
The Potential of Secondary Metabolites from Plants as Drugs or Leads against Protozoan Neglected Diseases—Part III: In-Silico Molecular Docking Investigations



Abstract
:
1. Introduction
2. Parasite Molecular Targets
3. Molecular Docking Studies
3.1. Leishmania and Trypanosoma Targets
3.2. Plasmodium Targets
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Murray, C.J.L.; Rosenfeld, L.C.; Lim, S.S.; Andrews, K.G.; Foreman, J.K.; Haring, D.; Fullman, N.; Naghavi, M.; Lozano, R.; Lopez, A.D. Global malaria mortality between 1980 and 2010: A systematic analysis. Lancet 2012, 379, 413–431. [Google Scholar] [CrossRef]
- Rassi, A., Jr.; Rassi, A.; Marin-Neto, J.A. Chagas disease. Lancet 2010, 375, 1388–1402. [Google Scholar] [CrossRef]
- Brun, R.; Blum, J.; Chappuis, F.; Burri, C. Human African trypanosomiasis. Lancet 2010, 375, 148–159. [Google Scholar] [CrossRef] [Green Version]
- Alvar, J.; Vélez, I.D.; Bern, C.; Herrero, M.; Desjeux, P.; Cano, J.; Jannin, J.; den Boer, M.; The WHO Leishmaniasis Control Team. Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 2012, 7, e35671. [Google Scholar] [CrossRef] [PubMed]
- Burrows, J.N.; Waterson, D. Discovering new medicines to control and eradicate malaria. Top. Med. Chem. 2011, 7, 125–180. [Google Scholar]
- Coura, J.R.; de Castro, S.L. A critical review on Chagas disease chemotherapy. Mem. Inst. Oswaldo Cruz 2002, 97, 3–24. [Google Scholar] [CrossRef]
- Fairlamb, A.H. Chemotherapy of human African trypanosomiasis: Current and future prospects. Trends Parasitol. 2003, 19, 488–494. [Google Scholar] [CrossRef] [PubMed]
- Croft, S.L.; Olliaro, P. Leishmaniasis chemotherapy—Challenges and opportunities. Clin. Microbiol. Infect. 2011, 17, 1478–1483. [Google Scholar] [CrossRef] [PubMed]
- Harvey, A.L.; Edrada-Ebal, R.A.; Quinn, R.J. The re-emergence of natural products for drug discovery in the genomics era. Nat. Rev. Drug Discov. 2015, 14, 111–129. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.A.; Biavatti, M.W.; Brun, R.; da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.G.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part I. Curr. Med. Chem. 2012, 19, 2128–2175. [Google Scholar] [PubMed]
- Schmidt, T.J.; Khalid, S.A.; Romanha, A.J.; Alves, T.M.A.; Biavatti, M.W.; Brun, R.; da Costa, F.B.; de Castro, S.L.; Ferreira, V.F.; de Lacerda, M.V.G.; et al. The potential of secondary metabolites from plants as drugs or leads against protozoan neglected diseases—Part II. Curr. Med. Chem. 2012, 19, 2176–2228. [Google Scholar] [CrossRef] [PubMed]
- Annang, F.; Genilloud, O.; Vicente, F. Contribution of natural products to drug discovery in tropical diseases. In Comprehensive Analysis of Parasite Biology: From Metabolism to Drug Discovery; Müller, S., Cerdan, R., Radulescu, O., Eds.; Wiley-VCH: Weinheim, Germany, 2016; pp. 75–104. [Google Scholar]
- Rosén, J.; Gottfries, J.; Muresan, S.; Backlund, A.; Oprea, T.I. Novel chemical space exploration via natural products. J. Med. Chem. 2009, 52, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Feher, M.; Schmidt, J.M. Property distributions: Differences between drugs, natural products, and molecules from combinatorial chemistry. J. Chem. Inform. Comput. Sci. 2003, 43, 218–227. [Google Scholar] [CrossRef] [PubMed]
- Ganesan, A. The impoact of natural products upon modern drug discovery. Curr. Opin. Chem. Biol. 2008, 12, 306–317. [Google Scholar] [CrossRef] [PubMed]
- Keller, T.H.; Pichota, A.; Yin, Z. A practical view of ‘druggability’. Curr. Opin. Chem. Biol. 2006, 10, 357–361. [Google Scholar] [CrossRef] [PubMed]
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.L.; Ullman, B.; Brennan, R.G.; Hill, C.P. Crystal structures of adenine phosphoribosyltransferase from Leishmania donovani. EMBO J. 1999, 18, 3533–3545. [Google Scholar] [CrossRef] [PubMed]
