
NHC Backbone Configuration in Ruthenium-Catalyzed Olefin Metathesis


Abstract
:1. Introduction

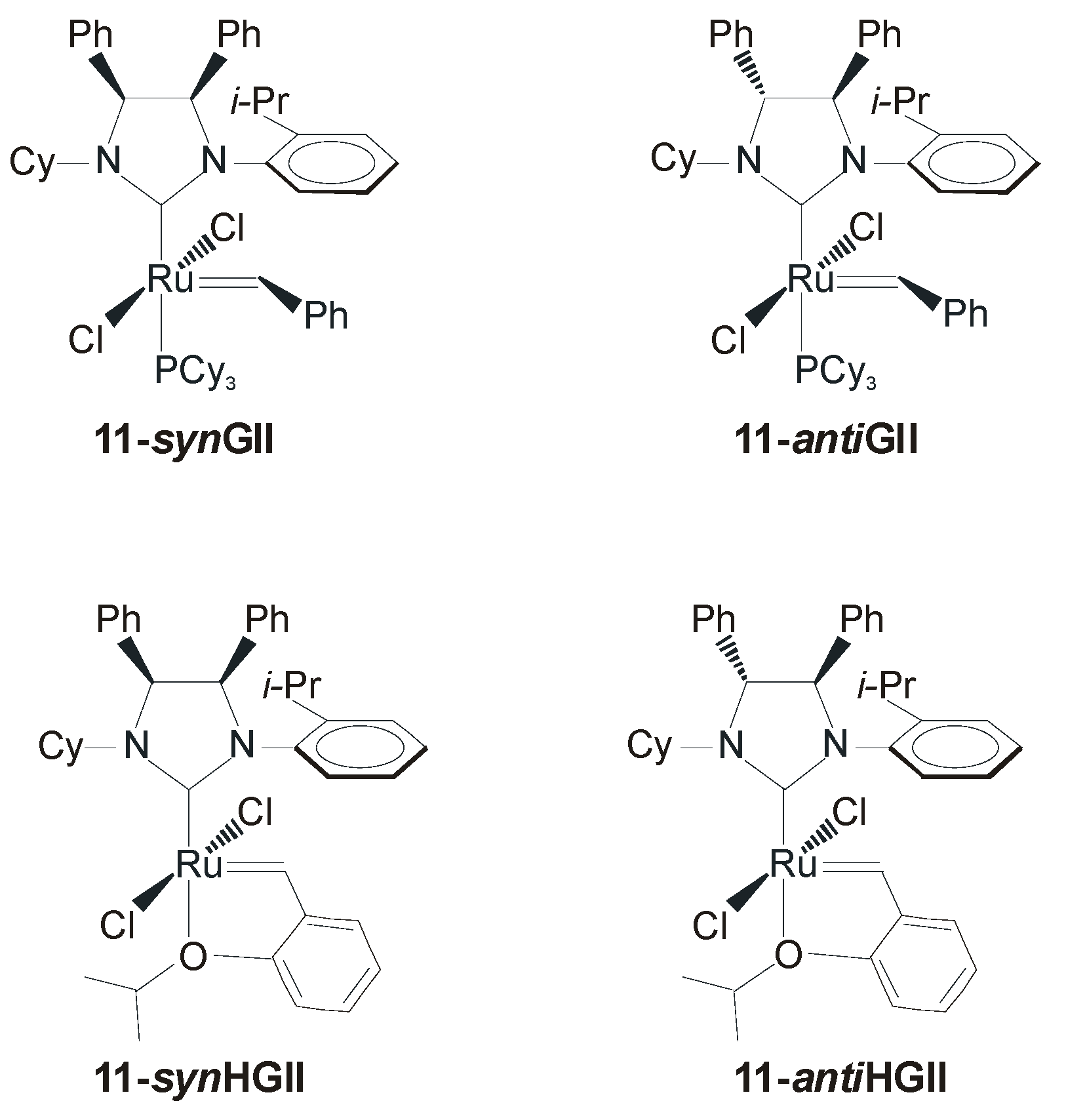
2. Ruthenium Catalysts Bearing Symmetrically N-Substituted NHCs with syn or anti Backbone Configurations
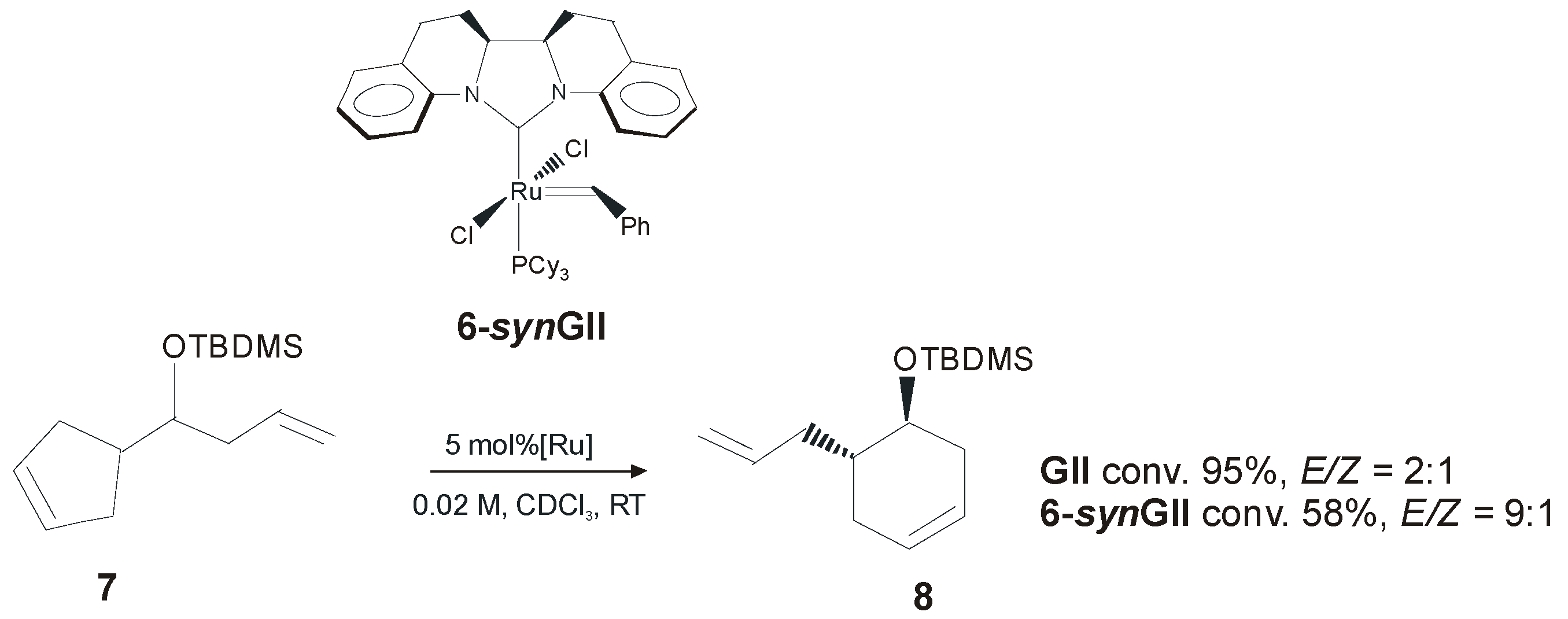
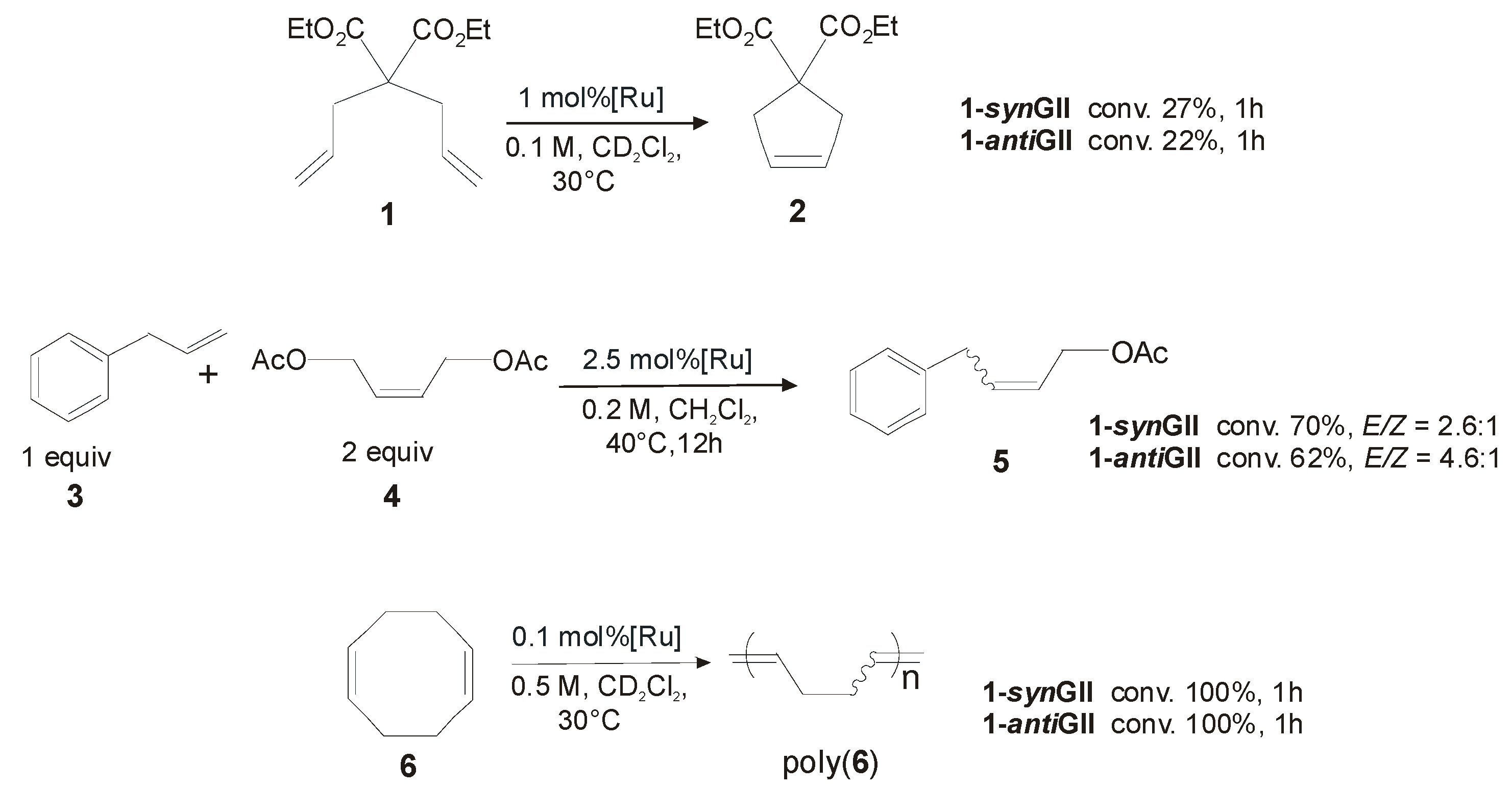
2.1. Non-Aromatic N-Substituents


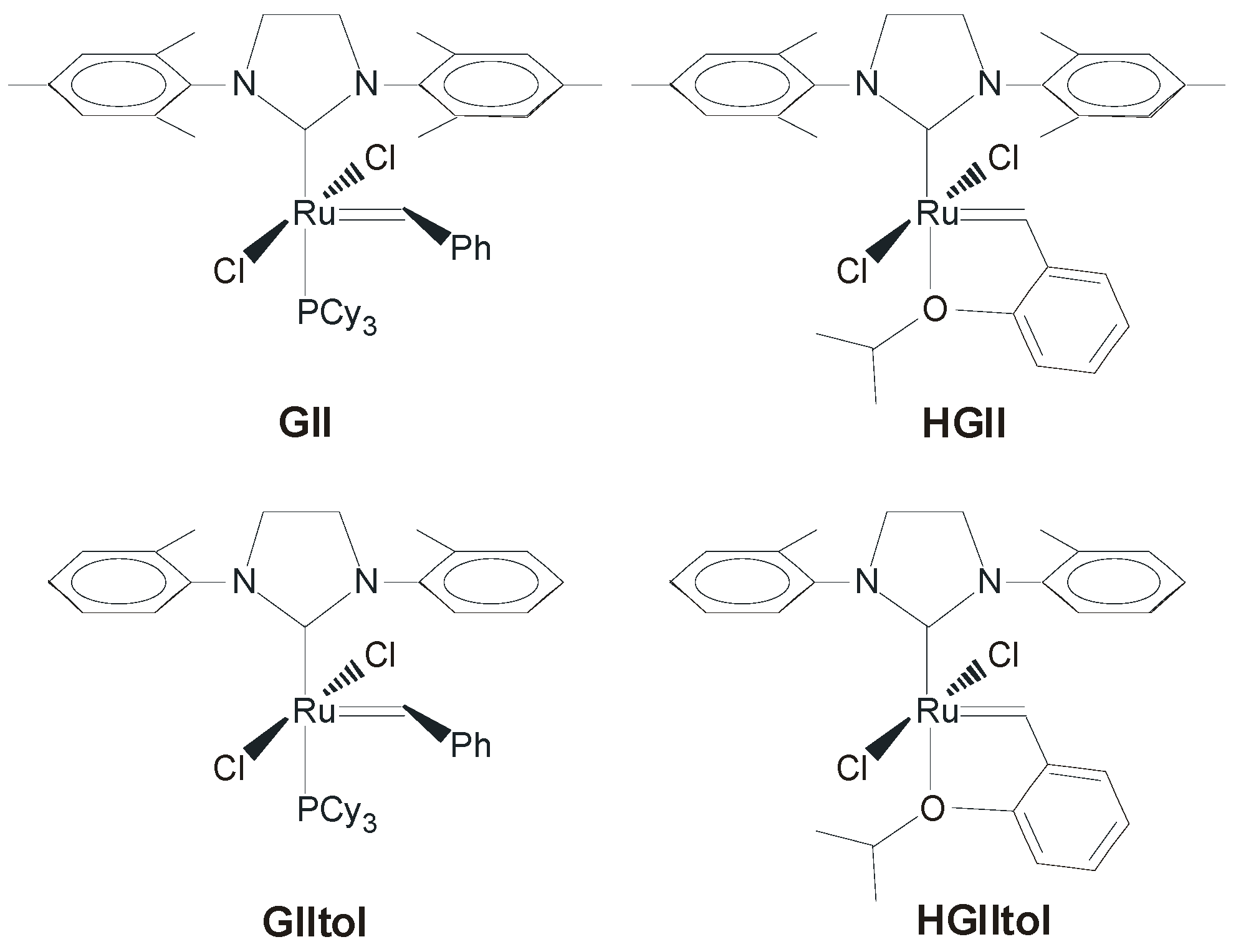
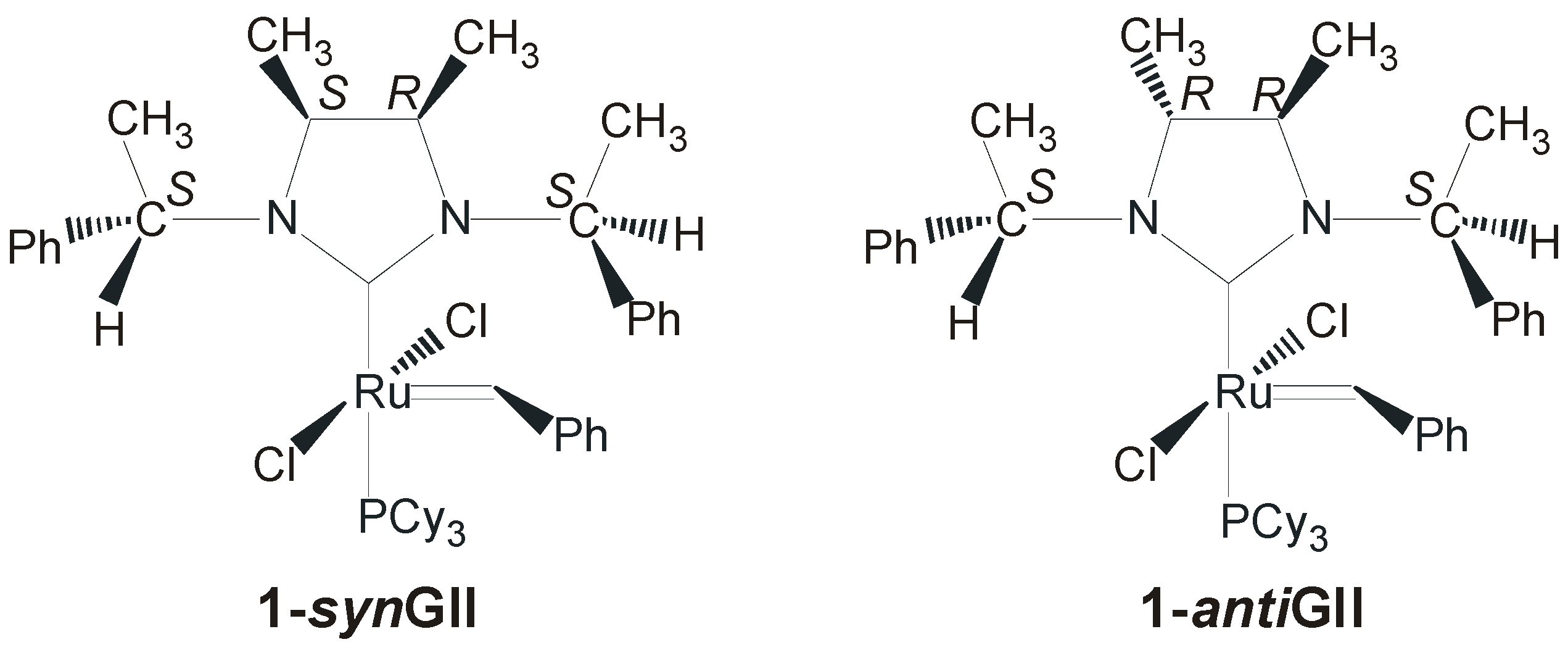
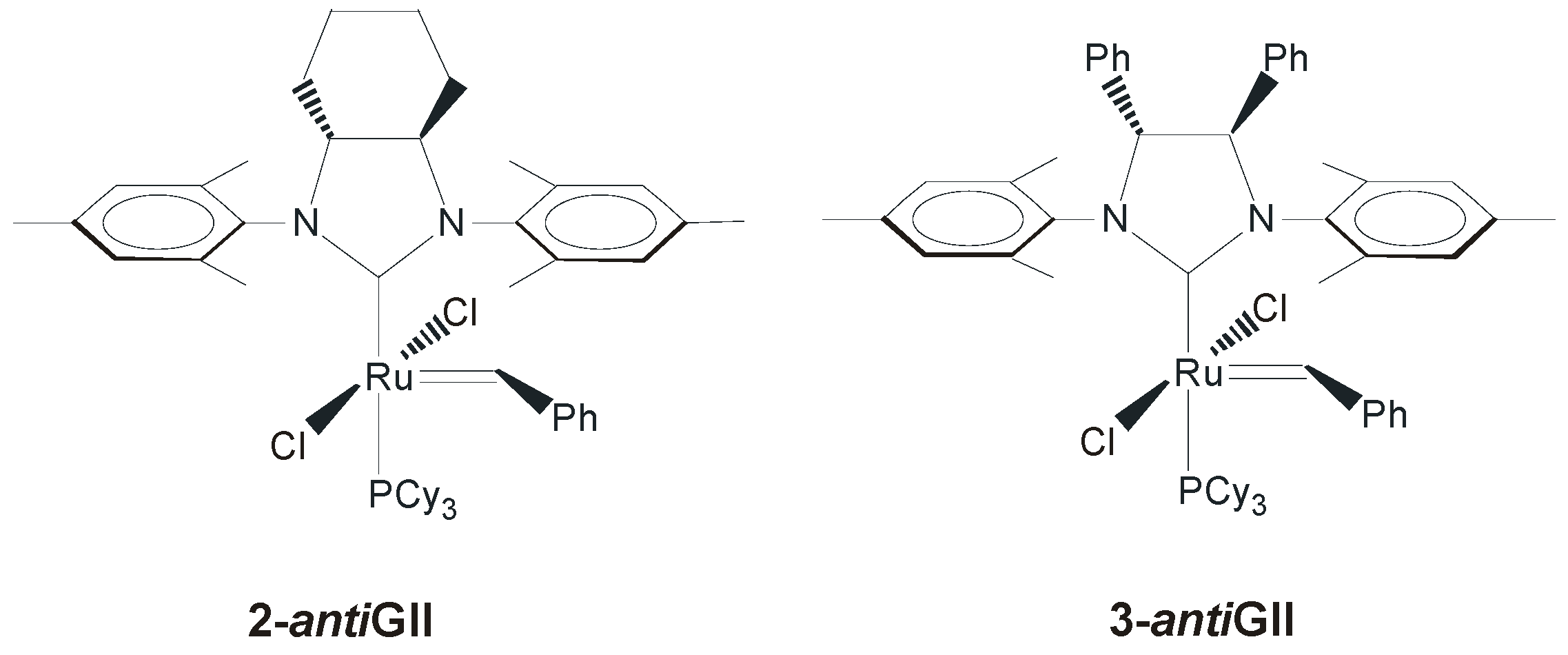
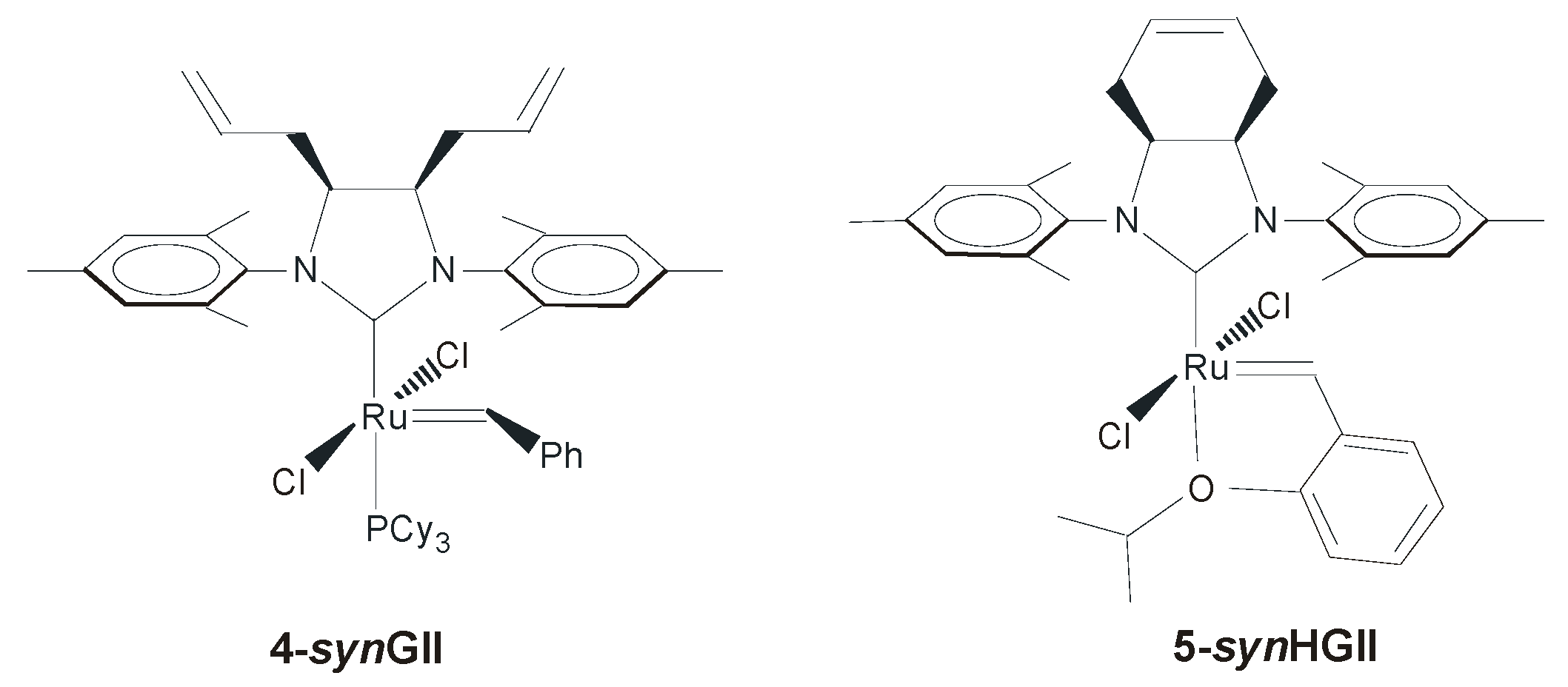
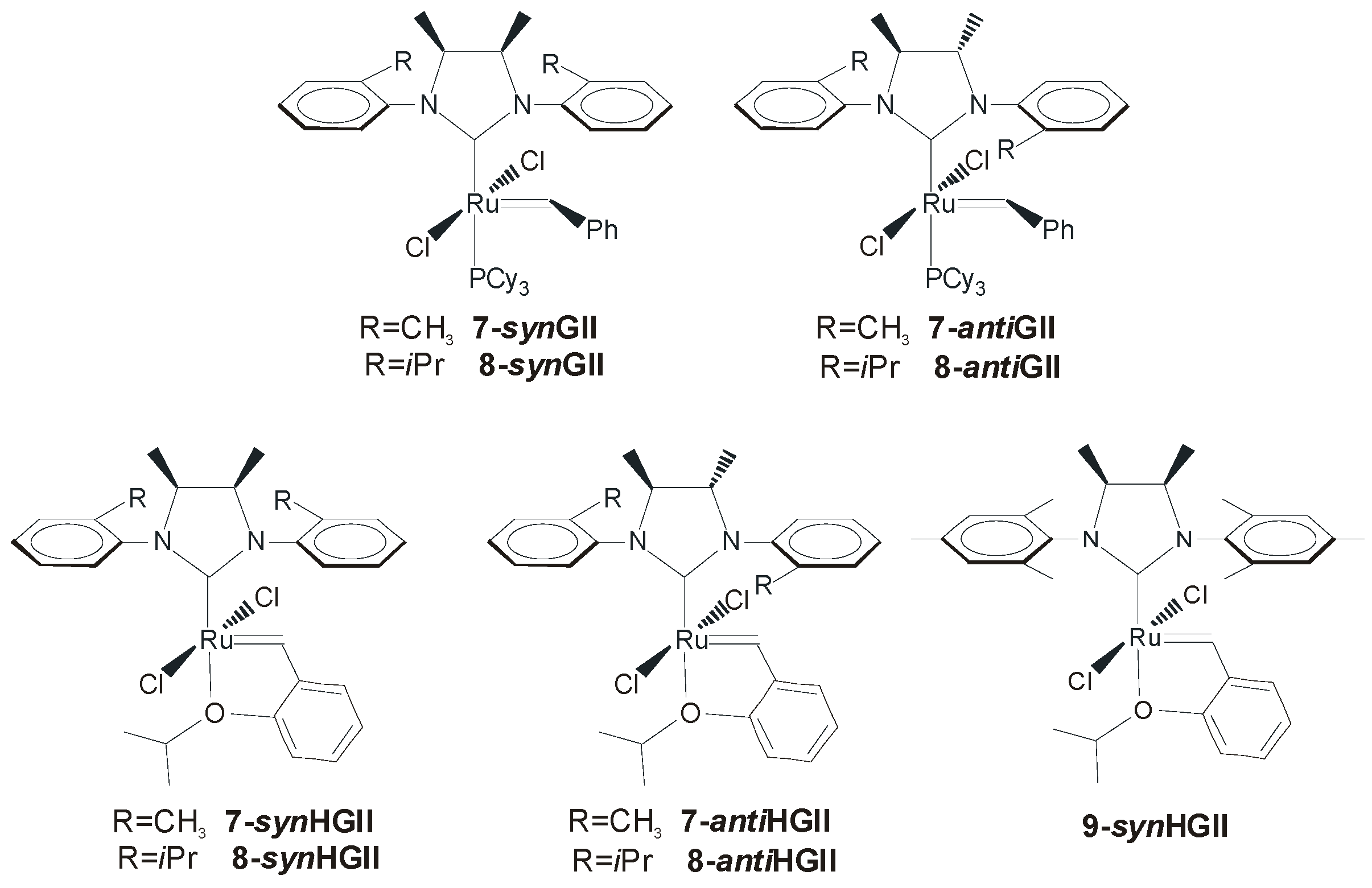
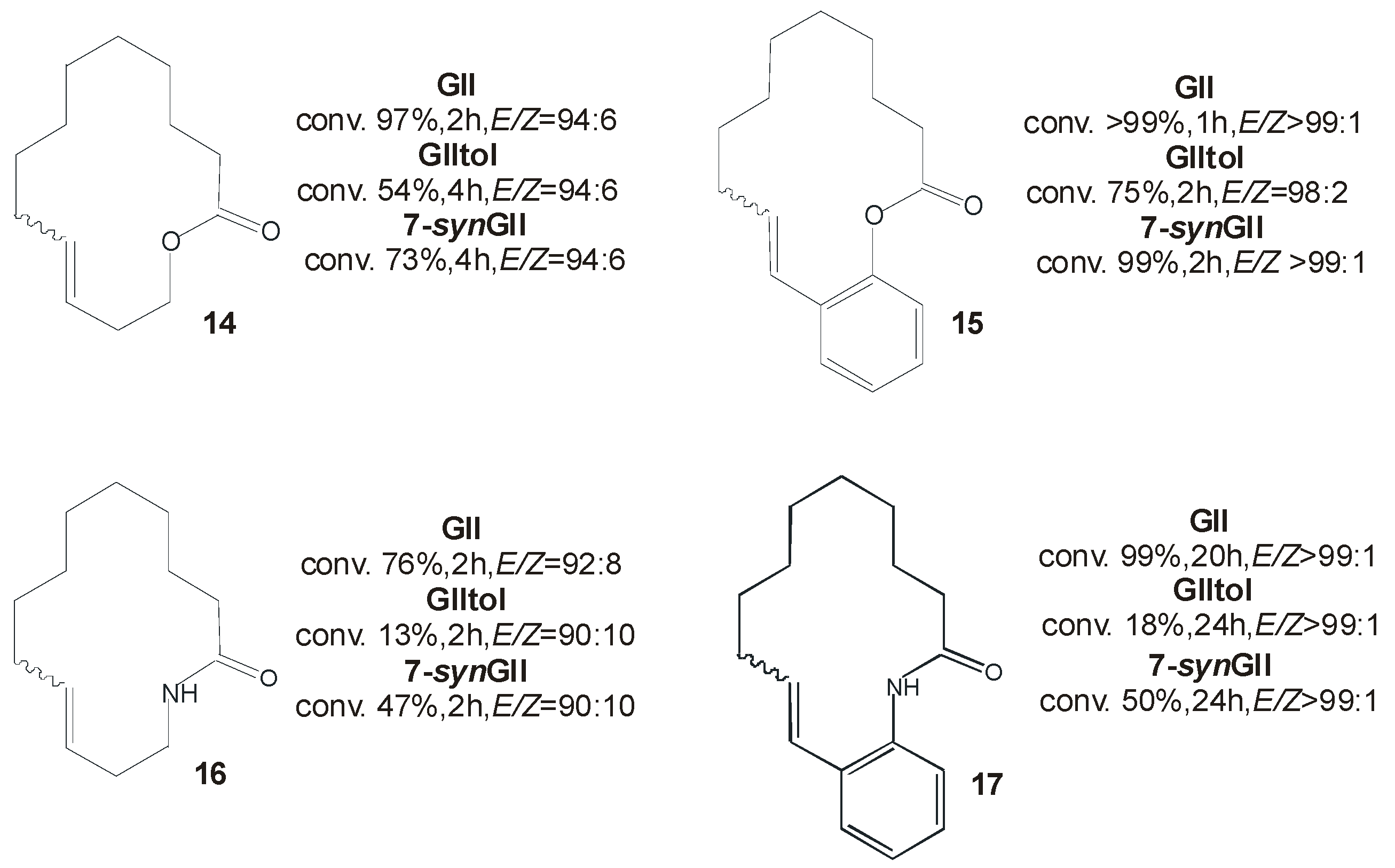
2.2. N-Aryl Substituents






| Catalyst | t (min) | Yield (%) a | Ref. | Catalyst | t (min) | Yield (%) b | Ref. |
|---|---|---|---|---|---|---|---|
| GII | 40 | >98 | [34] | HGIItol | 3 | >99 | [39] |
| GIItol | 60 | 98 | [26] | 7-synHGII | 4 | >99 | [26] |
| 7-synGII | 60 | 98 | [26] | 7-antiHGII | 5 | >99 | [27] |
| 7-antiGII | 60 | 92 | [26] | 8-synHGII | 3 | >99 | [27] |
| 8-synGII | 13 | 100 | [27] | 8-antiHGII | 6 | >99 | [27] |
| 8-antiGII | 60 | >96 | [27] |

| Catalyst | t (min) | Yield (%) a | Ref. | Catalyst | t (min) | Yield (%) b | Ref. |
|---|---|---|---|---|---|---|---|
| GII | 60 | 98 | [34] | HGIItol | 8 | >99 | [39] |
| GIItol | 60 | 84 | [26] | 7-synHGII | 9 | >99 | [27] |
| 7-synGII | 60 | 90 | [26] | 7-antiHGII | 16 | >99 | [27] |
| 7-antiGII | 60 | 80 | [26] | 8-synHGII | 6 | >99 | [27] |
| 8-synGII | 20 | 95 | [27] | 8-antiHGII | 12 | >99 | [27] |
| 8-antiGII | 60 | 83 | [27] |

| Catalyst | t (h) | Yield (%) a | Ref. | Catalyst | t (h) | Yield (%) b | Ref. |
|---|---|---|---|---|---|---|---|
| GII | 96 | 17 | [34] | HGIItol | 0.5 | >95 | [39] |
| GIItol | 1 | 70 | [26] | 7-synHGII | 0.5 | >95 | [27] |
| 7-synGII | 1 | 82 | [26] | 7-antiHGII | 1 | 78 | [27] |
| 7-antiGII | 1 | 47 | [26] | 8-synHGII | 1 | 85 | [27] |
| 8-synGII | 1 | 70 | [27] | 8-antiHGII | 1 | 59 | [27] |
| 8-antiGII | 1 | 35 | [27] | 9-synHGII | 72 | 55 | [22] |




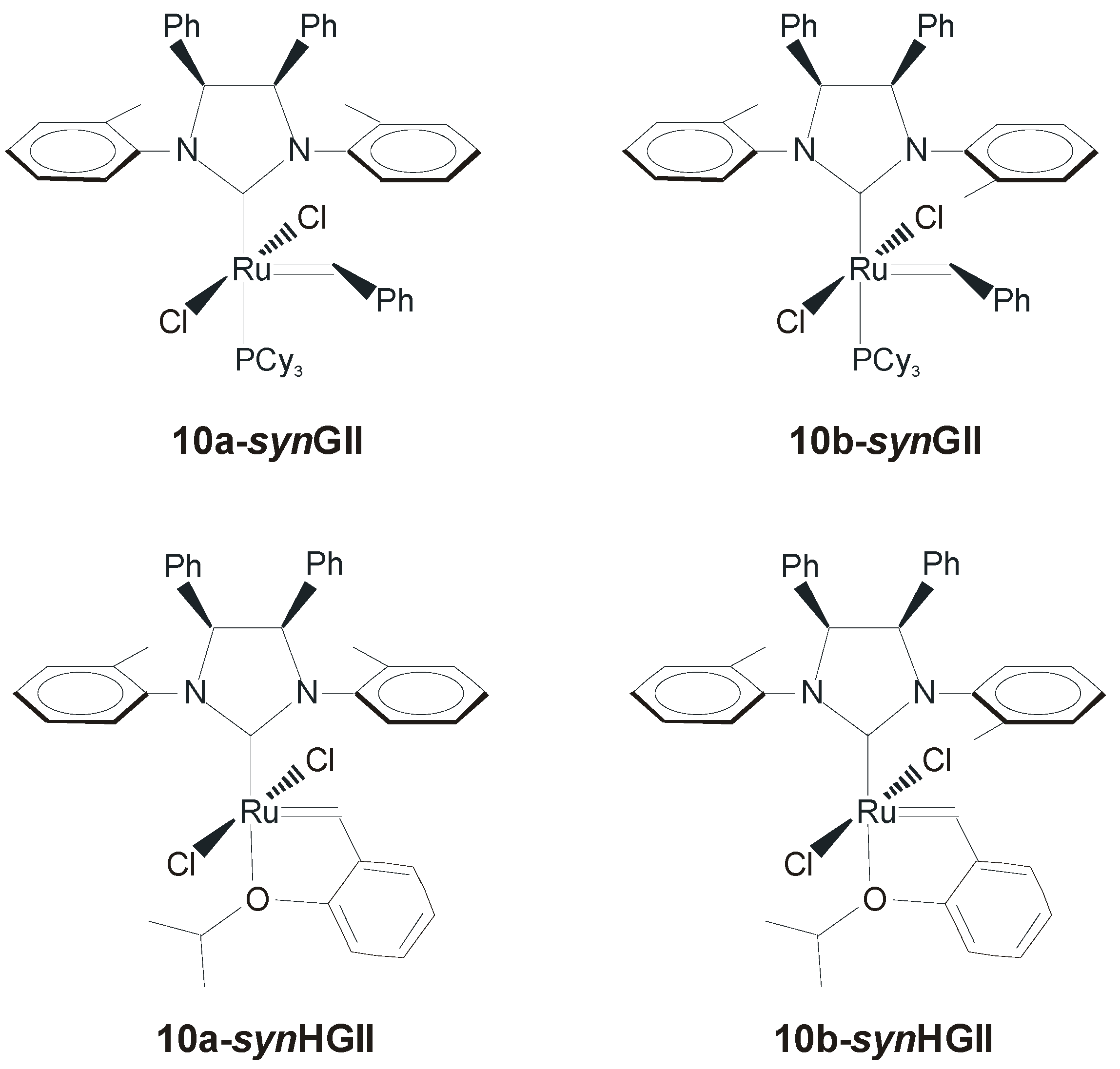
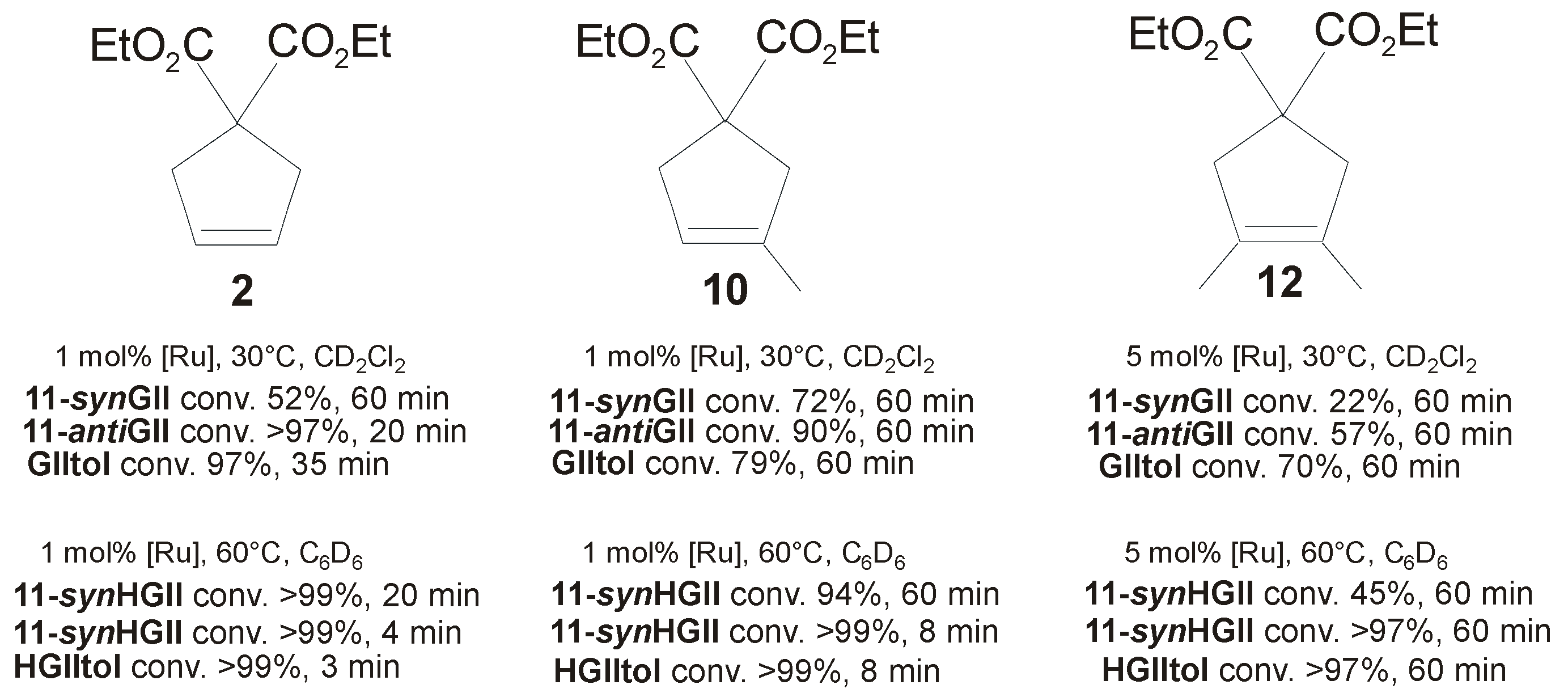
| Diene Substrate | RCM Product | Catalyst a (mol %) | t (min) | Yield (%) | Catalyst b (mol %) | t (min) | Yield (%) b |
