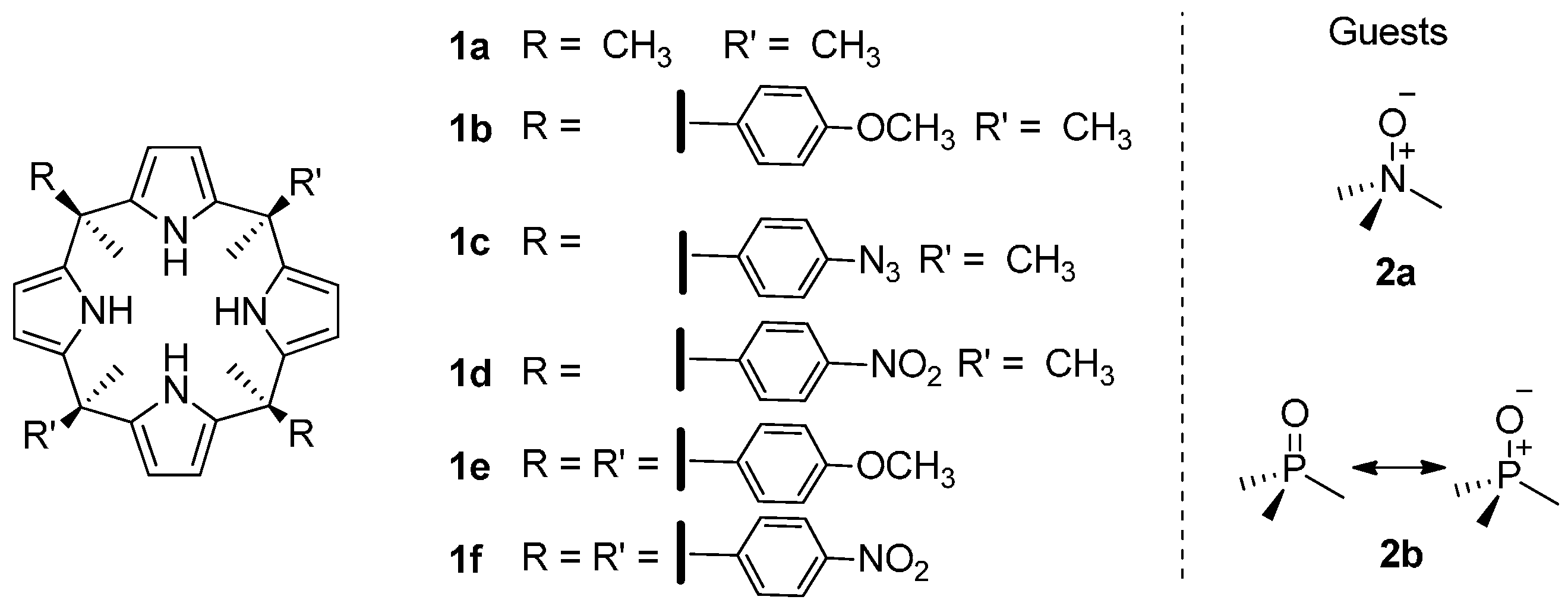
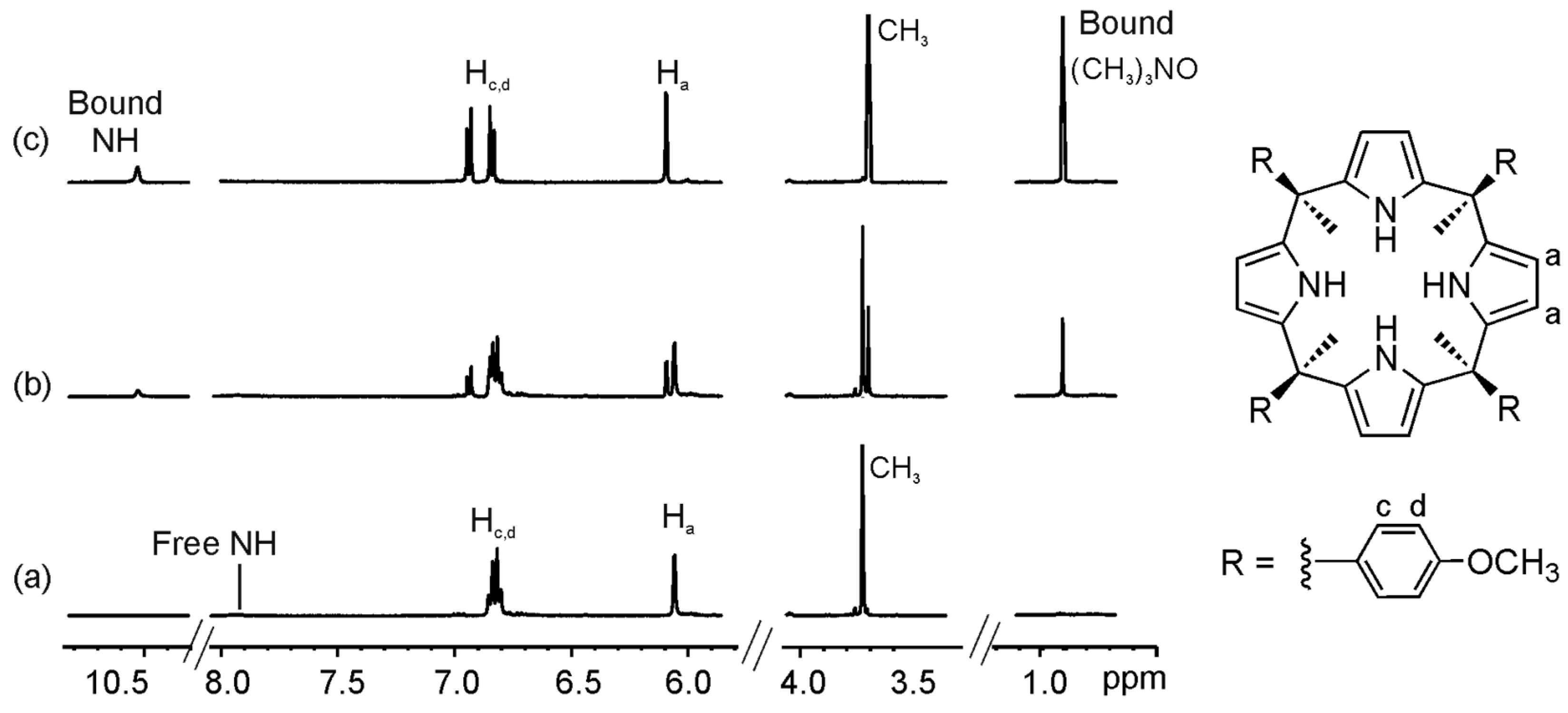
2.1. Binding of Octamethyl Calix[4]pyrrole 1a with Guests 2a and 2b. Definition of the Reference System

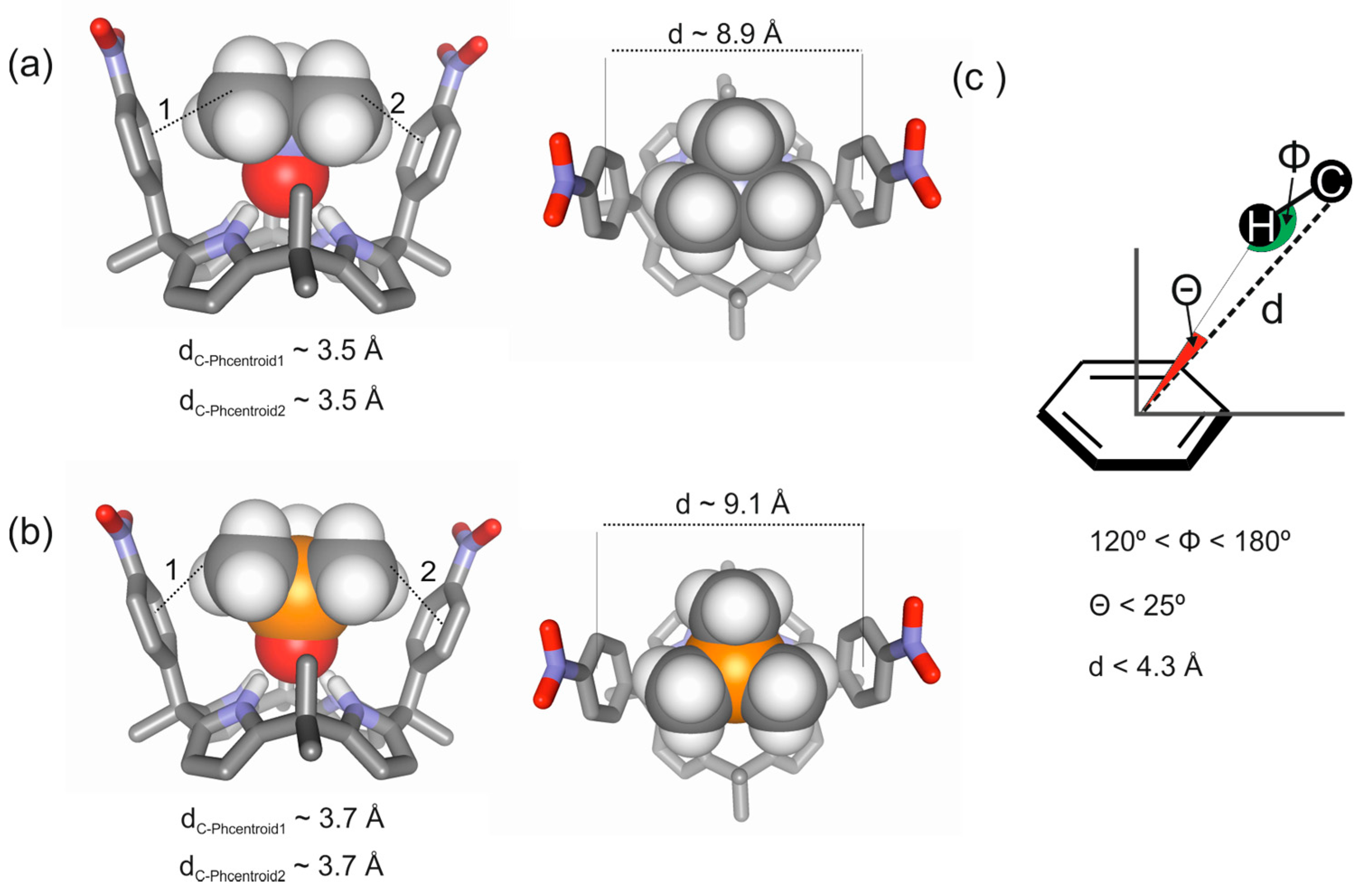
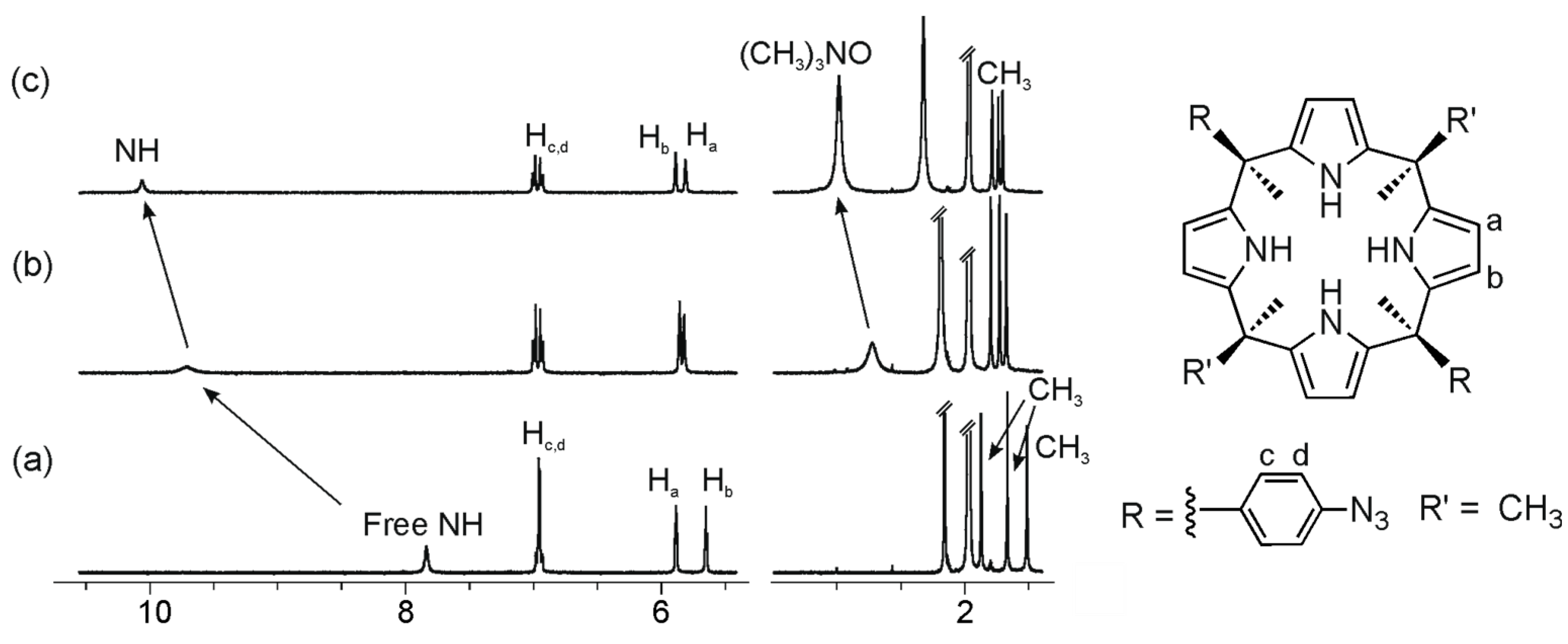
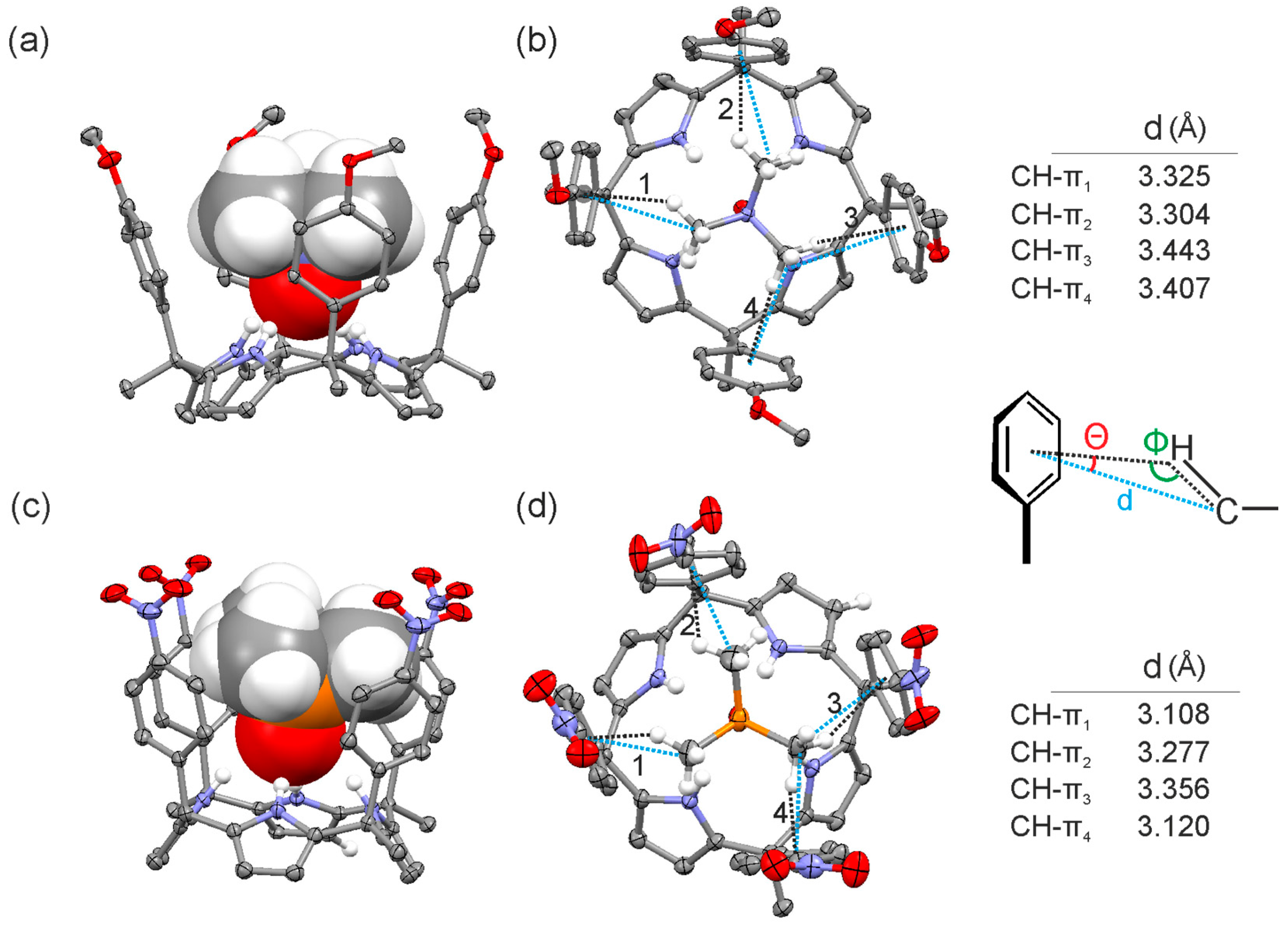
