
Microreactors—A Powerful Tool to Synthesize Peroxycarboxylic Esters


Abstract
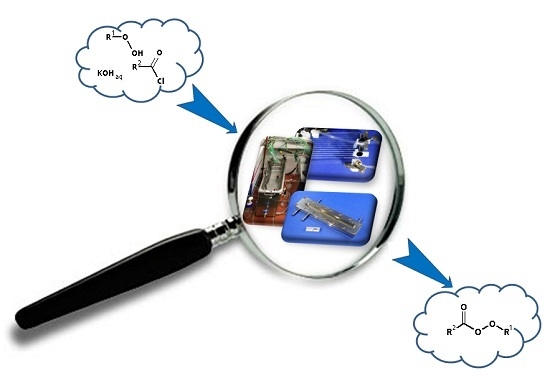
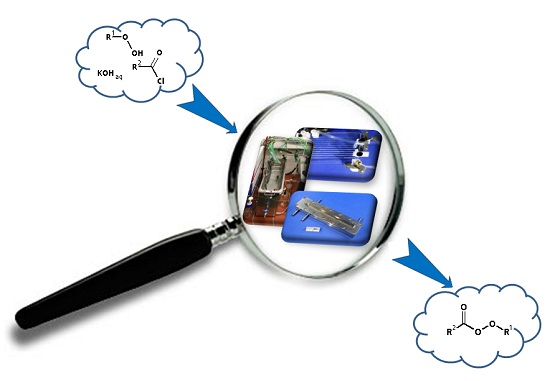
:
1. Introduction
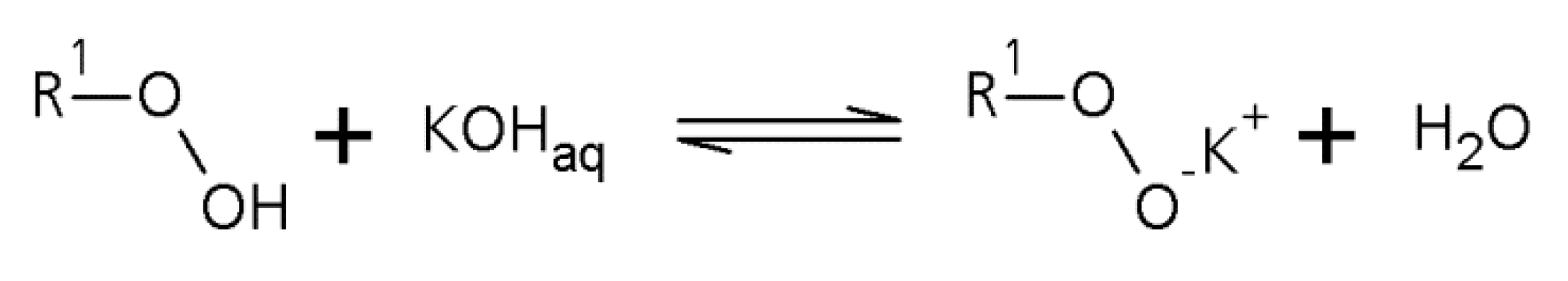
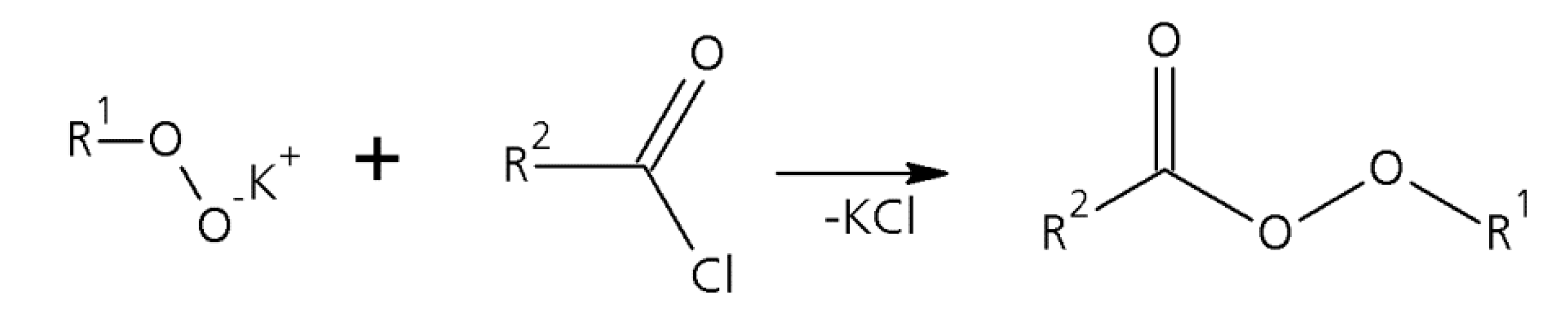