|---|---|---|---|---|---|---|---|
| 1 | 2 | 10a-synGII (1.0) | 30 | >98 | 10a-synHGII (1.0) | 5 | >99 |
| 1 | 2 | 10b-synGII (1.0) | 60 | 70 | 10b-synHGII (1.0) | 12 | >99 |
| 9 | 10 | 10a-synGII (1.0) | 35 | >95 | 10a-synHGII (1.0) | 6 | >99 |
| 9 | 10 | 10b-synGII (1.0) | 60 | 66 | 10b-synHGII (1.0) | 60 | 95 |
| 11 | 12 | 10a-synGII (5.0) | 30 | 92 | 10a-synHGII (5.0) | 30 | >99 |
| 11 | 12 | 10b-synGII (5.0) | 60 | 44 | 10b-synHGII (5.0) | 120 | 94 |

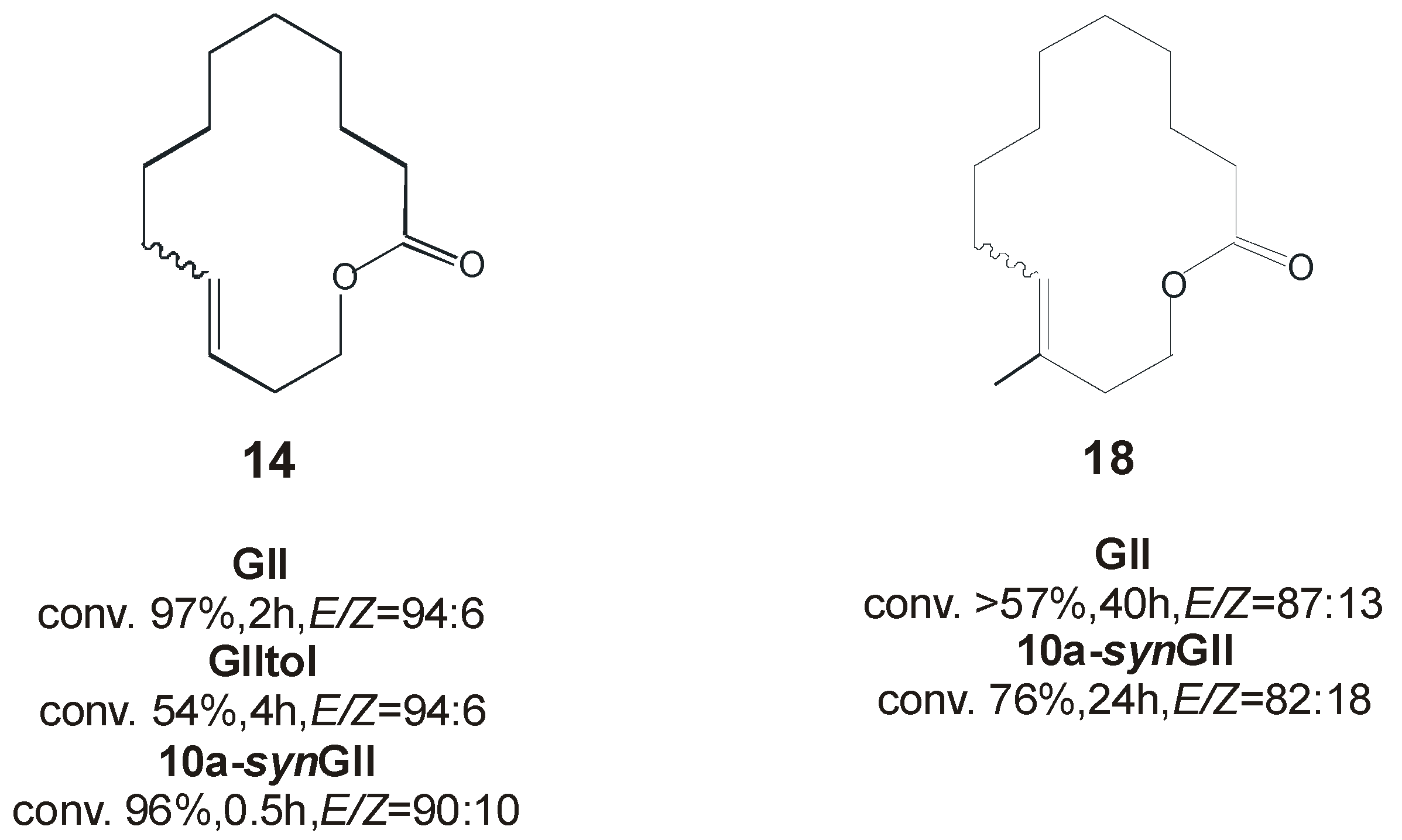
3. Ruthenium Catalysts Bearing Unsymmetrically N-Substituted NHCs with syn and anti Backbone Configurations


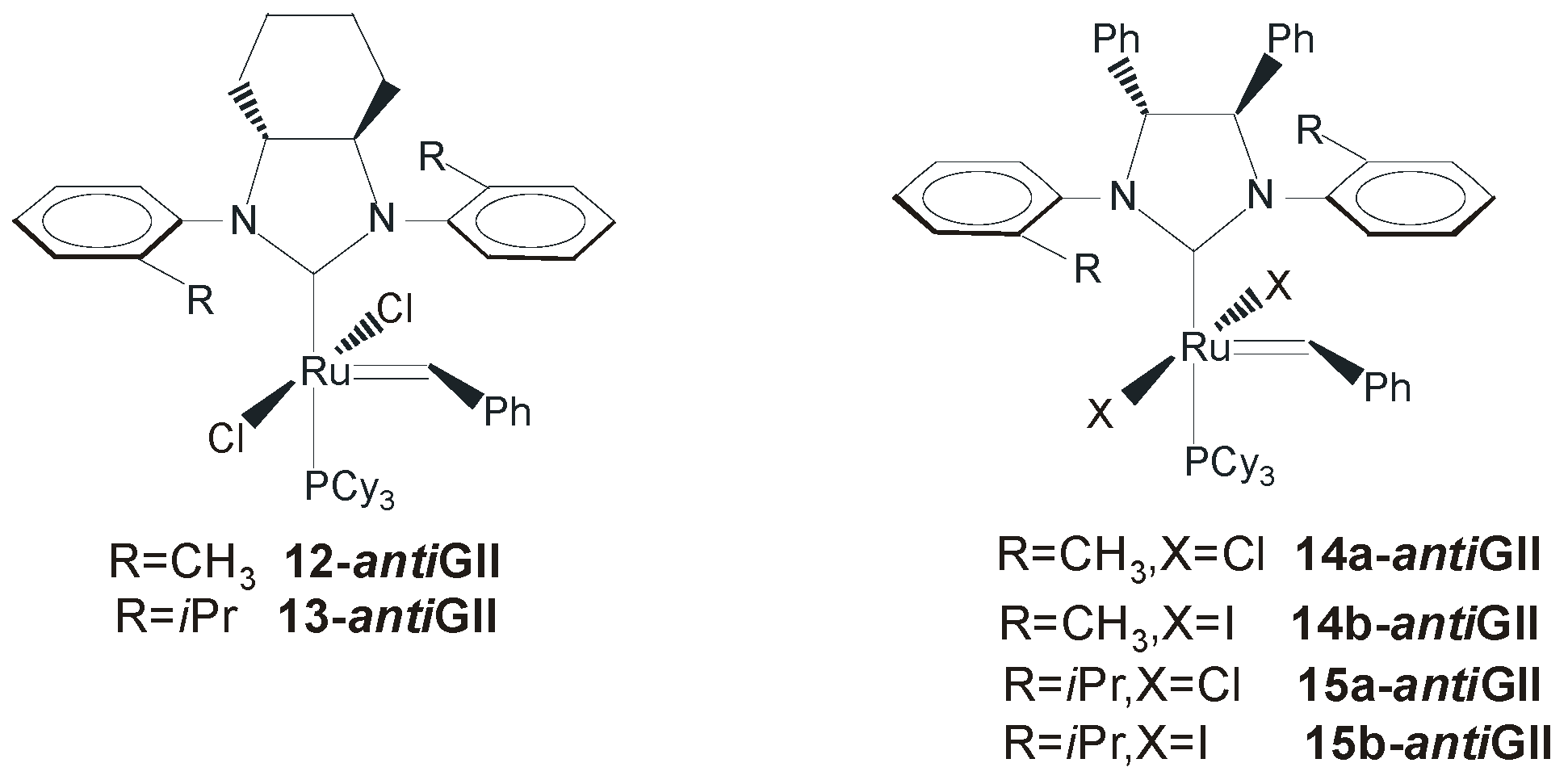
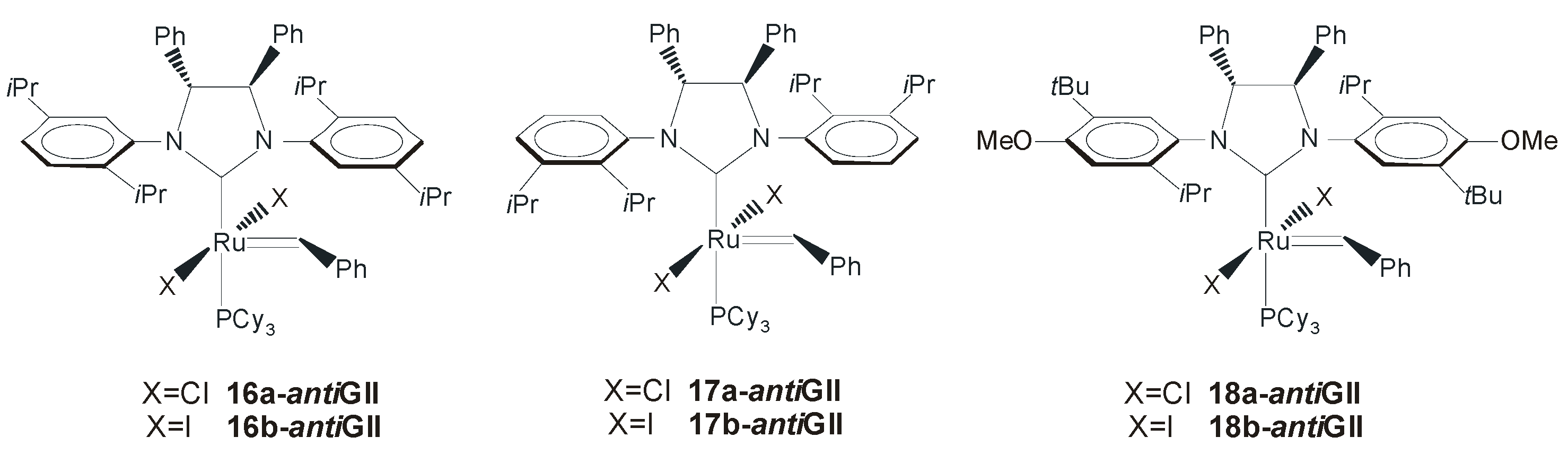
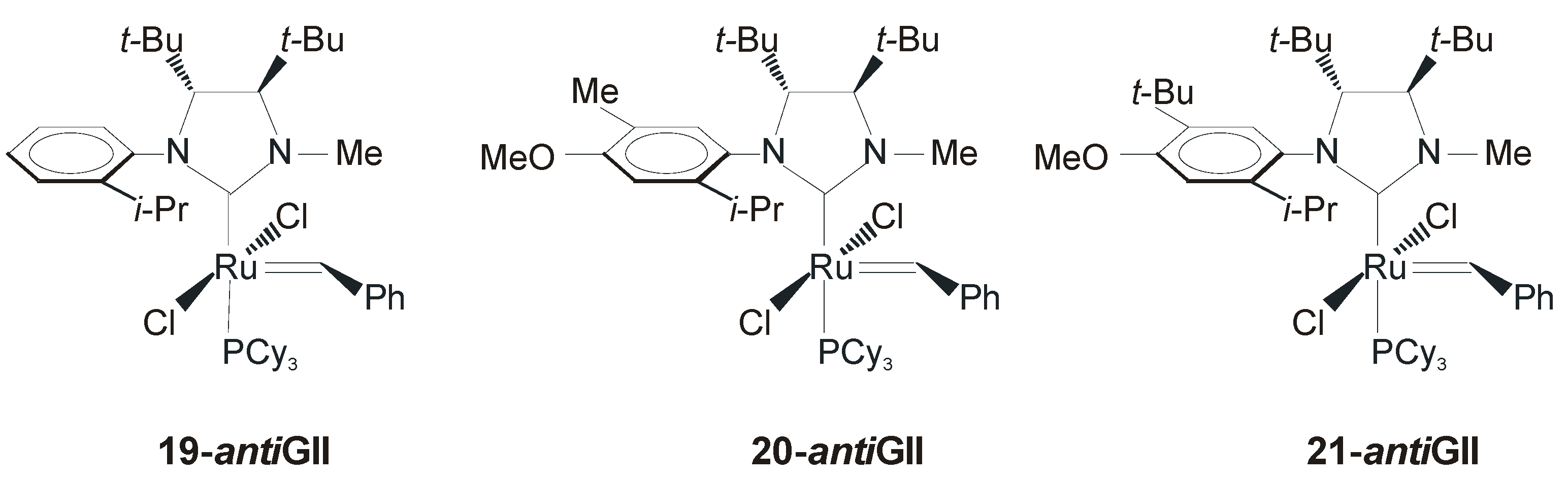
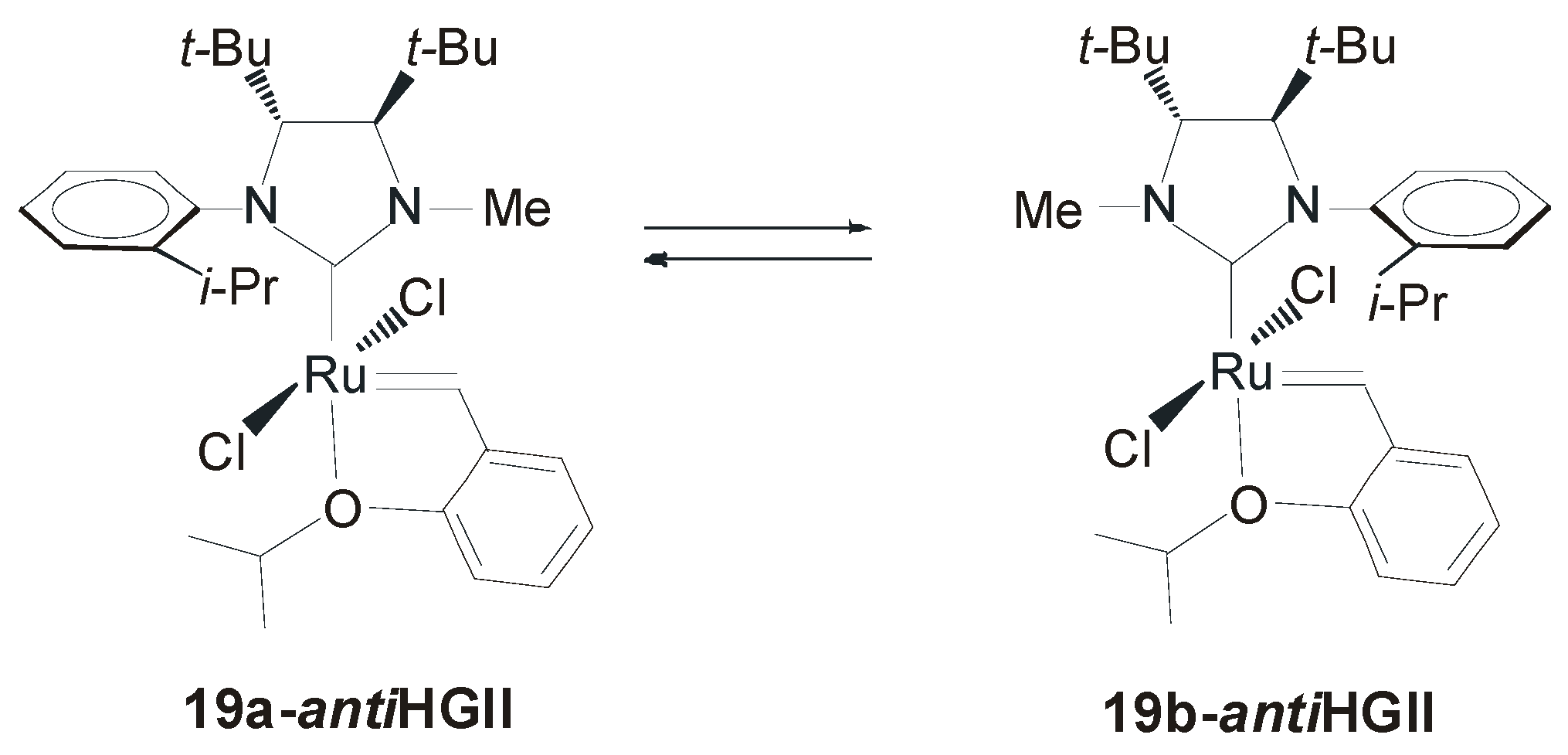
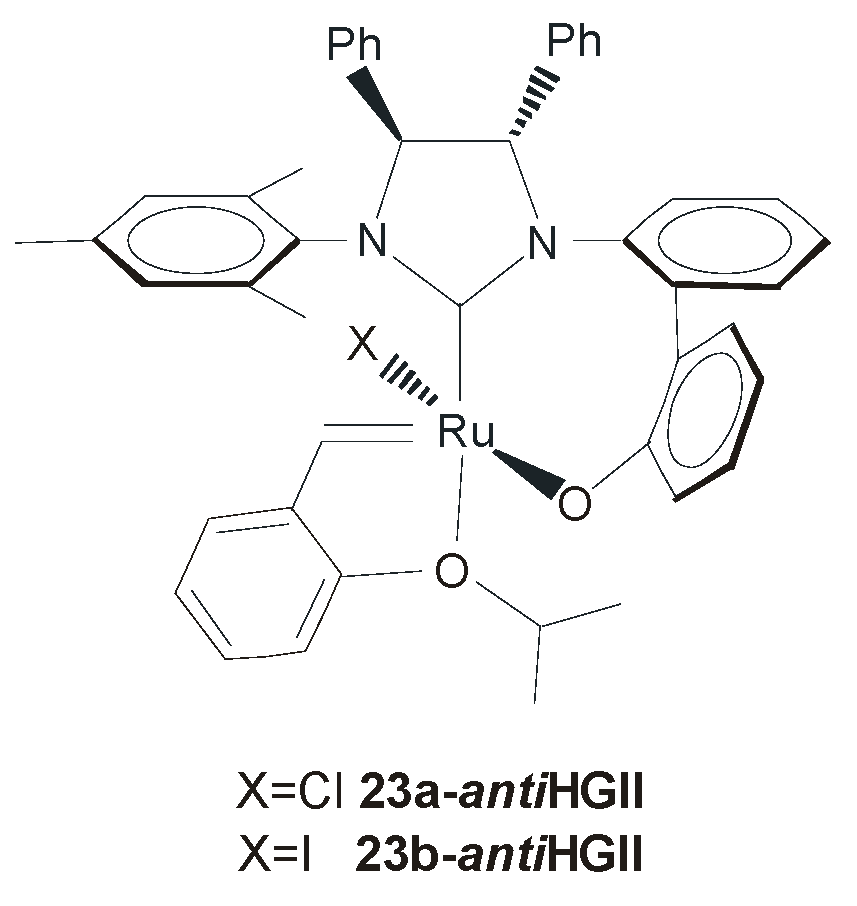
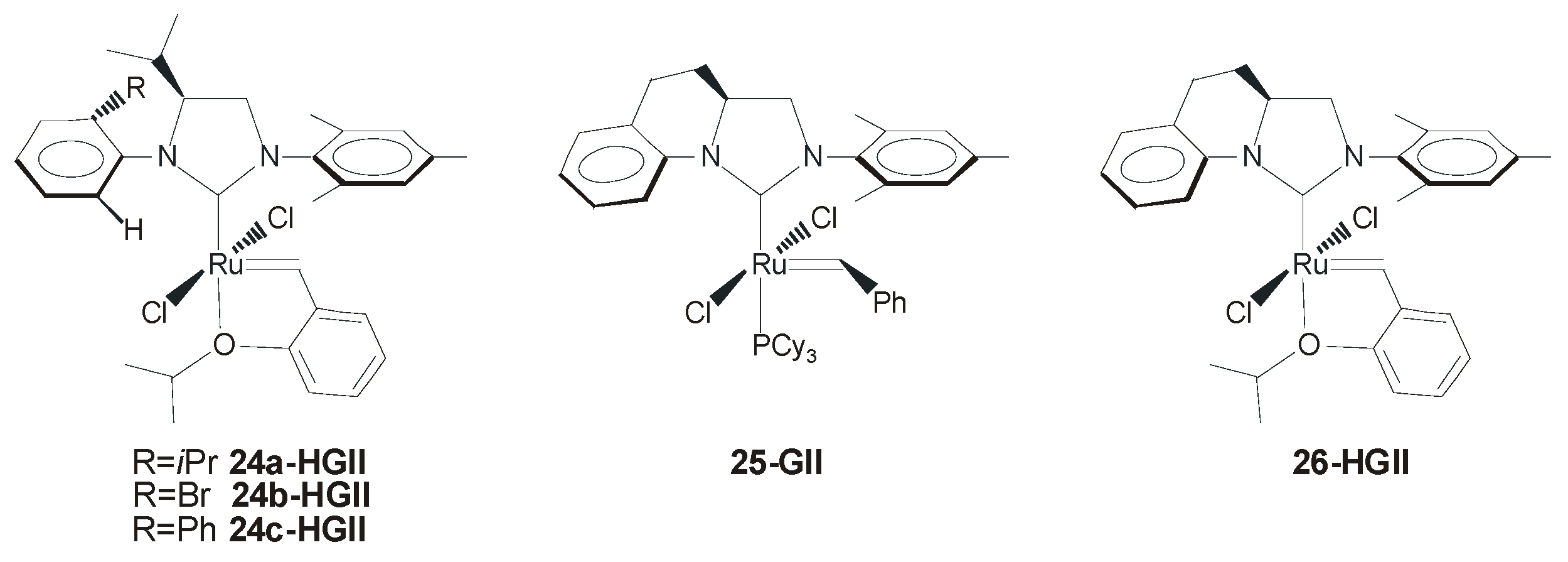
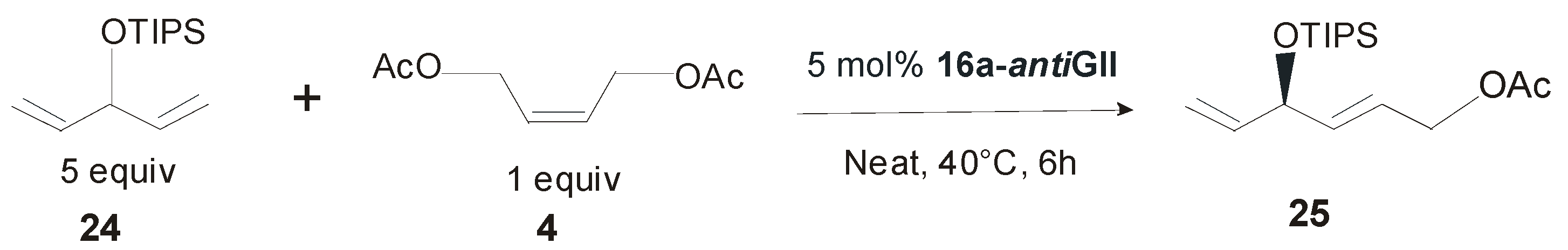
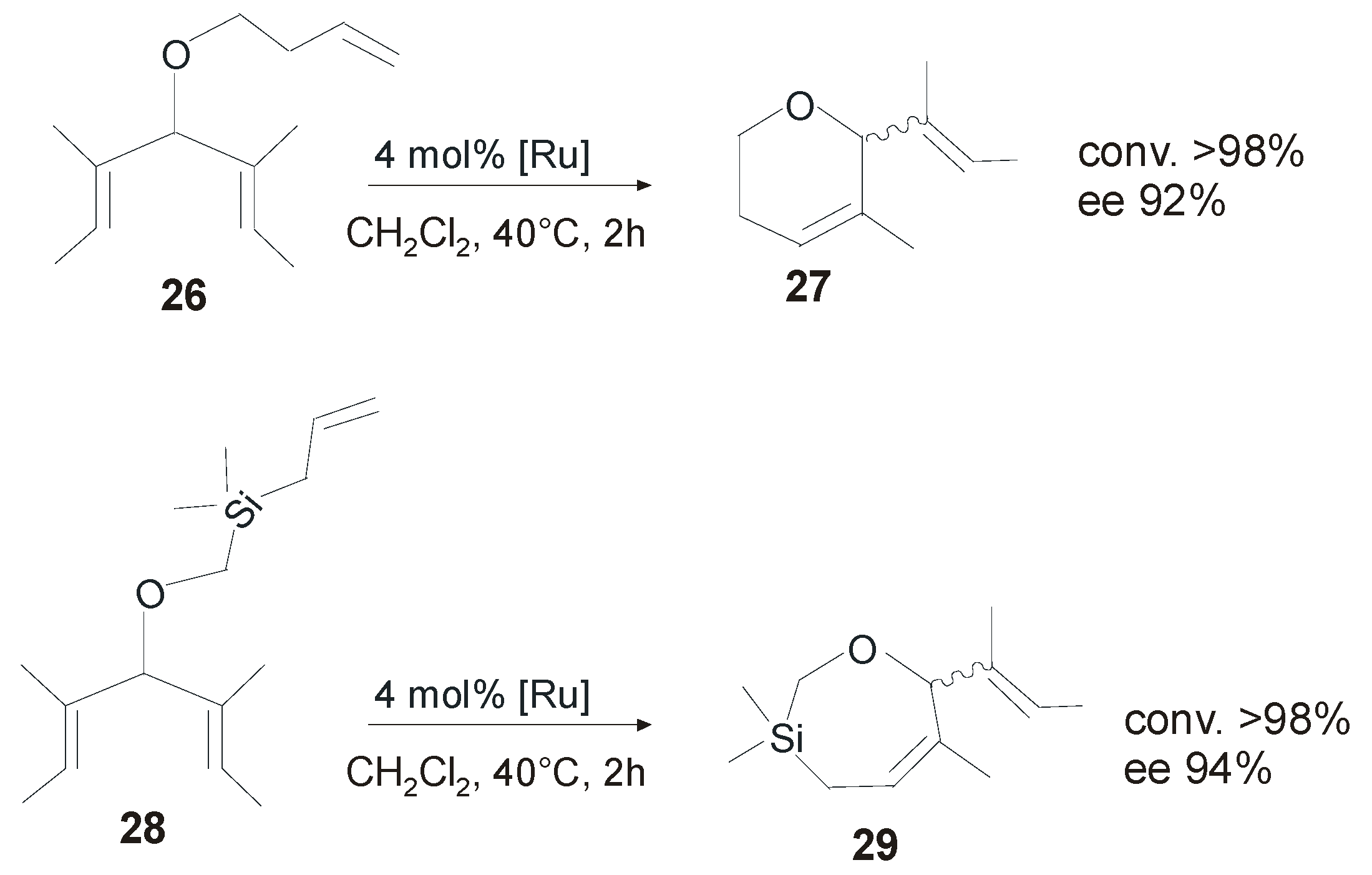
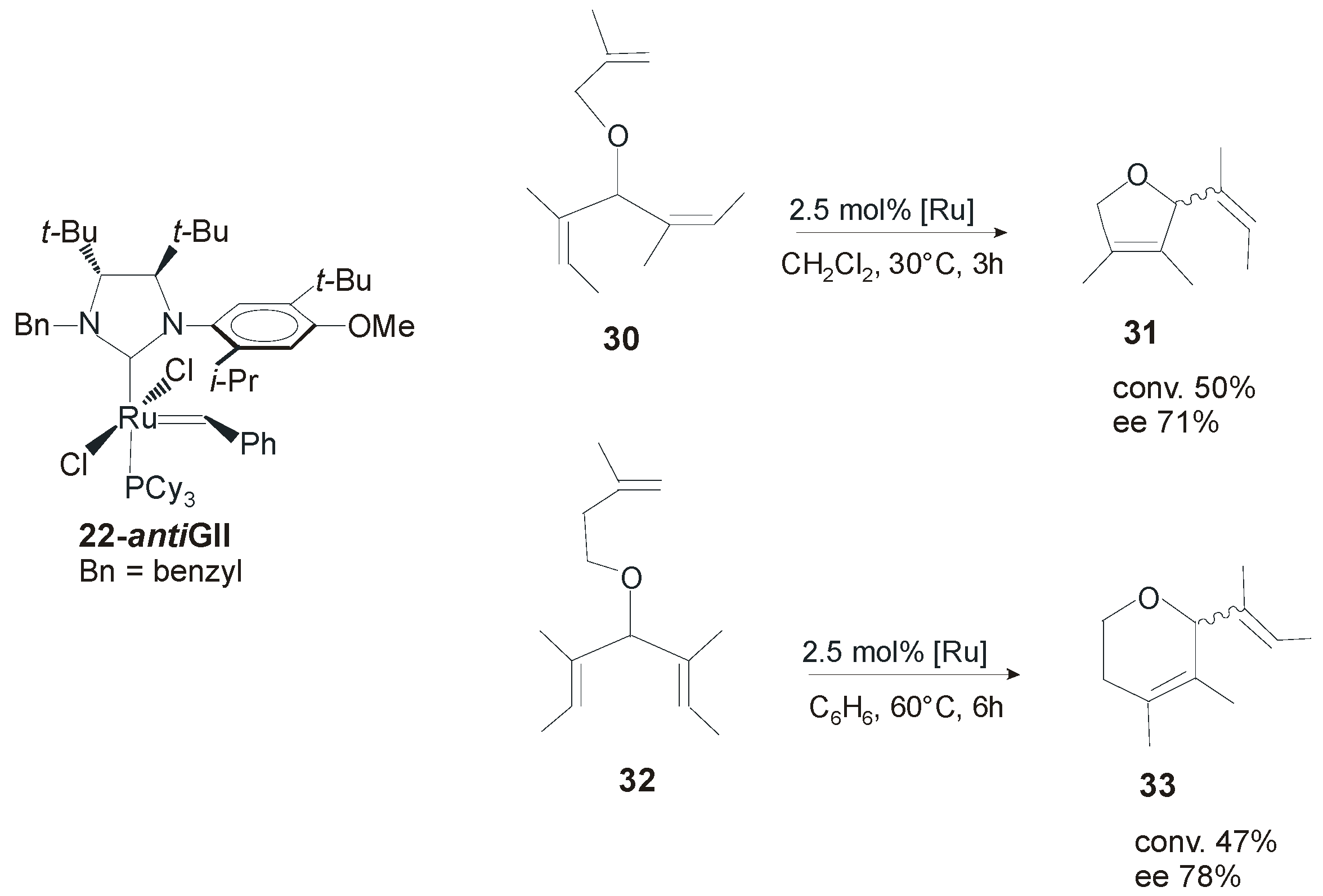
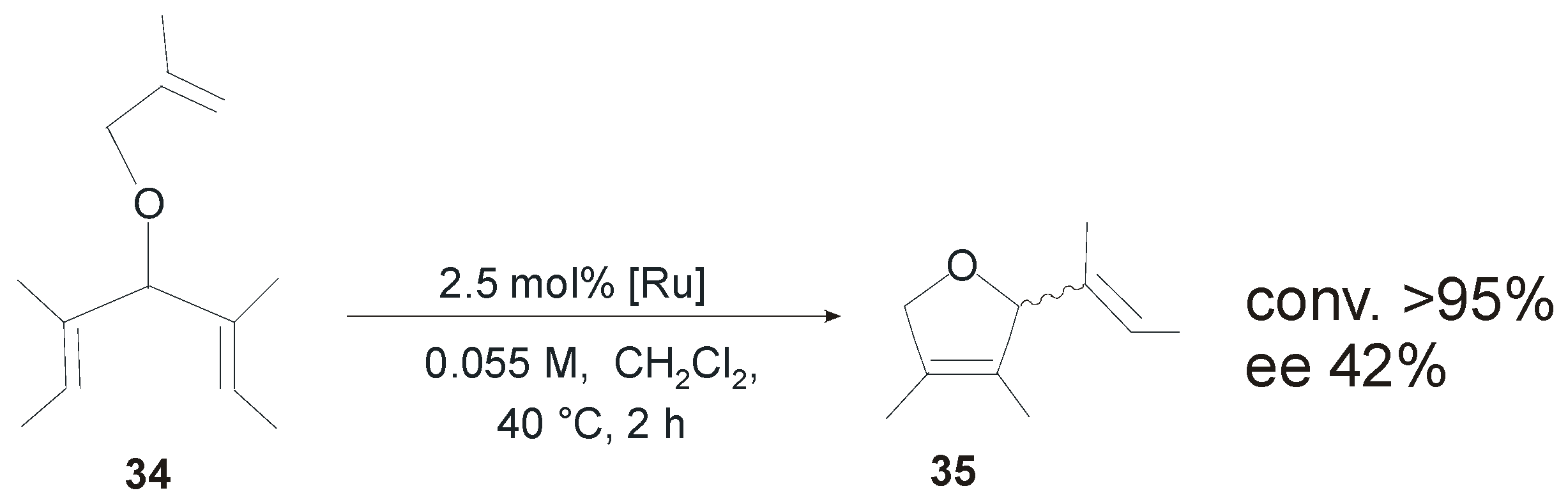
4. Ruthenium Complexes Bearing NHCs with anti Backbone Configuration in Asymmetric Metathesis
4.1. Symmetrical N-Substituents





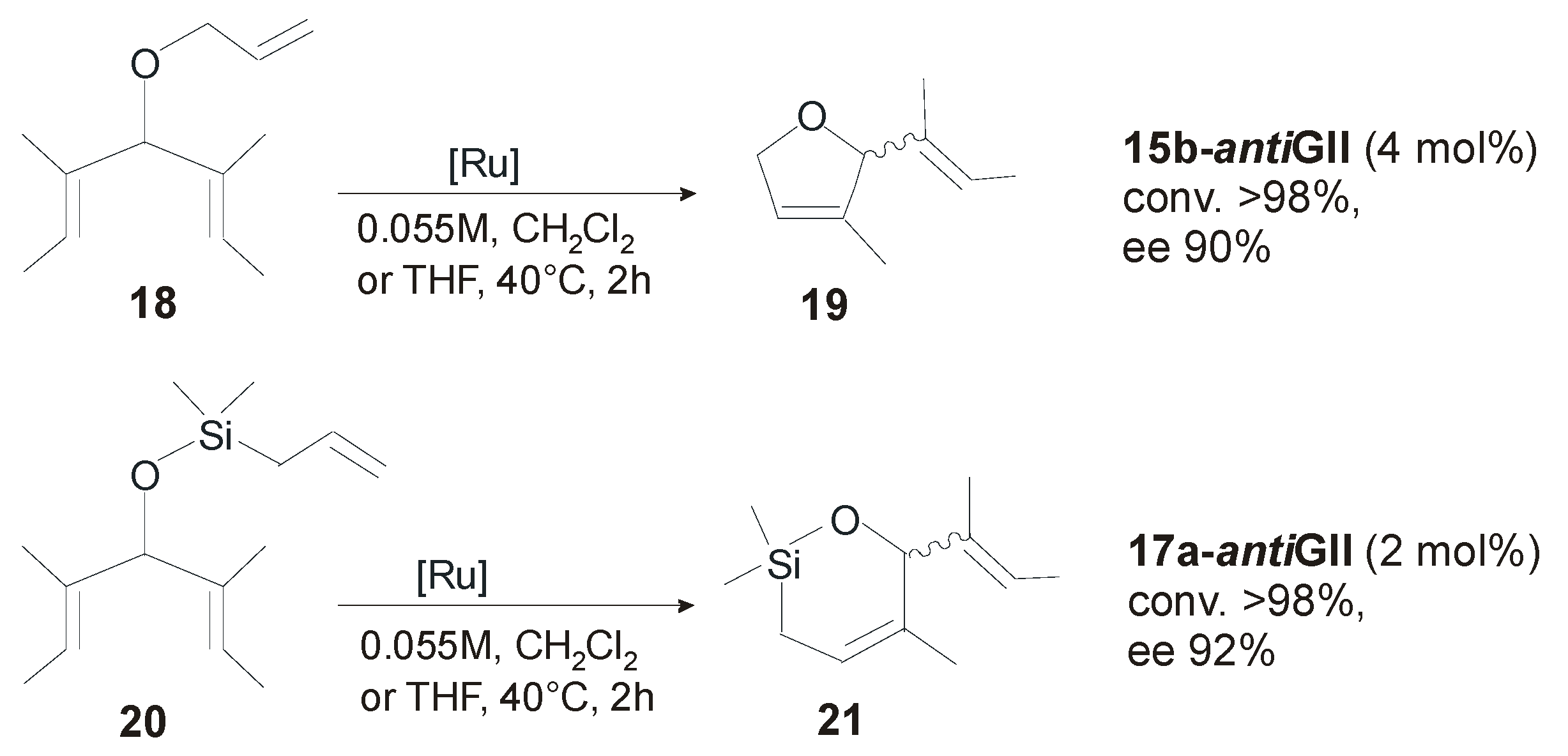
4.2. Unsymmetrical N-Substituents


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
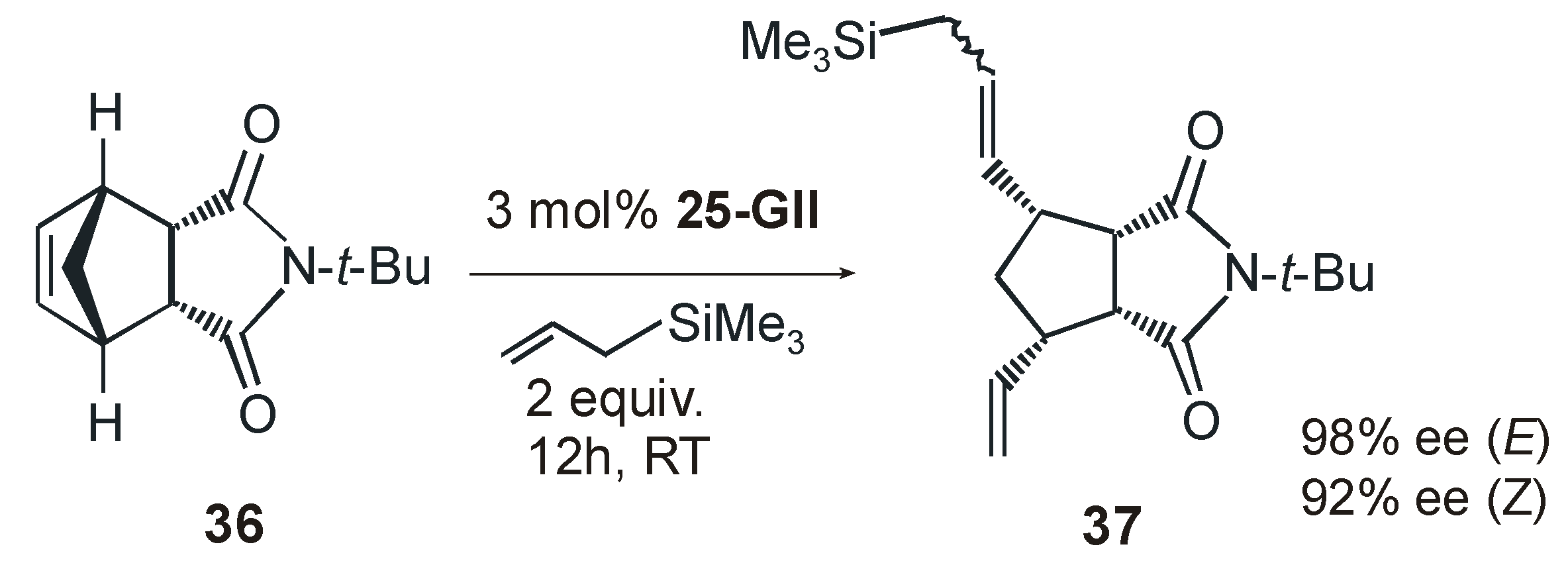{kind=link}
| Catalyst | ee (%) | Conv. (%) | Ref. |
|---|---|---|---|
| 19-antiGII a | 82 | >98 | [53] |
| 19-antiGII b | 48 | >98 | [53] |
| 15a-antiGII a | 35 | >98 | [51] |
| 15b-antiGII b | 90 | >98 | [51] |
| 17a-antiGII a | 46 | >98 | [51] |
| 17b-antiGII b | 90 | >98 | [51] |





5. Chiral Ruthenium Complexes Bearing Backbone-Monosubstituted NHCs


6. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Grubbs, R.H. Handbook of Metathesis; Wiley-VCH: Weinheim, Germany, 2003; Volume 1–3. [Google Scholar]
- Connon, S.J.; Blechert, S. Ruthenium Catalysts and Fine Chemistry; Dixneuf, P.H., Bruneau, C., Eds.; Springer: Heidelberg, Germany, 2004; Volume 11, pp. 93–124. [Google Scholar]
- Fürstner, A. Olefin Metathesis and Beyond. Angew. Chem. Int. Ed. 2000, 39, 3012–3043. [Google Scholar] [CrossRef]