2.2. Binding of “Two-Wall” Calix[4]pyrroles with Guests 2a and 2b



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ESP a | 2a | 2b | |||||
|---|---|---|---|---|---|---|---|
| Ka | −ΔG | −ΔΔG | Ka | −ΔG | −ΔΔG | ||
| 1a | - | 7.22 ± 1.4 × 101 b | 2.53 | - | 8.91 ± 1.8 | 1.30 | - |
| 1b | −20.7 | 5.15 ± 1.3 × 103 b | 5.06 | 2.51 ± 0.19 | 8.21 ± 0.9 × 101 b | 2.61 | 1.32 ± 0.13 |
| 1c | −12.9 | 3.87 ± 0.4 × 103 b | 4.89 | 2.36 ± 0.12 | 7.00 ± 0.8 × 101 b | 2.52 | 1.22 ± 0.14 |
| 1d | −2.0 | 4.72 ± 0.9 × 103 b | 5.01 | 2.52 ± 0.16 | 7.98 ± 1.6 × 101 b | 2.59 | 1.30 ± 0.17 |
| 1e | −20.7 | 1.41 ± 0.1 × 105 c | 7.02 | 4.49 ± 0.12 | 9.60 ± 0.9 × 101 b | 2.70 | 1.41 ± 0.13 |
| 1f | −2.0 | 1.06 ± 0.1 × 105 c | 6.85 | 4.32 ± 0.12 | 2.57 ± 0.3 × 102 b | 3.29 | 2.00 ± 0.14 |

2.3. Binding Studies Using “Four-Wall” Calix[4]pyrrole Receptors and Guests 2a and 2b


3. Experimental Section
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References and Notes
- Nishio, M.; Hirota, M.; Umezawa, Y. The CH/Pi Interaction. Evidence, Nature, and Consequences; Wiley-VCH: New York, NY, USA, 1998. [Google Scholar]
- Tsuzuki, S.; Fujii, A. Nature and physical origin of ch/pi interaction: Significant difference from conventional hydrogen bonds. Phys. Chem. Chem. Phys. 2008, 10, 2584–2594. [Google Scholar] [CrossRef] [PubMed]
- In literature values between 0.5 to 2 kcal/mol have been described.