1.1. A Short Survey of the History of the Perester Synthesis
1.2. Microreactors—A Powerful Tool for Organic Peroxide Production
2. Results and Discussion
2.1. Peroxycarboxylic Ester Synthesis Using Different Types of Microreactors and Concepts for Emulsification


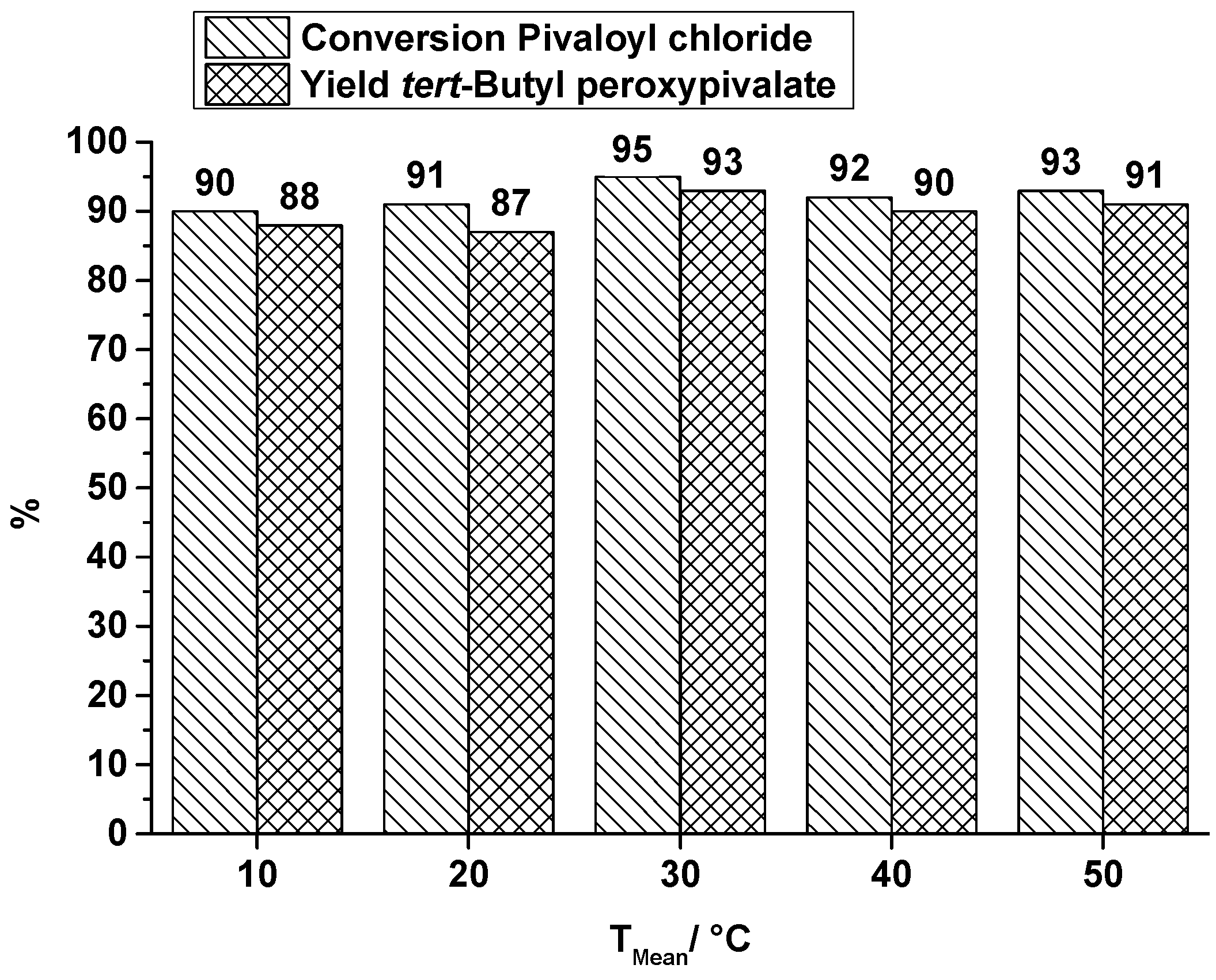
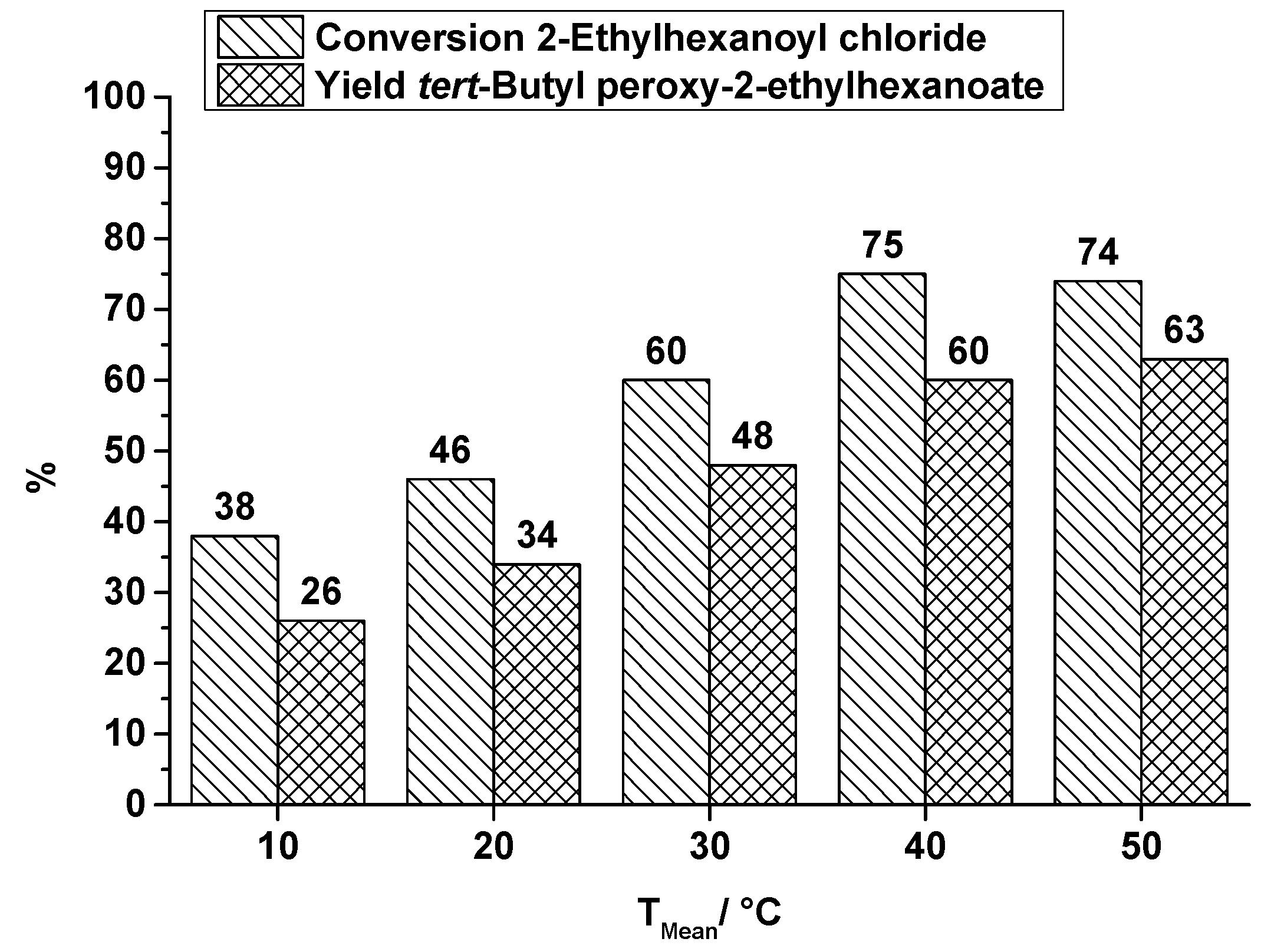
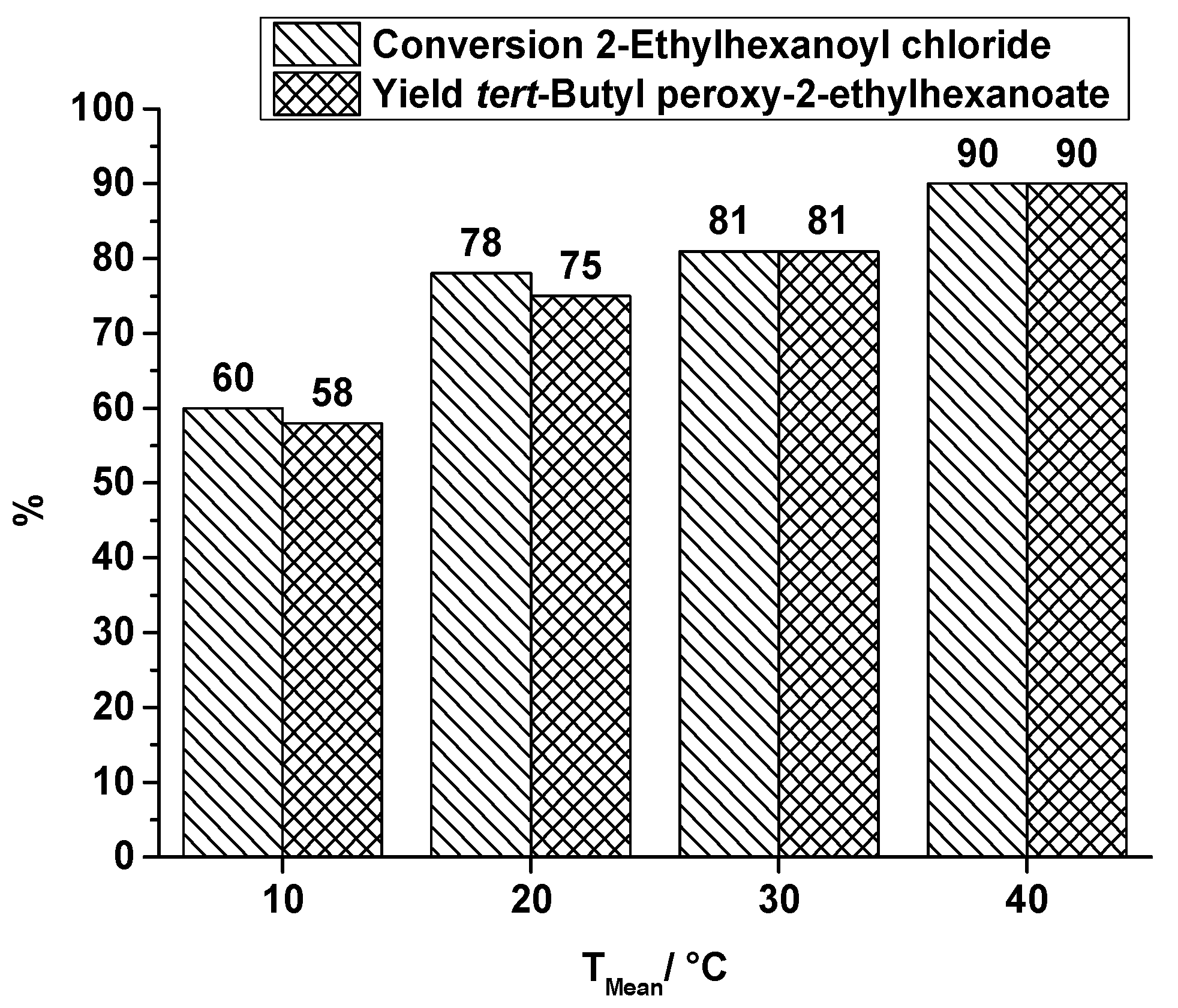
2.1.1. Orifice Microreactor

- A small and compact design which should be scalable to higher throughputs.