- Hoveyda, A.H.; Zhugralin, A.R. The remarkable metal-catalysed olefin metathesis reaction. Nature 2007, 450, 243–251. [Google Scholar] [CrossRef] [PubMed]
- Deshmuhk, P.H.; Blechert, S. Alkene metathesis: The search for better catalysts. Dalton Trans. 2007, 38, 2479–2491. [Google Scholar] [CrossRef] [PubMed]
- Astruc, D. The metathesis reactions: From a historical perspective to recent developments. New J. Chem. 2005, 29, 42–56. [Google Scholar] [CrossRef]
- Lozano-Vila, A.M.; Monsaert, S.; Bajek, A.; Verpoort, F. Ruthenium-Based Olefin Metathesis Catalysts Derived from Alkynes. Chem. Rev. 2010, 110, 4865–4909. [Google Scholar] [CrossRef] [PubMed]
- Grela, K. Olefin Metathesis Theory and Practice; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014. [Google Scholar]
- Hopkinson, M.N.; Richter, C.; Schedler, M.; Glorius, F. An overview of N-heterocyclic carbenes. Nature 2014, 510, 485–496. [Google Scholar] [CrossRef] [PubMed]
- Nolan, S.P. N-Heterocyclic Carbenes: Effective Tools for Organometallic Synthesis; Wiley-VCH: Weinheim, Germany, 2014. [Google Scholar]
- Vougioukalakis, G.C.; Grubbs, R.H. Ruthenium-Based Heterocyclic Carbene-Coordinated Olefin Metathesis Catalysts. Chem. Rev. 2010, 110, 1746–1787. [Google Scholar] [CrossRef] [PubMed]
- Samojlowicz, C.; Bieniek, M.; Grela, K. Ruthenium-Based Olefin Metathesis Catalysts Bearing N-Heterocyclic Carbene Ligands. Chem. Rev. 2009, 109, 3708–3742. [Google Scholar] [CrossRef] [PubMed]
- Tornatzky, J.; Kannenberg, A.; Blechert, S. New catalysts with unsymmetrical N-heterocyclic carbene ligands. Dalton Trans. 2012, 41, 8215–8225. [Google Scholar] [CrossRef] [PubMed]
- Hamad, F.B.; Sun, T.; Xiao, S.; Verpoort, F. Olefin metathesis ruthenium catalysts bearing unsymmetrical heterocylic carbenes. Coord. Chem. Rev. 2013, 257, 2274–2292. [Google Scholar] [CrossRef]
- Shahane, S.; Bruneau, C.; Fischmeister, C. Z Selectivity: Recent Advances in one of the Current Major Challenges of Olefin Metathesis. ChemCatChem 2013, 5, 3436–3459. [Google Scholar] [CrossRef]
- Seiders, T.J.; Ward, D.W.; Grubbs, R.H. Enantioselective Ruthenium-Catalyzed Ring-Closing Metathesis. Org. Lett. 2001, 3, 3225–3228. [Google Scholar] [CrossRef] [PubMed]