- Kuettel, S.; Greenwald, J.; Kostrewa, D.; Ahmed, S.; Scapozza, L.; Perozzo, R. Crystal structures of T. b. rhodesiense adenosine kinase complexed with inhibitor and activator: Implications for catalysis and hyperactivation. PLoS Negl. Trop. Dis. 2011, 5, e1164. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timm, J.; González-Pacanowska, D.; Wilson, K.S. Structures of adenosine kinase from Trypanosoma brucei brucei. Acta Crystallogr. F Struct. Biol. Commun. 2014, 70, 34–39. [Google Scholar] [CrossRef] [PubMed]
- Eaazhisai, K.; Jayalakshmi, R.; Gayathri, P.; Anand, R.P.; Sumathy, K.; Balaram, H.; Murthy, M.R. Crystal structure of fully ligated adenylosuccinate synthetase from Plasmodium falciparum. J. Mol. Biol. 2004, 335, 1251–1264. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.K.; Osman, K.T.; Loppnau, P.; Arrowsmith, C.H.; Edwards, A.M.; Bountra, C.; Hui, R.; Lin, Y.H. Crystal Structure of an M17 aminopeptidase from Trypanosoma brucei, Tb427tmp.02.4440. Unpublished work. 2012; doi:10.2210/pdb4efd/pdb. [Google Scholar]
- Vulliez-Le Normand, B.; Tonkin, M.L.; Lamarque, M.H.; Langer, S.; Hoos, S.; Roques, M.; Saul, F.A.; Faber, B.W.; Bentley, G.A.; Boulanger, M.J.; et al. Structural and functional insights into the malaria parasite moving junction complex. PLoS Pathog. 2012, 8, e1002755. [Google Scholar] [CrossRef] [PubMed]
- D’Antonio, E.L.; Ullman, B.; Roberts, S.C.; Dixit, U.G.; Wilson, M.E.; Hai, Y.; Christianson, D.W. Crystal structure of arginase from Leishmania mexicana and implications for the inhibition of polyamine biosynthesis in parasitic infections. Arch. Biochem. Biophys. 2013, 535, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Dowling, D.P.; Ilies, M.; Olszewski, K.L.; Portugal, S.; Mota, M.M.; Llinás, M.; Christianson, D.W. Crystal structure of arginase from Plasmodium falciparum and implications for l-arginine depletion in malarial infection. Biochemistry 2010, 49, 5600–5608. [Google Scholar] [CrossRef] [PubMed]
- Ilies, M.; Di Costanzo, L.; Dowling, D.P.; Thorn, K.J.; Christianson, D.W. Binding of α,α-disubstituted amino acids to arginase suggests new avenues for inhibitor design. J. Med. Chem. 2011, 54, 5432–5443. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, P.; Haouz, A.; Pereira, C.A.; Aguilar, C.; Alzari, P.M. The crystal structure of Trypanosoma cruzi arginine kinase. Proteins 2007, 69, 209–212. [Google Scholar] [CrossRef] [PubMed]
- Wrenger, C.; Müller, I.B.; Schifferdecker, A.J.; Jain, R.; Jordanova, R.; Groves, M.R. Specific inhibition of the aspartate aminotransferase of Plasmodium falciparum. J. Mol. Biol. 2011, 405, 956–971. [Google Scholar] [CrossRef] [PubMed]
- Hain, A.U.; Weltzer, R.R.; Hammond, H.; Jayabalasingham, B.; Dinglasan, R.R.; Graham, D.R.; Colquhoun, D.R.; Coppens, I.; Bosch, J. Structural characterization and inhibition of the Plasmodium Atg8-Atg3 interaction. J. Struct. Biol. 2012, 180, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Wu, P.; Marion-Tsukamaki, R.; Mackey, Z.B.; Brinen, L.S. Crystal Structures of TbCatB and rhodesain, potential chemotherapeutic targets and major cysteine proteases of Trypanosoma brucei. PLoS Negl. Trop. Dis. 2010, 4, e701. [Google Scholar] [CrossRef] [PubMed]
- Koopmann, R.; Cupelli, K.; Redecke, L.; Nass, K.; Deponte, D.P.; White, T.A.; Stellato, F.; Rehders, D.; Liang, M.; Andreasson, J.; et al. In vivo protein crystallization opens new routes in structural biology. Nat. Methods 2012, 9, 259–262. [Google Scholar] [CrossRef] [PubMed]