- Nishio, M. CH/π hydrogen bonds in crystals. CrystEngComm 2004, 6, 130–158. [Google Scholar] [CrossRef]
- Zondlo, N.J. Aromatic-proline interactions: Electronically tunable ch/π interactions. Acc. Chem. Res. 2013, 46, 1039–1049. [Google Scholar] [CrossRef] [PubMed]
- Nishio, M.; Umezawa, Y.; Fantini, J.; Weiss, M.S.; Chakrabarti, P. CH/π hydrogen bonds in biological macromolecules. Phys. Chem. Chem. Phys. 2014, 16, 12648–12683. [Google Scholar] [CrossRef] [PubMed]
- Ballester, P.; Biros, S.M. CH-π and π-π interactions as contributors to the guest binding in reversible inclusion and encapsulation complexes. In The Importance of Pi-Interactions in Crystal Engineering; John Wiley & Sons, Ltd: Chichester, West Sussex, UK, 2012; pp. 79–107. [Google Scholar]
- Andreetti, G.D.; Ungaro, R.; Pochini, A. Crystal and molecular-structure of cyclo(quarter[(5-tert-butyl-2-hydroxy-1,3-phenylene)methylene]) toluene (1-1) clathrate. J. Chem. Soc. Chem. Commun. 1979, 1005–1007. [Google Scholar] [CrossRef]
- Umezawa, Y.; Tsuboyama, S.; Takahashi, H.; Uzawa, J.; Nishio, M. Ch/pi interaction in the conformation of organic compounds. A database study. Tetrahedron 1999, 55, 10047–10056. [Google Scholar] [CrossRef]
- Paliwal, S.; Geib, S.; Wilcox, C.S. Chemistry of synthetic receptors and functional-group arrays 24. Molecular torsion balance for weak molecular recognition forces—Effects of tilted-t edge-to-face aromatic interactions on conformational selection and solid-state structure. J. Am. Chem. Soc. 1994, 116, 4497–4498. [Google Scholar] [CrossRef]
- Hof, F.; Scofield, D.M.; Schweizer, W.B.; Diederich, F. A weak attractive interaction between organic fluorine and an amide group. Angew. Chem. Int. Ed. 2004, 43, 5056–5059. [Google Scholar] [CrossRef] [PubMed]
- Kim, E.; Paliwal, S.; Wilcox, C.S. Measurements of molecular electrostatic field effects in edge-to-face aromatic interactions and CH-pi interactions with implications for protein folding and molecular recognition. J. Am. Chem. Soc. 1998, 120, 11192–11193. [Google Scholar] [CrossRef]
- Cockroft, S.L.; Hunter, C.A. Desolvation tips the balance: Solvent effects on aromatic interactions. Chem. Commun. 2006, 3806–3808. [Google Scholar] [CrossRef] [PubMed]
- Santana, A.G.; Jimenez-Moreno, E.; Gomez, A.M.; Corzana, F.; Gonzalez, C.; Jimenez-Oses, G.; Jimenez-Barbero, J.; Asensio, J.L. A dynamic combinatorial approach for the analysis of weak carbohydrate/aromatic complexes: Dissecting facial selectivity in CH/pi stacking interactions. J. Am. Chem. Soc. 2013, 135, 3347–3350. [Google Scholar] [CrossRef] [PubMed]
- Jimenez-Moreno, E.; Gomez, A.M.; Bastida, A.; Corzana, F.; Jimenez-Oses, G.; Jimenez-Barbero, J.; Asensio, J.L. Modulating weak interactions for molecular recognition: A dynamic combinatorial analysis for assessing the contribution of electrostatics to the stability of CH-pi bonds in water. Angew. Chem. Int. Ed. 2015, 54, 4344–4348. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ramirez, G.; Escudero-Adan, E.C.; Benet-Buchholz, J.; Ballester, P. Quantitative evaluation of anion-pi interactions in solution. Angew. Chem. Int. Ed. 2008, 47, 4114–4118. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, L.; Gil-Ramírez, G.; Frontera, A.; Quiñonero, D.; Escudero-Adán, E.C.; Ballester, P. Thermodynamic characterization of halide-π interactions in solution using “two-wall” aryl extended calix[4]pyrroles as model system. J. Am. Chem. Soc. 2014, 136, 3208–3218. [Google Scholar] [CrossRef] [PubMed]