- Ritter, T.; Day, M.W.; Grubbs, R.H. Rate Acceleration in Olefin Metathesis through a Fluorine-Ruthenium Interaction. J. Am. Chem. Soc. 2006, 128, 11768–11769. [Google Scholar] [CrossRef] [PubMed]
- Stewart, I.C.; Ung, T.; Pletnev, A.A.; Berlin, J.M.; Grubbs, R.H.; Schrodi, Y. Highly Efficient Ruthenium Catalysts for the Formation of Tetrasubstituted Olefins via Ring-Closing Metathesis. Org. Lett. 2007, 9, 1589–1592. [Google Scholar] [CrossRef] [PubMed]
- Berlin, J.M.; Campbell, K.; Ritter, T.; Funk, T.W.; Chlenov, A.; Grubbs, R.H. Ruthenium-Catalyzed Ring-Closing Metathesis to Form Tetrasubstituted Olefins. Org. Lett. 2007, 9, 1339–1342. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.K.; Grubbs, R.H. Olefin Metathesis Catalyst: Stabilization Effect of Backbone Substitutions of N-Heterocyclic Carbene. Org. Lett. 2008, 10, 2693–2696. [Google Scholar] [CrossRef] [PubMed]
- Grisi, F.; Costabile, C.; Gallo, E.; Mariconda, A.; Tedesco, C.; Longo, P. Ruthenium-Based Complexes Bearing Saturated Chiral N-Heterocyclic Carbene Ligands: Dynamic Behavior and Catalysis. Organometallics 2008, 27, 4649–4656. [Google Scholar] [CrossRef]
- Kuhn, K.M.; Bourg, J.-B.; Chung, C.K.; Virgil, S.C.; Grubbs, R.H. Effects of NHC-Backbone Substitution on Efficiency in Ruthenium-Based Olefin Metathesis. J. Am. Chem. Soc. 2009, 131, 5313–5320. [Google Scholar] [CrossRef] [PubMed]
- Savoie, T.; Stenne, B.; Collins, S.K. Improved Chiral Olefin Metathesis Catalysts: Increasing the Thermal and Solution Stability via Modification of a C1-Symmetrical N-Heterocyclic Carbene Ligand. Adv. Synth. Catal. 2009, 351, 1826–1832. [Google Scholar] [CrossRef]
- Vieille-Petit, L.; Luan, X.; Gatti, M.; Blumentritt, S.; Linden, A.; Clavier, H.; Nolan, S.P.; Dorta, R. Improving Grubbs’ II type ruthenium catalysts by appropriately modifying the N-heterocyclic carbene ligand. Chem. Commun. 2009, 18, 3783–3785. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatti, M.; Vieille-Petit, L.; Luan, X.; Mariz, R.; Drinkel, E.; Linden, A.; Dorta, R. Impact of NHC Ligand Conformation and Solvent Concentration on the Ruthenium-Catalyzed Ring-Closing Metathesis Reaction. J. Am. Chem. Soc. 2009, 131, 9498–9499. [Google Scholar] [CrossRef] [PubMed]