- Combination of both reaction steps into one microreactor.
- The heat exchanger must be located below the reaction plate to enable the measurement of the heat profile along the top of the reaction plate which is covering the reaction channel.
- The reaction channel must be accessible for cleaning.
- For re-emulsification five orifices are used.
- Two caterpillar micromixers are used for generating a homogeneous mixture (first step) and for generating a pre-emulsion (second step).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Section | L/m | A/m2 | V/m3 | |
|---|---|---|---|---|
| KTBP-Formation | Mixer 1–Mixer 2 | 0.38 | 1.1 × 10−3 | 1.9 × 10−7 |
| TBPP-Formation | Mixer 2–Orifice 1 | 0.15 | 4.2 × 10−4 | 7.5 × 10−8 |
| Orifice 1–Orifice 2 | 0.20 | 5.8 × 10−4 | 1.0 × 10−7 | |
| Orifice 2–Orifice 3 | 0.16 | 4.6 × 10−4 | 8.2 × 10−8 | |
| Orifice 3–Orifice 4 | 0.09 | 2.5 × 10−4 | 4.4 × 10−8 | |
| Orifice 4–Orifice 5 | 0.06 | 1.7× 10−4 | 3.1 × 10−8 | |
| Orifice 5–Outlet | 0.03 | 9.6 × 10−5 | 1.7 × 10−8 | |
| Sum | 1.08 | 3.1 × 10−3 | 5.4 × 10−7 | |



| Entry | Trct./°C | Qges/mL·min−1 | Δp/Pa | Ev/kJ·m−3 | Conversion % | Yield % | τsect. 2/s | τges/s | |
|---|---|---|---|---|---|---|---|---|---|
| TBHP | PivCl | TBPP | |||||||
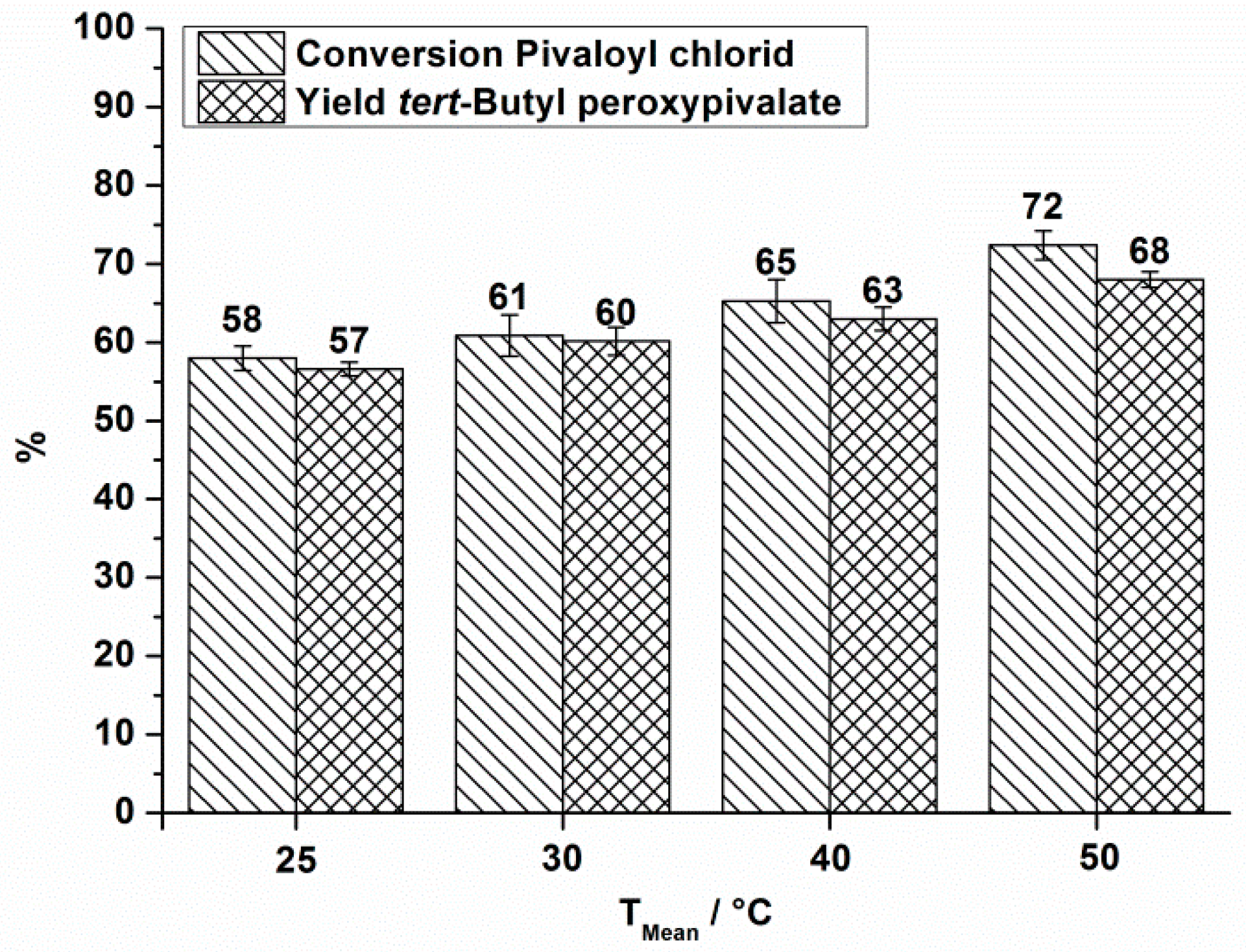
| 1 | 25 | 10.5 | 180,000 | 180 | 54 | 58 | 57 | 2.1 | 3.5 |
| 2 | 30 | 10.5 | 150,000 | 150 | 56 | 61 | 60 | 2.1 | 3.5 |
| 3 | 40 | 10.5 | 140,000 | 140 | 59 | 65 | 63 | 2.1 | 3.5 |
| 4 | 50 | 10.5 | 120,000 | 120 | 66 | 72 | 68 | 2.1 | 3.5 |
| Entry | Trct./°C | Qges/mL·min−1 | Δp/Pa | Ev/kJ·m−3 | Conversion % | Yield % | τsect. 2/s | τges/s | |
|---|---|---|---|---|---|---|---|---|---|
| TBHP | PivCl | TBPP | |||||||
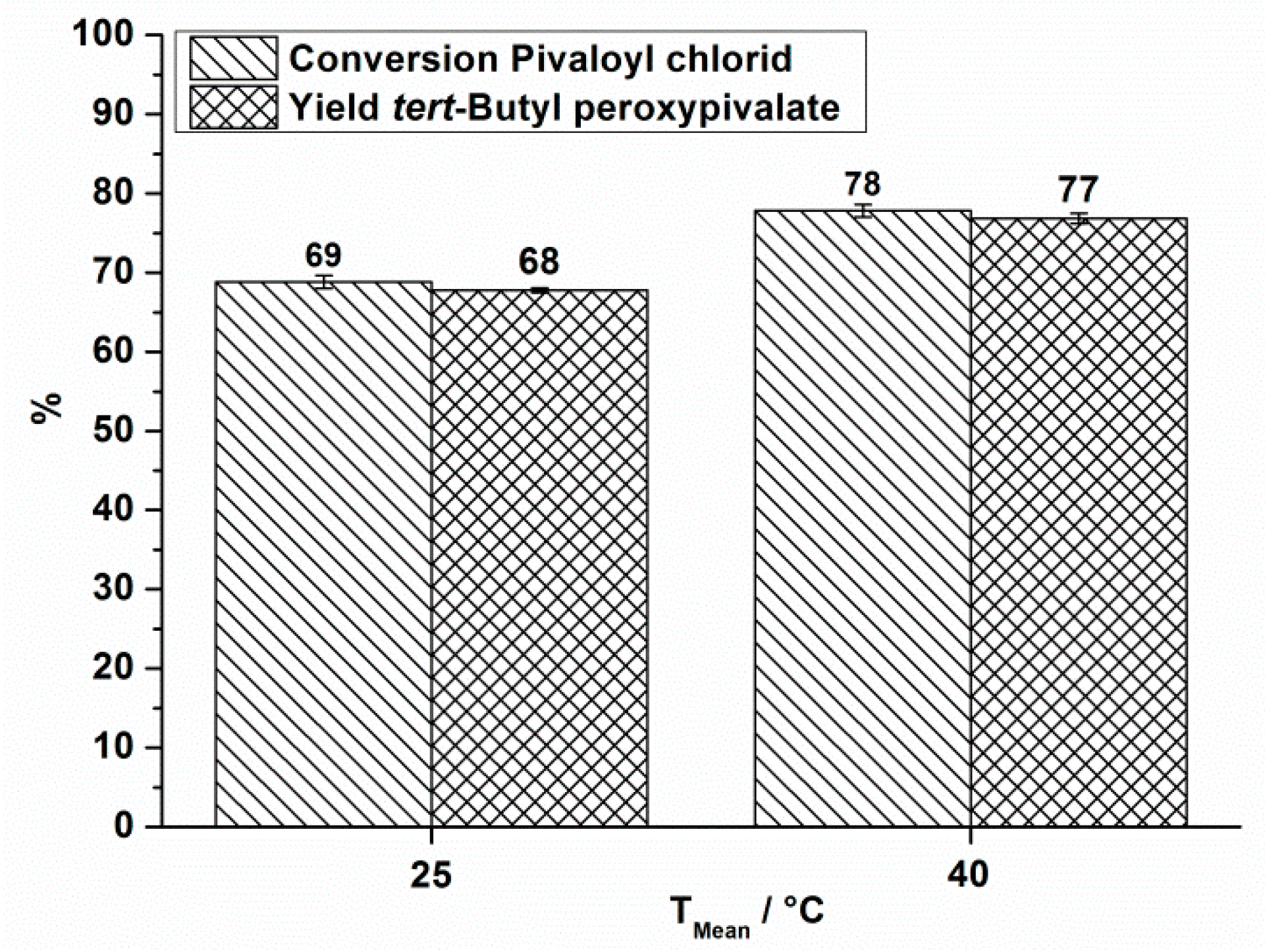
| 1 | 25 | 18.5 | 380,000 | 380 | 71 | 69 | 68 | 1.2 | 2 |