- Redecke, L.; Nass, K.; DePonte, D.P.; White, T.A.; Rehders, D.; Barty, A.; Stellato, F.; Liang, M.; Barends, T.R.; Boutet, S.; et al. Natively inhibited Trypanosoma brucei cathepsin B structure determined by using an X-ray laser. Science 2013, 339, 227–230. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.K.; Pizarro, J.C.; Artz, J.D.; Amaya, M.F.; Xiao, T.; Lew, J.; Wasney, G.; Senesterra, G.; Kozieradzki, I.; Cossar, D.; et al. Crystal structure of choline kinase from Plasmodium Falciparum, PF14_0020. Unpublished work. 2009; doi:10.2210/pdb3fi8/pdb. [Google Scholar]
- Gillmor, S.A.; Craik, C.S.; Fletterick, R.J. Structural determinants of specificity in the cysteine protease cruzain. Protein Sci. 1997, 6, 1603–1611. [Google Scholar] [CrossRef] [PubMed]
- Brinen, L.S.; Hansell, E.; Cheng, J.; Roush, W.R.; McKerrow, J.H.; Fletterick, R.J. A target within the target: Probing cruzain’s P1’ site to define structural determinants for the Chagas’ disease protease. Structure 2000, 8, 831–840. [Google Scholar] [CrossRef]
- Huang, L.; Brinen, L.S.; Ellman, J.A. Crystal structures of reversible ketone-based inhibitors of the cysteine protease cruzain. Bioorg. Med. Chem. 2003, 11, 21–92. [Google Scholar] [CrossRef]
- Choe, Y.; Brinen, L.S.; Price, M.S.; Engel, J.C.; Lange, M.; Grisostomi, C.; Weston, S.G.; Pallai, P.V.; Cheng, H.; Hardy, L.W.; et al. Development of α-keto-based inhibitors of cruzain, a cysteine protease implicated in Chagas disease. Bioorg. Med. Chem. 2005, 13, 2141–2156. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Lee, J.H.; Farady, C.J.; Marion, R.; Rickert, M.; Sajid, M.; Pandey, K.C.; Caffrey, C.R.; Legac, J.; Hansell, E.; et al. Vinyl sulfones as antiparasitic agents and a structural basis for drug design. J. Biol. Chem. 2009, 284, 25697–25703. [Google Scholar] [CrossRef] [PubMed]
- Bryant, C.; Kerr, I.D.; Debnath, M.; Ang, K.K.; Ratnam, J.; Ferreira, R.S.; Jaishankar, P.; Zhao, D.; Arkin, M.R.; McKerrow, J.H.; et al. Novel non-peptidic vinylsulfones targeting the S2 and S3 subsites of parasite cysteine proteases. Bioorg. Med. Chem. Lett. 2009, 19, 6218–6221. [Google Scholar] [CrossRef] [PubMed]
- Mott, B.T.; Ferreira, R.S.; Simeonov, A.; Jadhav, A.; Ang, K.K.; Leister, W.; Shen, M.; Silveira, J.T.; Doyle, P.S.; Arkin, M.R.; et al. Identification and optimization of inhibitors of Trypanosomal cysteine proteases: Cruzain, rhodesain, and TbCatB. J. Med. Chem. 2010, 53, 52–60. [Google Scholar] [CrossRef] [PubMed]
- Brak, K.; Kerr, I.D.; Barrett, K.T.; Fuchi, N.; Debnath, M.; Ang, K.; Engel, J.C.; McKerrow, J.H.; Doyle, P.S.; Brinen, L.S.; et al. Nonpeptidic tetrafluorophenoxymethyl ketone cruzain inhibitors as promising new leads for Chagas disease chemotherapy. J. Med. Chem. 2010, 53, 1763–1773. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.T.; Brinen, L.S.; Kerr, I.D.; Hansell, E.; Doyle, P.S.; McKerrow, J.H.; Roush, W.R. In vitro and in vivo studies of the trypanocidal properties of WRR-483 against Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2010, 4, e825. [Google Scholar] [CrossRef] [PubMed]
- Wiggers, H.J.; Rocha, J.R.; Fernandes, W.B.; Sesti-Costa, R.; Carneiro, Z.A.; Cheleski, J.; da Silva, A.B.; Juliano, L.; Cezari, M.H.; Silva, J.S.; et al. Non-peptidic cruzain inhibitors with trypanocidal activity discovered by virtual screening and in vitro assay. PLoS Negl. Trop. Dis. 2013, 7, e2370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brinen, L.S.; Gillmor, S.A.; Fletterick, R.J. Crystal structures of cruzain bound to three different substrates. Unpublished work. 2003; doi:10.2210/pdb1ewl/pdb, doi:10.2210/pdb1ewm/pdb, doi:10.2210/pdb1ewo/pdb. [Google Scholar]