- Grisi, F.; Mariconda, A.; Costabile, C.; Bertolasi, V.; Longo, P. Influence of syn and anti Configurations of NHC Backbone on Ru-Catalyzed Olefin Metathesis. Organometallics 2009, 28, 4988–4995. [Google Scholar] [CrossRef]
- Costabile, C.; Mariconda, A.; Cavallo, L.; Longo, P.; Bertolasi, V.; Ragone, F.; Grisi, F. The Pivotal Role of Symmetry in the Ruthenium-Catalyzed Ring-Closing Metathesis of Olefins. Chem. Eur. J. 2011, 17, 8618–8629. [Google Scholar] [CrossRef] [PubMed]
- Stenne, B.; Timperio, J.; Savoie, J.; Dudding, T.; Collins, S.K. Desymmetrizations Forming Tetrasubstituted Olefins Using Enantioselective Olefin Metathesis. Org. Lett. 2010, 12, 2032–2035. [Google Scholar] [CrossRef] [PubMed]
- Tiede, S.; Berger, A.; Schlesiger, D.; Rost, D.; Lühl, A.; Blechert, S. Highly Active Chiral Ruthenium-Based Metathesis Catalysts through a Monosubstitution in the N-Heterocyclic Carbene. Angew. Chem. Int. Edit. 2010, 49, 3972–3975. [Google Scholar] [CrossRef] [PubMed]
- Dinger, M.B.; Nieczypor, P.; Mol, J.C. Adamantyl-Substituted N-Heterocyclic Carbene Ligands in Second-Generation Grubbs-Type Metathesis Catalysts. Organometallics 2003, 22, 5291–5296. [Google Scholar] [CrossRef]
- Ledoux, N.; Allaert, B.; Pattyn, S.; Mierde, H.V.; Vercaemst, C.; Verpoort, F. N,N′-Dialkyl- and N-Alkyl-N-mesityl-Substituted N-Heterocyclic Carbenes as Ligands in Grubbs Catalysts. Chem. Eur. J. 2006, 12, 4654–4661. [Google Scholar] [CrossRef] [PubMed]
- Louie, J.; Grubbs, R.H. Highly Active Metathesis Catalysts Generated in Situ from Inexpensive and Air-Stable Precursors. Angew. Chem. Int. Ed. 2001, 40, 247–249. [Google Scholar] [CrossRef]
- Ledoux, N.; Linden, A.; Allaert, B.; Vander Mierde, H.; Verpoort, F. Comparative Investigation of Hoveyda–Grubbs Catalysts bearing Modified N-Heterocyclic Carbene Ligands. Adv. Synth. Catal. 2007, 349, 1692–1700. [Google Scholar] [CrossRef]
- Ritter, T.; Hejl, A.; Wenzel, A.G.; Funk, T.W.; Grubbs, R.H. A Standard System of Characterization for Olefin Metathesis Catalysts. Organometallics 2006, 25, 5740–5745. [Google Scholar] [CrossRef]
- Scholl, M.; Ding, S.; Lee, C.W.; Grubbs, R.H. Synthesis and Activity of a New Generation of Ruthenium-Based Olefin Metathesis Catalysts Coordinated with 1,3-Dimesityl-4,5-dihydroimidazol-2-ylidene Ligands. Org. Lett. 1999, 1, 953–956. [Google Scholar] [CrossRef] [PubMed]