- Adriaenssens, L.; Estarellas, C.; Jentzsch, A.V.; Belmonte, M.M.; Matile, S.; Ballester, P. Quantification of nitrate-pi interactions and selective transport of nitrate using calix[4]pyrroles with two aromatic walls. J. Am. Chem. Soc. 2013, 135, 8324–8330. [Google Scholar] [CrossRef] [PubMed]
- Gil-Ramírez, G.; Chas, M.; Ballester, P. Selective pairwise encapsulation using directional interactions. J. Am. Chem. Soc. 2010, 132, 2520–2521. [Google Scholar] [CrossRef] [PubMed]
- Hunter, C.A. Quantifying intermolecular interactions: Guidelines for the molecular recognition toolbox. Angew. Chem. Int. Ed. 2004, 43, 5310–5324. [Google Scholar] [CrossRef] [PubMed]
- The α and β values for pyrrole, and CH3CN can be found in literature, ref 20 of this manuscript. The corresponding α and β values for guests 2a and 2b were kindly provided to us by Prof. C.A. Hunter (University of Cambridge, personal communication).
- Spartan’02 (Wavefuntion, Inc., Irvine, CA, USA). The molecules were optimized by using AM1, and the maxima and minima in the molecular electrostatic potential surface were determined.
- Structures were energy minimized at the B3LYP/6-31G* level of theory using the Spartan’02 (Wavefuntion, Inc., Irvine, CA, USA) software. ESPs were computed at the same level of theory and plotted on the 0.002 isodensity surface corresponding to the van der Waals surface.
- The ESP values of the studied guest molecules and the aromatic rings were also calculated in a continuum model of acetonitrile. The minima ESP values obtained for the guests were located at the oxygen atom: 2a: −71.3 kcal·mol−1, 2b: −67.5 kcal·mol−1 In the case of the meso-phenyl substituents of the hosts we report the ESP values at their center: 1b: −24.3 kcal·mol−1, 1c: −13.2 kcal·mol−1 and 1d: −0.62 kcal·mol−1. As could be expected, there are small changes in the magnitudes of the ESP values calculated in vacuum or in an acetonitrile continuum solvent model, however both series of data followed an identical trend.
- Gans, P.; Sabatini, A.; Vacca, A. HypNMR 2008, version 4.0.66; Protonic Software GmbH: Hanau, Germany, 2006. [Google Scholar]
- Ahlrichs, R.; Bar, M.; Haser, M.; Horn, H.; Kolmel, C. Electronic-structure calculations on workstation computers—The program system turbomole. Chem. Phys. Lett. 1989, 162, 165–169. [Google Scholar] [CrossRef]
- It is worthy to point out that similar titrations were performed using calix[4]pyrroles with a positive ESP value at the center of the aromatic ring (e.g., pentafluorophenyl or 3,5-dinitro substituted aromatic walls). We decided not to include these results as accurate association constants determination was not possible for these systems due to the fact that other species with higher stoichiometry were detected when more than 1 equivalent was added, probably 1:2 complexes.