- Venugopal, V.; Datta, A.K.; Bhattacharyya, D.; Dasgupta, D.; Banerjee, R. Structure of cyclophilin from Leishmania donovani bound to cyclosporin at 2.6 Å resolution: Correlation between structure and thermodynamic data. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 1187–1195. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, T.L.; Merritt, E.A. Cyclophilin from Leishmania major. Unpublished work. 2006; doi:10.2210/pdb2hqj/pdb. [Google Scholar]
- Peterson, M.R.; Hall, D.R.; Berriman, M.; Nunes, J.A.; Leonard, G.A.; Fairlamb, A.H.; Hunter, W.N. The three-dimensional structure of a Plasmodium falciparum cyclophilin in complex with the potent anti-malarial cyclosporin A. J. Mol. Biol. 2000, 298, 123–133. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, P.K.; Westrop, G.D.; Ramos, T.; Müller, S.; Coombs, G.H.; Hunter, W.N. Structure of Leishmania major cysteine synthase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2012, 68, 738–743. [Google Scholar] [CrossRef] [PubMed]
- Hemsworth, G.R.; Moroz, O.V.; Fogg, M.J.; Scott, B.; Bosch-Navarrete, C.; González-Pacanowska, D.; Wilson, K.S. The crystal structure of the Leishmania major deoxyuridine triphosphate nucleotidohydrolase in complex with nucleotide analogues, dUMP, and deoxyuridine. J. Biol. Chem. 2011, 286, 16470–16481. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, J.L.; Leal, I.; Nguyen, C.; Kasinathan, G.; Bell, E.; Jones, A.F.; Berry, C.; Benito, A.; Turkenburg, J.P.; Dodson, E.J.; et al. dUTPase as a platform for antimalarial drug design: Structural basis for the selectivity of a class of nucleoside inhibitors. Structure 2005, 13, 329–338. [Google Scholar] [CrossRef] [PubMed]
- Baragaña, B.; McCarthy, O.; Sánchez, P.; Bosch-Navarrete, C.; Kaiser, M.; Brun, R.; Whittingham, J.L.; Roberts, S.M.; Zhou, X.X.; Wilson, K.S.; et al. β-Branched acyclic nucleoside analogues as inhibitors of Plasmodium falciparum dUTPase. Bioorg. Med. Chem. 2011, 19, 2378–2391. [Google Scholar] [CrossRef] [PubMed]
- Hampton, S.E.; Baragaña, B.; Schipani, A.; Bosch-Navarrete, C.; Musso-Buendía, J.A.; Recio, E.; Kaiser, M.; Whittingham, J.L.; Roberts, S.M.; Shevtsov, M.; et al. Design, synthesis, and evaluation of 5′-diphenyl nucleoside analogues as inhibitors of the Plasmodium falciparum dUTPase. ChemMedChem 2011, 6, 1816–1831. [Google Scholar] [CrossRef] [PubMed]
- Hemsworth, G.R.; González-Pacanowska, D.; Wilson, K.S. On the catalytic mechanism of dimeric dUTPases. Biochem. J. 2013, 456, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Harkiolaki, M.; Dodson, E.J.; Bernier-Villamor, V.; Turkenburg, J.P.; González-Pacanowska, D.; Wilson, K.S. The crystal structure of Trypanosoma cruzi dUTPase reveals a novel dUTP/dUDP binding fold. Structure 2004, 12, 41–53. [Google Scholar] [CrossRef] [PubMed]
- Almo, S.C.; Bonanno, J.B.; Sauder, J.M.; Emtage, S.; Dilorenzo, T.P.; Malashkevich, V.; Wasserman, S.R.; Swaminathan, S.; Eswaramoorthy, S.; Agarwal, R.; et al. Structural genomics of protein phosphatases. J. Struct. Funct. Genom. 2007, 8, 121–140. [Google Scholar] [CrossRef] [PubMed]
- Yuvaniyama, J.; Chitnumsub, P.; Kamchonwongpaisan, S.; Vanichtanankul, J.; Sirawaraporn, W.; Taylor, P.; Walkinshaw, M.D.; Yuthavong, Y. Insights into antifolate resistance from malarial DHFR-TS structures. Nat. Struct. Biol. 2003, 10, 357–365. [Google Scholar] [CrossRef] [PubMed]
- Dasgupta, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Maneeruttanarungroj, C.; Nichols, S.E.; Lyons, T.M.; Tirado-Rives, J.; Jorgensen, W.L.; Yuthavong, Y.; Anderson, K.S. Exploiting structural analysis, in silico screening, and serendipity to identify novel inhibitors of drug-resistant falciparum malaria. ACS Chem. Biol. 2009, 4, 29–40. [Google Scholar] [CrossRef] [PubMed]