- Weigl, K.; Köhler, K.; Dechert, S.; Meyer, F. Synthesis and Structure of N-Heterocyclic Carbene Complexes with Tethered Olefinic Groups: Application of the Ruthenium Catalyst in Olefin Metathesis. Organometallics 2005, 24, 4049–4056. [Google Scholar] [CrossRef]
- Vehlow, K.; Gessler, S.; Blechert, S. Deactivation of Ruthenium Olefin Metathesis Catalysts through Intramolecular Carbene-Arene Bond Formation. Angew. Chem. Int. Ed. 2007, 46, 8082–8085. [Google Scholar] [CrossRef] [PubMed]
- Chooi, S.Y.M.; Leung, P.; Ng, S.; Quek, G.H.; Sim, K.Y. A simple route to optically pure 2,3-diaminobutane. Tetrahedron Asymmetry 1991, 2, 981–982. [Google Scholar] [CrossRef]
- Perfetto, A.; Costabile, C.; Longo, P.; Grisi, F. Ruthenium Olefin Metathesis Catalysts with Frozen NHC Ligand Conformations. Organometallics 2014, 33, 2747–2759. [Google Scholar] [CrossRef]
- Mariconda, A.; Grisi, F.; Granito, A.; Longo, P. Stereoselective Ring-Opening Metathesis Polymerization of 7-tert-Butoxy-bicyclo[2,2,1]hepta-2,5-diene by NHC–Ruthenium Catalysts. Macromol. Chem. Phys. 2013, 214, 1973–1979. [Google Scholar] [CrossRef]
- Jafarpour, L.; Stevens, E.D.; Nolan, S.P. A sterically demanding nucleophilic carbene: 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene). Thermochemistry and catalytic application in olefin metathesis. J. Organomet. Chem. 2000, 606, 49–54. [Google Scholar]
- Fürstner, A.; Ackermann, L.; Gabor, B.; Goddard, R.; Lehmann, C.W.; Mynott, R.; Stelzer, F.; Thiel, O.R. Comparative Investigation of Ruthenium-Based Metathesis Catalysts Bearing N-Heterocyclic Carbene (NHC) Ligands. Chem. Eur. J. 2001, 7, 3236–3253. [Google Scholar] [CrossRef]
- Dinger, M.B.; Mol, J.C. High Turnover Numbers with Ruthenium-Based Metathesis Catalysts. Adv. Synth. Catal. 2002, 344, 671–677. [Google Scholar] [CrossRef]
- Courchay, F.C.; Sworen, J.C.; Wagener, K.B. Metathesis Activity and Stability of New Generation Ruthenium Polymerization Catalysts. Macromolecules 2003, 36, 8231–8239. [Google Scholar] [CrossRef]