| 2 | 40 | 18.5 | 300,000 | 300 | 79 | 78 | 77 | 1.2 | 2 |
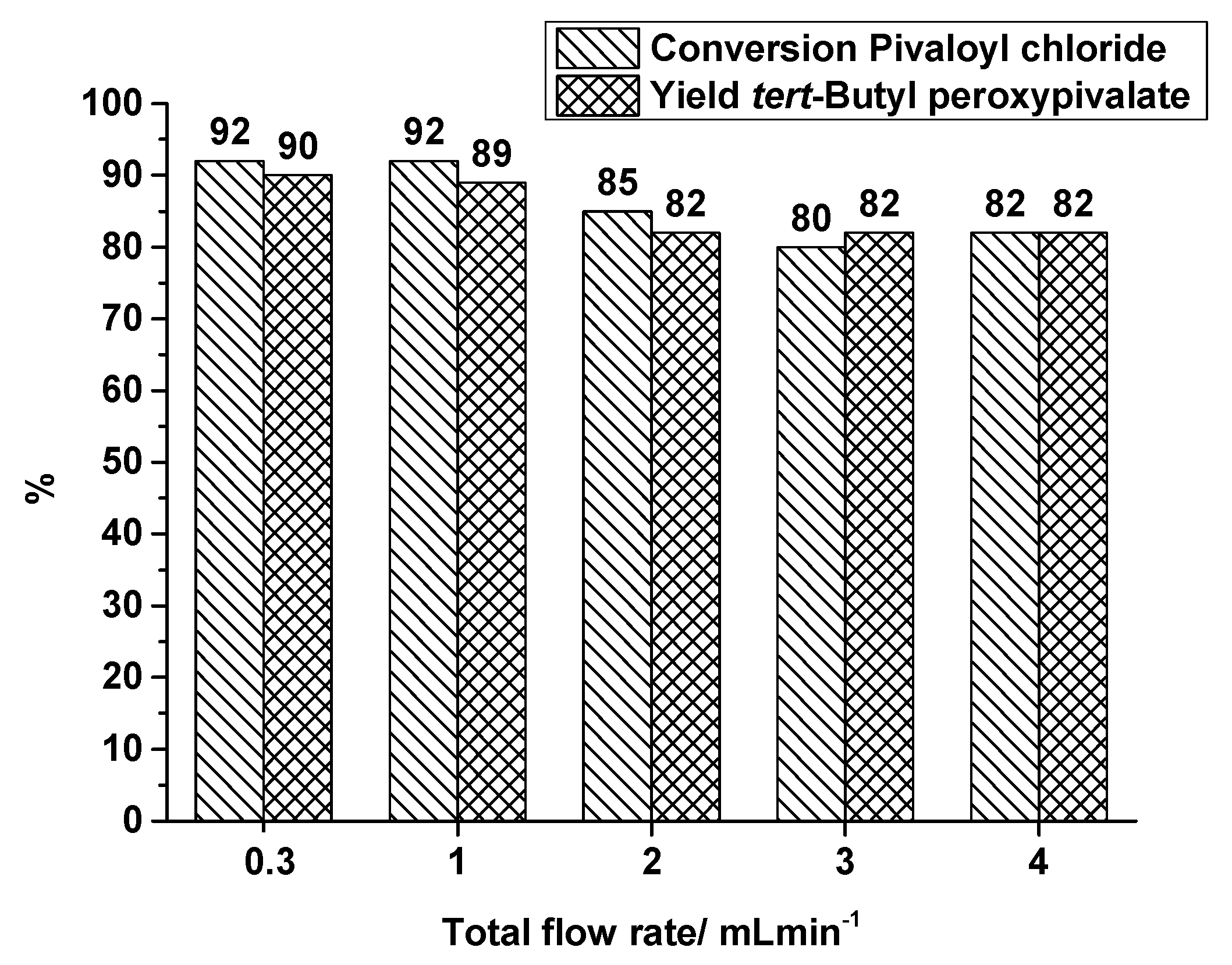
2.1.2. Split and Recombine Microreactor



| Entry | Trct./°C | Qges/mL·min−1 | Δp/Pa | Ev/kJ·m−3 | Conversion % | Yield % | τges/s |
|---|---|---|---|---|---|---|---|
| PivCl | TBPP | ||||||
| 1 | 40 | 1 | 124,226 | 124 | 92 | 89 | 19.2 |
| 2 | 40 | 2 | 146,917 | 147 | 85 | 82 | 9.6 |
| 3 | 40 | 3 | 174,491 | 174 | 80 | 82 | 6.4 |
| 4 | 40 | 4 | 205,021 | 205 | 82 | 82 | 4.8 |
2.1.3. Capillary Tube Reactor in Combination with Ultrasonication

3. Experimental Section
3.1. Test Procedure and Set-Up Using the Orifice Microreactor
3.2. Test Procedure and Set-Up Using the Split and Recombine Reactor and a Capillary Tube Reactor Combined with Ultrasonication
3.3. Preparation of the Intermediate tert-Butyl Potassium Peroxide (TBKP) in Semi-Batch Mode
3.4. Chemicals
3.5. Analytical Part
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Dittmeyer, R.; Keim, W.; Kreysa, G.; Oberholz, A. Winnacker-Küchler-Chemische Technik. In Prozesse und Produkte, Band 3 Anorganische Grundstoffe, Zwischenprodukte; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2005. [Google Scholar]
- Klenk, H.; Götz, P.; Siegmeier, R.; Mayr, W. Ullmanns Encyclopedia of Industrial Chemistry—Peroxy Compounds, Organic; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2000. [Google Scholar]
- Sanchez, J.; Myers, T.N. Peroxides and Peroxide Compounds, Organic Peroxides. Kirk-Othmer Encycl. Chem. Technol. 2000. [Google Scholar] [CrossRef]
- Hordijk, A.; de Groot, J. Experimental data on the thermal kinetics of organic peroxides. Thermochim. Acta 1986, 101, 45–63. [Google Scholar] [CrossRef]
- Ho, T.; Duh, Y.; Chen, J.R. Case studies of incidents in runaway reactions and emerging relief. Process Saf. Prog. 1998, 17, 259–262. [Google Scholar] [CrossRef]