- Vanichtanankul, J.; Taweechai, S.; Yuvaniyama, J.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Yuthavong, Y. Trypanosomal dihydrofolate reductase reveals natural antifolate resistance. ACS Chem. Biol. 2011, 6, 905–911. [Google Scholar] [CrossRef] [PubMed]
- Vanichtanankul, J.; Taweechai, S.; Uttamapinant, C.; Chitnumsub, P.; Vilaivan, T.; Yuthavong, Y.; Kamchonwongpaisan, S. Combined spatial limitation around residues 16 and 108 of Plasmodium falciparum dihydrofolate reductase explains resistance to cycloguanil. Antimicrob. Agents Chemother. 2012, 56, 3928–3935. [Google Scholar] [CrossRef] [PubMed]
- Yuthavong, Y.; Tarnchompoo, B.; Vilaivan, T.; Chitnumsub, P.; Kamchonwongpaisan, S.; Charman, S.A.; McLennan, D.N.; White, K.L.; Vivas, L.; Bongard, E.; et al. Malarial dihydrofolate reductase as a paradigm for drug development against a resistance-compromised target. Proc. Natl. Acad. Sci. USA 2012, 109, 16823–16828. [Google Scholar] [CrossRef] [PubMed]
- Schormann, N.; Senkovich, O.; Walker, K.; Wright, D.L.; Anderson, A.C.; Rosowsky, A.; Ananthan, S.; Shinkre, B.; Velu, S.; Chattopadhyay, D. Structure-based approach to pharmacophore identification, in silico screening, and three-dimensional quantitative structure-activity relationship studies for inhibitors of Trypanosoma cruzi dihydrofolate reductase function. Proteins 2008, 73, 889–901. [Google Scholar] [CrossRef] [PubMed]
- Senkovich, O.; Schormann, N.; Chattopadhyay, D. Structures of dihydrofolate reductase-thymidylate synthase of Trypanosoma cruzi in the folate-free state and in complex with two antifolate drugs, trimetrexate and methotrexate. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 704–716. [Google Scholar] [CrossRef] [PubMed]
- Schormann, N.; Velu, S.E.; Murugesan, S.; Senkovich, O.; Walker, K.; Chenna, B.C.; Shinkre, B.; Desai, A.; Chattopadhyay, D. Synthesis and characterization of potent inhibitors of Trypanosoma cruzi dihydrofolate reductase. Bioorg. Med. Chem. 2010, 18, 4056–4066. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, T.L.; Merritt, E.A.; Ullman, B.; Yates, P.A. Crystal structure of dihydroorotate dehydrogenase from Leishmania donovani. Unpublished work. 2008; doi:10.2210/pdb3c61/pdb. [Google Scholar]
- Cheleski, J.; Rocha, J.R.; Pinheiro, M.P.; Wiggers, H.J.; da Silva, A.B.; Nonato, M.C.; Montanari, C.A. Novel insights for dihydroorotate dehydrogenase class 1A inhibitors discovery. Eur. J. Med. Chem. 2010, 45, 5899–5909. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.T.; Feliciano, P.R.; Pinheiro, M.P.; Nonato, M.C. Crystal structure of dihydroorotate dehydrogenase from Leishmania major. Biochimie 2012, 94, 1739–1748. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, M.P.; Emery, F.S.; Nonato, M.C. Target sites for the design of anti-trypanosomatid drugs based on the structure of dihydroorotate dehydrogenase. Curr. Pharm. Des. 2013, 19, 2615–2627. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.T.; Feliciano, P.R.; Nonato, M.C. Crystal structure of Leishmania major dihydroorotate dehydrogenase. Unpublished work. 2010; doi:10.2210/pdb3gye/pdb, doi:10.2210/pdb3gz3/pdb. [Google Scholar]
- Hurt, D.E.; Widom, J.; Clardy, J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Crystallogr. D Biol. Crystallogr. 2006, 62, 312–323. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Gujjar, R.; El Mazouni, F.; Kaminsky, W.; Malmquist, N.A.; Goldsmith, E.J.; Rathod, P.K.; Phillips, M.A. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J. Biol. Chem. 2009, 284, 26999–27009. [Google Scholar] [CrossRef] [PubMed]
- Booker, M.L.; Bastos, C.M.; Kramer, M.L.; Barker, R.H.; Skerlj, R.; Sidhu, A.B.; Deng, X.; Celatka, C.; Cortese, J.F.; Guerrero Bravo, J.E.; et al. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J. Biol. Chem. 2010, 285, 33054–33064. [Google Scholar] [CrossRef] [PubMed]