- Urbina-Blanco, C.A.; Leitgeb, A.; Slugovc, C.; Bantreil, X.; Clavier, H.; Slawin, A.M.Z.; Nolan, S.P. Olefin Metathesis Featuring Ruthenium Indenylidene Complexes with a Sterically Demanding NHC Ligand. Chem. Eur. J. 2011, 17, 5045–5053. [Google Scholar] [CrossRef] [PubMed]
- Grisi, F.; Costabile, C.; Grimaldi, A.; Viscardi, C.; Saturnino, C.; Longo, P. Synthesis of Unsaturated Macrocycles by Ru-Catalyzed Ring-Closing Metathesis: A Comparative Study. Eur. J. Org. Chem. 2012, 2012, 5928–5934. [Google Scholar] [CrossRef]
- Perfetto, A.; Costabile, C.; Longo, P.; Bertolasi, V.; Grisi, F. Probing the Relevance of NHC Ligand Conformations in the Ru-Catalysed Ring-Closing Metathesis Reaction. Chem. Eur. J. 2013, 19, 10492–10496. [Google Scholar] [CrossRef] [PubMed]
- Stewart, I.C.; Benitez, D.; O’Leary, D.J.; Tkatchouk, E.; Day, M.W.; GoddardIII, W.A.; Grubbs, R.H. Conformations of N-Heterocyclic Carbene Ligands in Ruthenium Complexes Relevant to Olefin Metathesis. J. Am. Chem. Soc. 2009, 131, 1931–1938. [Google Scholar] [CrossRef] [PubMed]
- Paradiso, V.; Bertolasi, V.; Grisi, F. Novel Olefin Metathesis Ruthenium Catalysts Bearing Backbone-Substituted Unsymmetrical NHC Ligands. Organometallics 2014, 33, 5932–5935. [Google Scholar] [CrossRef]
- Costabile, C.; Cavallo, L. Origin of Enantioselectivity in the Asymmetric Ru-Catalyzed Metathesis of Olefins. J. Am. Chem. Soc. 2004, 126, 9592–9600. [Google Scholar] [CrossRef] [PubMed]
- Funk, T.W.; Berlin, J.M.; Grubbs, R.H. Highly Active Chiral Ruthenium Catalysts for Asymmetric Ring-Closing Olefin Metathesis. J. Am. Chem. Soc. 2006, 128, 1840–1846. [Google Scholar] [CrossRef] [PubMed]
- Berlin, J.M.; Goldberg, S.D.; Grubbs, R.H. Highly Active Chiral Ruthenium Catalysts for Asymmetric Cross- and Ring-Opening Cross-Metathesis. Angew. Chem. Int. Ed. 2006, 45, 7591–7595. [Google Scholar] [CrossRef] [PubMed]
- Fournier, P.; Collins, S.K. A Highly Active Chiral Ruthenium-Based Catalyst for Enantioselective Olefin Metathesis. Organometallics 2007, 26, 2945–2949. [Google Scholar] [CrossRef]
- Fournier, P.; Savoie, J.; Stenne, B.; Bédard, M.; Grandbois, A.; Collins, S.K. Mechanistically Inspired Catalysts for Enantioselective Desymmetrizations by Olefin Metathesis. Chem. Eur. J. 2008, 14, 8690–8695. [Google Scholar] [CrossRef] [PubMed]