- Duh, Y.; Wu, X.H.; Kao, C. Hazard ratings for organic peroxides. Process Saf. Prog. 2008, 27, 89–99. [Google Scholar] [CrossRef]
- Roßmer, D.; Wobst, S. Verfahren zur Kontinuierlichen Herstellung von Perestern. DD128663 (A1), 30 November 1977. [Google Scholar]
- Winter, H.; Schmid, E. Verfahren zur Herstellung von Percarbonsaeure-tert.-butylestern. DE1518740 (B1), 29 June 1972. [Google Scholar]
- Kohn, P.M. Continuous peroxyester route to make its commercial debut. Chem. Eng. 1978, 7, 88–89. [Google Scholar]
- Milas, N.A.; Surgenor, D.M. Studies in organic peroxides IX. tert-butyl peroxyesters. J. Am. Chem. Soc. 1946, 68, 205–208. [Google Scholar] [CrossRef]
- Hessel, V.; Hardt, S.; Löwe, H. Chemical Micro-Process Engineering—Fundamentals, Modelling, and Reactions; Wiley-VCH: Weinheim, Germany, 2004. [Google Scholar]
- CoPIRIDE Project. Available online: http://www.copiride.eu/about_copiride.html (accessed on 22 July 2015).
- F3 Factory Project. Available online: http://www.f3factory.com/scripts/pages/en/home.php (accessed on 22 July 2015).
- POLYCAT Project. Available online: http://www.polycat-fp7.eu/about_copiride.html (accessed on 22 July 2015).
- Chemie auf Rädern. Available online: http://corporate.evonik.de/de/produkte/product-stories/Pages/chemie-auf-raedern.aspx (accessed on 22 July 2015).
- INVITE Facility. Available online: http://www.invite-research.com/leistungen/leistungsportfolio.html (accessed on 22 July 2015).
- Azzawi, A.; Mehesch, H.E.; Zadow, E.; Frose, S.; Rhede, M. Method for the Production of Organic Peroxides by Means of a Microreaction Technique. WO2007042313 A3, 29 April 2007. [Google Scholar]