- Coteron, J.M.; Marco, M.; Esquivias, J.; Deng, X.; White, K.L.; White, J.; Koltun, M.; El Mazouni, F.; Kokkonda, S.; Katneni, K.; et al. Structure-guided lead optimization of triazolopyrimidine-ring substituents identifies potent Plasmodium falciparum dihydroorotate dehydrogenase inhibitors with clinical candidate potential. J. Med. Chem. 2011, 54, 5540–5561. [Google Scholar] [CrossRef] [PubMed]
- Ross, L.S.; Javier Gamo, F.; Lafuente-Monasterio, M.J.; Singh, O.M.; Rowland, P.; Wiegand, R.C.; Wirth, D.F. In vitro resistance selections for Plasmodium falciparum dihydroorotate dehydrogenase inhibitors give mutants with multiple point mutations in the drug-binding site and altered growth. J. Biol. Chem. 2014, 289, 17980–17995. [Google Scholar] [CrossRef] [PubMed]
- Arakaki, T.L.; Buckner, F.S.; Gillespie, J.R.; Malmquist, N.A.; Phillips, M.A.; Kalyuzhniy, O.; Luft, J.R.; Detitta, G.T.; Verlinde, C.L.; van Voorhis, W.C.; et al. Characterization of Trypanosoma brucei dihydroorotate dehydrogenase as a possible drug target; structural, kinetic and RNAi studies. Mol. Microbiol. 2008, 68, 37–50. [Google Scholar] [CrossRef] [PubMed]
- Pinheiro, M.P.; Iulek, J.; Cristina Nonato, M. Crystal structure of Trypanosoma cruzi dihydroorotate dehydrogenase from Y strain. Biochem. Biophys. Res. Commun. 2008, 369, 812–817. [Google Scholar] [CrossRef] [PubMed]
- Inaoka, D.K.; Sakamoto, K.; Shimizu, H.; Shiba, T.; Kurisu, G.; Nara, T.; Aoki, T.; Kita, K.; Harada, S. Structures of Trypanosoma cruzi dihydroorotate dehydrogenase complexed with substrates and products: Atomic resolution insights into mechanisms of dihydroorotate oxidation and fumarate reduction. Biochemistry 2008, 47, 10881–10891. [Google Scholar] [CrossRef] [PubMed]
- Inaoka, D.K.; Shimizu, H.; Sakamoto, K.; Shiba, T.; Kurisu, G.; Nara, T.; Aoki, T.; Harada, S.; Kita, K. Crystal structures of Trypanosoma cruzi dihydroorotate dehydrogenase. Unpublished work. 2008; doi:10.2210/pdb2e68/pdb, doi:10.2210/pdb2e6a/pdb, doi:10.2210/pdb2e6f/pdb, doi:10.2210/pdb2djl/pdb, doi:10.2210/pdb2djx/pdb. [Google Scholar]
- Inaoka, D.K.; Iida, M.; Tabuchi, T.; Lee, N.; Matsuoka, S.; Shiba, T.; Sakamoto, K.; Suzuki, S.; Balogun, E.O.; Nara, T.; et al. Structures of Trypanosoma cruzi dihydroorotate dehydrogenase. Unpublished work. 2013; doi:10.2210/pdb3w1a/pdb, doi:10.2210/pdb3w1l/pdb, doi:10.2210/pdb3w1m/pdb, doi:10.2210/pdb3w1n/pdb, doi:10.2210/pdb3w1p/pdb, doi:10.2210/pdb3w1q/pdb, doi:10.2210/pdb3w1r/pdb, doi:10.2210/pdb3w1t/pdb, doi:10.2210/pdb3w1ut/pdb, doi:10.2210/pdb3w1x/pdb, doi:10.2210/pdb3w22/pdb, doi:10.2210/pdb3w23/pdb, doi:10.2210/pdb3w2j/pdb, doi:10.2210/pdb3w2k/pdb, doi:10.2210/pdb3w2l/pdb, doi:10.2210/pdb3w2m/pdb, doi:10.2210/pdb3w2n/pdb, doi:10.2210/pdb3w2u/pdb. [Google Scholar]
- Inaoka, D.K.; Iida, M.; Tabuchi, T.; Lee, N.; Matsuoka, S.; Shiba, T.; Sakamoto, K.; Suzuki, S.; Rocha, J.R.; Balogun, E.O.; et al. Structure of Trypanosoma cruzi dihydroorotate dehydrogenase in complex with MII-4-053. Unpublished work. 2014; doi:10.2210/pdb3w3o/pdb. [Google Scholar]
- Inaoka, D.K.; Iida, M.; Tabuchi, T.; Lee, N.; Hashimoto, S.; Matsuoka, S.; Kuranaga, T.; Shiba, T.; Sakamoto, K.; Suzuki, S.; et al. Structures of Trypanosoma cruzi dihydroorotate dehydrogenase. Unpublished work. 2014; doi:10.2210/pdb3w6y/pdb, doi:10.2210/pdb3w70/pdb, doi:10.2210/pdb3w71/pdb, doi:10.2210/pdb3w72/pdb, doi:10.2210/pdb3w73/pdb, doi:10.2210/pdb3w74/pdb, doi:10.2210/pdb3w75/pdb, doi:10.2210/pdb3w76/pdb, doi:10.2210/pdb3w7c/pdb, doi:10.2210/pdb3w7d/pdb, doi:10.2210/pdb3w7e/pdb, doi:10.2210/pdb3w7g/pdb, doi:10.2210/pdb3w7h/pdb, doi:10.2210/pdb3w7i/pdb, doi:10.2210/pdb3w7j/pdb, doi:10.2210/pdb3w7k/pdb, doi:10.2210/pdb3w7l/pdb, doi:10.2210/pdb3w7m/pdb, doi:10.2210/pdb3w7n/pdb, doi:10.2210/pdb3w7o/pdb, doi:10.2210/pdb3w7p/pdb, doi:10.2210/pdb3w7q/pdb, doi:10.2210/pdb4jd4/pdb, doi:10.2210/pdb4jdb/pdb. [Google Scholar]