- These results also pointed out that the change in the pka of the pyrrolic NHs caused by the variation on the substituents of the aromatic rings does not have a significant influence on the binding constants as already suggested in other works (reference [17]).
- The slope of the plot of the free energy values vs. β described in ref 13 was determined considering that initially the calix[4]pyrrole is in the 1,3-alternate conformation with two acetonitrile molecules hydrogen bonded and thus establishing two CH-π interactions. Upon guest binding, the calix[4]pyrrole is locked in the cone conformation and the guest can engage in two CH-π interactions with the aromatic walls. Consequently, the slope (different from 0) is determined by (2 × α − 2 × αs) which is −1.7 and −1.2 for 2a and 2b, respectively.
- Bhayana, B.; Wilcox, C.S. A minimal protein folding model to measure hydrophobic and CH-pi effects on interactions between nonpolar surfaces in water. Angew. Chem. Int. Ed. 2007, 46, 6833–6836. [Google Scholar] [CrossRef] [PubMed]
- Bruno, G.; Cafeo, G.; Kohnke, F.H.; Nicolo, F. Tuning the anion binding properties of calixpyrroles by means of p-nitrophenyl substituents at their meso-positions. Tetrahedron 2007, 63, 10003–10010. [Google Scholar] [CrossRef]
- Park, J.S.; Yoon, K.Y.; Kim, D.S.; Lynch, V.M.; Bielawski, C.W.; Johnston, K.P.; Sessler, J.L. Chemoresponsive alternating supramolecular copolymers created from heterocomplementary calix[4]pyrroles. Proc. Natl. Acad. Sci. USA 2011, 108, 20913–20917. [Google Scholar] [CrossRef] [PubMed]
- Anzenbacher, P., Jr.; Jursikova, K.; Lynch, V.M.; Gale, P.A.; Sessler, J.L. Calix[4]pyrroles containing deep cavities and fixed walls. Synthesis, structural studies, and anion binding properties of the isomeric products derived from the condensation of p-hydroxyacetophenone and pyrrole. J. Am. Chem. Soc. 1999, 121, 11020–11021. [Google Scholar] [CrossRef]
- Wiseman, T.; Williston, S.; Brandts, J.F.; Lin, L.-N. Rapid measurement of binding constants and heats of binding using a new titration calorimeter. Anal. Biochem. 1989, 179, 131–137. [Google Scholar] [CrossRef]
- Sample Availability: Samples of the compounds 1a–1f, 2a and 2b are available from the authors. However, these compounds do not have an extended shelf stable and they need to be purified just before use.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aragay, G.; Hernández, D.; Verdejo, B.; Escudero-Adán, E.C.; Martínez, M.; Ballester, P. Quantification of CH-π Interactions Using Calix[4]pyrrole Receptors as Model Systems. Molecules 2015, 20, 16672-16686. https://doi.org/10.3390/molecules200916672
Aragay G, Hernández D, Verdejo B, Escudero-Adán EC, Martínez M, Ballester P. Quantification of CH-π Interactions Using Calix[4]pyrrole Receptors as Model Systems. Molecules. 2015; 20(9):16672-16686. https://doi.org/10.3390/molecules200916672
Chicago/Turabian StyleAragay, Gemma, Daniel Hernández, Begoña Verdejo, Eduardo C. Escudero-Adán, Marta Martínez, and Pablo Ballester. 2015. "Quantification of CH-π Interactions Using Calix[4]pyrrole Receptors as Model Systems" Molecules 20, no. 9: 16672-16686. https://doi.org/10.3390/molecules200916672
APA StyleAragay, G., Hernández, D., Verdejo, B., Escudero-Adán, E. C., Martínez, M., & Ballester, P. (2015). Quantification of CH-π Interactions Using Calix[4]pyrrole Receptors as Model Systems. Molecules, 20(9), 16672-16686. https://doi.org/10.3390/molecules200916672