- Ebrahimi, F.; Kolehmainen, E.; Oinas, P.; Hietapelto, V.; Turunen, I. Production of unstable percarboxylic acids in a microstructured reactor. Chem. Eng. J. 2011, 167, 713–717. [Google Scholar] [CrossRef]
- Ebrahimi, F.; Kolehmainen, E.; Turunen, I. Safety Advantages of on-side microprocesses. Org. Proc. Res. Dev. 2009, 13, 965–969. [Google Scholar] [CrossRef]
- Wu, W.; Qian, G.; Zhou, X.G.; Yuan, W.K. Peroxidation of methyl ethyl ketone in a microchannel reactor. Chem. Eng. Sci. 2007, 62, 5127–5132. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, W.; Qian, G.; Zhou, X.G. Continuous synthesis of methyl ethyl ketone peroxide in a microreaction system with concentrated hydrogen peroxide. J. Hazard. Mat. 2010, 181, 1024–1030. [Google Scholar] [CrossRef] [PubMed]
- Illg, T.; Hessel, V.; Löb, P.; Schouten, J.C. Continuous synthesis if tert-butyl peroxypivalate using a single-channel microreactor equipped with orifices as emulsification units. ChemSusChem 2011, 4, 392–398. [Google Scholar] [CrossRef] [PubMed]
- Illg, T.; Hessel, V.; Löb, P.; Schouten, J.C. Orifice microreactor for the production of an organic peroxide-non reactive and reactive characterization. Green Chem. 2012, 14, 1420–1433. [Google Scholar] [CrossRef]
- Illg, T. Novel Process Windows for the Safe and Continuous Synthesis of Tert-Butyl Peroxypivalate with Micro Process Technology; Technische Universität Eindhoven: Eindhoven, The Netherlands, 2013. [Google Scholar]
- Fritzsche, L.; Knorr, A. Transformation of the 2nd step of a peroxyester synthesis from semi-batch to continuous mode. Chem. Eng. Process. Process Intensif. 2013, 70, 217–221. [Google Scholar] [CrossRef]
- U.S. Chemical Safety and Hazard Investigation Board. Case Study: Fire and Explosion: Hazards of Benzoyl Peroxide; No. 2003-03-C-OH; U.S. Chemical Safety and Hazard Investigation Board: Washington, DC, USA, 2003.