- Inaoka, D.K.; Iida, M.; Tabuchi, T.; Lee, N.; Hashimoto, S.; Matsuoka, S.; Kuranaga, T.; Shiba, T.; Sakamoto, K.; Suzuki, S.; et al. Structures of Trypanosoma cruzi dihydroorotate dehydrogenase. Unpublished work. 2014; doi:10.2210/pdb3w83/pdb, doi:10.2210/pdb3w84/pdb, doi:10.2210/pdb3w85/pdb. [Google Scholar]
- Inaoka, D.K.; Hashimoto, S.; Rocha, J.R.; Iida, M.; Tabuchi, T.; Lee, N.; Matsuoka, S.; Kuranaga, T.; Shiba, T.; Balogun, E.O.; et al. Structures of Trypanosoma cruzi dihydroorotate dehydrogenase. Unpublished work. 2014; doi:10.2210/pdb3w86/pdb, doi:10.2210/pdb3w87/pdb, doi:10.2210/pdb3w88/pdb. [Google Scholar]
- Bhatt, T.K.; Yogavel, M.; Wydau, S.; Berwal, R.; Sharma, A. Ligand-bound structures provide atomic snapshots for the catalytic mechanism of D-amino acid deacylase. J. Biol. Chem. 2010, 285, 5917–5930. [Google Scholar] [CrossRef] [PubMed]
- Yogavel, M.; Khan, S.; Bhatt, T.K.; Sharma, A. Structure of d-tyrosyl-tRNATyr deacylase using home-source Cu Kalpha and moderate-quality iodide-SAD data: Structural polymorphism and HEPES-bound enzyme states. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 584–592. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, S.; Routh, S.B.; Kamarthapu, V.; Chalissery, J.; Muthukumar, S.; Hussain, T.; Kruparani, S.P.; Deshmukh, M.V.; Sankaranarayanan, R. Mechanism of chiral proofreading during translation of the genetic code. Elife 2013, 2, e01519. [Google Scholar] [CrossRef] [PubMed]
- Da Silva Giotto, M.T.; Hannaert, V.; Vertommen, D.; de AS Navarro, M.V.; Rider, M.H.; Michels, P.A.; Garratt, R.C.; Rigden, D.J. The crystal structure of Trypanosoma brucei enolase: Visualisation of the inhibitory metal binding site III and potential as target for selective, irreversible inhibition. J. Mol. Biol. 2003, 331, 653–665. [Google Scholar] [CrossRef]
- De AS Navarro, M.V.; Gomes Dias, S.M.; Mello, L.V.; da Silva Giotto, M.T.; Gavalda, S.; Blonski, C.; Garratt, R.C.; Rigden, D.J. Structural flexibility in Trypanosoma brucei enolase revealed by X-ray crystallography and molecular dynamics. FEBS J. 2007, 274, 5077–5089. [Google Scholar] [CrossRef] [PubMed]
- Perozzo, R.; Kuo, M.; Sidhu, A.B.; Valiyaveettil, J.T.; Bittman, R.; Jacobs, W.R.; Fidock, D.A.; Sacchettini, J.C. Structural elucidation of the specificity of the antibacterial agent triclosan for malarial enoyl acyl carrier protein reductase. J. Biol. Chem. 2002, 277, 13106–13114. [Google Scholar] [CrossRef] [PubMed]
- Pidugu, L.S.; Kapoor, M.; Surolia, N.; Surolia, A.; Suguna, K. Structural basis for the variation in triclosan affinity to enoyl reductases. J. Mol. Biol. 2004, 343, 147–155. [Google Scholar] [CrossRef] [PubMed]
- Freundlich, J.S.; Anderson, J.W.; Sarantakis, D.; Shieh, H.M.; Yu, M.; Valderramos, J.C.; Lucumi, E.; Kuo, M.; Jacobs, W.R.; Fidock, D.A.; et al. Synthesis, biological activity, and X-ray crystal structural analysis of diaryl ether inhibitors of malarial enoyl acyl carrier protein reductase. Part 1: 4′-substituted triclosan derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 5247–5252. [Google Scholar] [CrossRef] [PubMed]
- Muench, S.P.; Prigge, S.T.; McLeod, R.; Rafferty, J.B.; Kirisits, M.J.; Roberts, C.W.; Mui, E.J.; Rice, D.W. Studies of Toxoplasma gondii and Plasmodium falciparum enoyl acyl carrier protein reductase and implications for the development of antiparasitic agents. Acta Crystallogr. D Biol. Crystallogr. 2007, 63, 328–338. [Google Scholar] [CrossRef] [PubMed]