- Grandbois, A.; Collins, S.K. Enantioselective Synthesis of [7]Helicene: Dramatic Effects of Olefin Additives and Aromatic Solvents in Asymmetric Olefin Metathesis. Chem. Eur. J. 2008, 14, 9323–9329. [Google Scholar] [CrossRef] [PubMed]
- Van Veldhuizen, J.J.; Campbell, J.E.; Giudici, R.E.; Hoveyda, A.H. A Readily Available Chiral Ag-Based N-Heterocyclic Carbene Complex for Use in Efficient and Highly Enantioselective Ru-Catalyzed Olefin Metathesis and Cu-Catalyzed Allylic Alkylation Reactions. J. Am. Chem. Soc. 2005, 127, 6877–6882. [Google Scholar] [CrossRef] [PubMed]
- Giudici, R.E.; Hoveyda, A.H. Directed Catalytic Asymmetric Olefin Metathesis. Selectivity Control by Enoate and Ynoate Groups in Ru-Catalyzed Asymmetric Ring-Opening/Cross-Metathesis. J. Am. Chem. Soc. 2007, 129, 3824–3825. [Google Scholar] [CrossRef] [PubMed]
- Kannenberg, A.; Rost, D.; Eibauer, S.; Tiede, S.; Blechert, S. A Novel Ligand for the Enantioselective Ruthenium-Catalyzed OlefinMetathesis. Angew. Chem. Int. Ed. 2011, 50, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Ragone, F.; Poater, A.; Cavallo, L. Flexibility of N-Heterocyclic Carbene Ligands in Ruthenium Complexes Relevant to Olefin Metathesis and Their Impact in the First Coordination Sphere of the Metal. J. Am. Chem. Soc. 2010, 132, 4249–4258. [Google Scholar] [CrossRef] [PubMed]
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paradiso, V.; Costabile, C.; Grisi, F. NHC Backbone Configuration in Ruthenium-Catalyzed Olefin Metathesis. Molecules 2016, 21, 117. https://doi.org/10.3390/molecules21010117
Paradiso V, Costabile C, Grisi F. NHC Backbone Configuration in Ruthenium-Catalyzed Olefin Metathesis. Molecules. 2016; 21(1):117. https://doi.org/10.3390/molecules21010117
Chicago/Turabian StyleParadiso, Veronica, Chiara Costabile, and Fabia Grisi. 2016. "NHC Backbone Configuration in Ruthenium-Catalyzed Olefin Metathesis" Molecules 21, no. 1: 117. https://doi.org/10.3390/molecules21010117
APA StyleParadiso, V., Costabile, C., & Grisi, F. (2016). NHC Backbone Configuration in Ruthenium-Catalyzed Olefin Metathesis. Molecules, 21(1), 117. https://doi.org/10.3390/molecules21010117