- Kommission für Anlagensicherheit (KAS), TRAS 410 Erkennen und Beherrschen Exothermer Chemischer Reaktionen, Ermittlung der Gefahren, Bewertung und Zusätzliche Maßnahmen, Commission on Process Safety (Germany) Technical Rules on Installation Safety 410 Identification and Control of Exothermic Chemical Reactions, October 2012, Published in BAnz AT 20.12.2012, A Former Version (TAA-GS-05 Guide for the Identification and Control of Exothermic Chemical Reactions) is available in English. Available online: http://www.kas-bmu.de/publikationen/tras/TRAS_410_09102012.pdf (accessed on 17 December 2015).
- Knorr, A. Anwendung der TRAS 410 auf die Sicherheitstechnische Beurteilung Einer Perestersynthese (Application of the Technical Rule TRAS 410 to the Safety Evaluation of a Peroxide Synthesis). Ph.D. Thesis, Technische Universität Berlin, Berlin, Germany, January 2006. [Google Scholar]
- Fritzsche, L.; Knorr, A. Specification of the limit temperature Texo for a peroxide synthesis using different estimation methods. Chem. Eng. Trans. 2014, 36, 121–126. [Google Scholar]
- Karbstein, H.; Schubert, H. Developments in the continuous mechanical production of oil-water macro-emulsions. Chem. Eng. Process. 1995, 34, 205–211. [Google Scholar] [CrossRef]
- Stang, M.; Schuchmann, H.; Schubert, H. Emulsification in High-Pressure Homogenizers. Eng. Life Sci. 2001, 4, 151–157. [Google Scholar] [CrossRef]
- Bošković, D.; Löbbecke, S.; Groß, A.; Köhler, M. Residence Time distribution studies in microfluidic mixing structures. Chem. Eng. Technol. 2011, 34, 361–370. [Google Scholar] [CrossRef]
- Rivas, D.F.; Ashokkumar, M.; Leong, T.; Yasui, K.; Tuziuti, T.; Kentish, S.; Lohse, D.; Gardeniers, H.J.G.E. Sonoluminescence and sonochemiluminescence from a microreactor. Ultrason. Sonochem. 2012, 19, 1252–1259. [Google Scholar] [CrossRef] [PubMed]
- Wandrey, P.-A.; Wehrstedt, K.-D. Sensitisation of liquid organic peroxides against detonation impact-an excessive or practice-orientated test method in Chemical Reactions. In Proceedings of the 35th Tutzing Symposium, Tutzing, Germany, 10–13 March 1997; Kreysa, G., Langer, O.-U., Pilz, V., Eds.; DECHEMA: Frankfurt am Main, Germany, 1997; pp. 133–146. [Google Scholar]
- UN Recommendations on the Transport of Dangerous Goods, Manual of Tests and Criteria, 5th ed.; United Nation: New York, NY, USA; Geneva, Switzerland, 2009.
- Sample Availability: Samples are not available because storage of peroxide compounds is limited to a short time/ to a specific time period. They decompose continuously, even if stored at low temperatures. The original composition changes, concentration of peroxide degrades and byproducts may be produced.
© 2015 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Illg, T.; Knorr, A.; Fritzsche, L. Microreactors—A Powerful Tool to Synthesize Peroxycarboxylic Esters. Molecules 2016, 21, 5. https://doi.org/10.3390/molecules21010005
Illg T, Knorr A, Fritzsche L. Microreactors—A Powerful Tool to Synthesize Peroxycarboxylic Esters. Molecules. 2016; 21(1):5. https://doi.org/10.3390/molecules21010005
Chicago/Turabian StyleIllg, Tobias, Annett Knorr, and Lutz Fritzsche. 2016. "Microreactors—A Powerful Tool to Synthesize Peroxycarboxylic Esters" Molecules 21, no. 1: 5. https://doi.org/10.3390/molecules21010005
APA StyleIllg, T., Knorr, A., & Fritzsche, L. (2016). Microreactors—A Powerful Tool to Synthesize Peroxycarboxylic Esters. Molecules, 21(1), 5. https://doi.org/10.3390/molecules21010005