- Freundlich, J.S.; Wang, F.; Tsai, H.C.; Kuo, M.; Shieh, H.M.; Anderson, J.W.; Nkrumah, L.J.; Valderramos, J.C.; Yu, M.; Kumar, T.R.; et al. X-ray structural analysis of Plasmodium falciparum enoyl acyl carrier protein reductase as a pathway toward the optimization of triclosan antimalarial efficacy. J. Biol. Chem. 2007, 282, 25436–25444. [Google Scholar] [CrossRef] [PubMed]
- Maity, K.; Bhargav, S.P.; Sankaran, B.; Surolia, N.; Surolia, A.; Suguna, K. X-ray crystallographic analysis of the complexes of enoyl acyl carrier protein reductase of Plasmodium falciparum with triclosan variants to elucidate the importance of different functional groups in enzyme inhibition. IUBMB Life 2010, 62, 467–476. [Google Scholar] [PubMed]
- Belluti, F.; Perozzo, R.; Lauciello, L.; Colizzi, F.; Kostrewa, D.; Bisi, A.; Gobbi, S.; Rampa, A.; Bolognesi, M.L.; Recanatini, M.; et al. Design, synthesis, and biological and crystallographic evaluation of novel inhibitors of Plasmodium falciparum enoyl-ACP-reductase (PfFabI). J. Med. Chem. 2013, 56, 7516–7526. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.X.; Pandey, K.C.; Somoza, J.R.; Sijwali, P.S.; Kortemme, T.; Brinen, L.S.; Fletterick, R.J.; Rosenthal, P.J.; McKerrow, J.H. Structural basis for unique mechanisms of folding and hemoglobin binding by a malarial protease. Proc. Natl. Acad. Sci. USA 2006, 103, 11503–11508. [Google Scholar] [CrossRef] [PubMed]
- Hogg, T.; Nagarajan, K.; Herzberg, S.; Chen, L.; Shen, X.; Jiang, H.; Wecke, M.; Blohmke, C.; Hilgenfeld, R.; Schmidt, C.L. Structural and functional characterization of Falcipain-2, a hemoglobinase from the malarial parasite Plasmodium falciparum. J. Biol. Chem. 2006, 281, 25425–25437. [Google Scholar] [CrossRef] [PubMed]
- Wang, S.X.; Pandey, K.C.; Scharfstein, J.; Whisstock, J.; Huang, R.K.; Jacobelli, J.; Fletterick, R.J.; Rosenthal, P.J.; Abrahamson, M.; Brinen, L.S.; et al. The structure of chagasin in complex with a cysteine protease clarifies the binding mode and evolution of an inhibitor family. Structure 2007, 15, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Kerr, I.D.; Lee, J.H.; Pandey, K.C.; Harrison, A.; Sajid, M.; Rosenthal, P.J.; Brinen, L.S. Structures of falcipain-2 and falcipain-3 bound to small molecule inhibitors: Implications for substrate specificity. J. Med. Chem. 2009, 52, 852–857. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.; Heitmann, A.; Witt, T.; Li, H.; Jiang, H.; Shen, X.; Heussler, V.T.; Rennenberg, A.; Hilgenfeld, R. Structural basis for the regulation of cysteine-protease activity by a new class of protease inhibitors in Plasmodium. Structure 2011, 19, 919–929. [Google Scholar] [CrossRef] [PubMed]
- Aripirala, S.; Gonzalez-Pacanowska, D.; Oldfield, E.; Kaiser, M.; Amzel, L.M.; Gabelli, S.B. Structural and thermodynamic basis of the inhibition of Leishmania major farnesyl diphosphate synthase by nitrogen-containing bisphosphonates. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 802–810. [Google Scholar] [CrossRef] [PubMed]
- Mao, J.; Mukherjee, S.; Zhang, Y.; Cao, R.; Sanders, J.M.; Song, Y.; Zhang, Y.; Meints, G.A.; Gao, Y.G.; Mukkamala, D.; et al. Solid-state NMR, crystallographic, and computational investigation of bisphosphonates and farnesyl diphosphate synthase-bisphosphonate complexes. J. Am. Chem. Soc. 2006, 128, 14485–14497. [Google Scholar] [CrossRef] [PubMed]
- Cao, R.; Chen, C.K.; Guo, R.T.; Wang, A.H.; Oldfield, E. Structures of a potent phenylalkyl bisphosphonate inhibitor bound to farnesyl and geranylgeranyl diphosphate synthases. Proteins 2008, 73, 431–439. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, R.; Yin, F.; Hudock, M.P.; Guo, R.T.; Krysiak, K.; Mukherjee, S.; Gao, Y.G.; Robinson, H.; Song, Y.; et al. Lipophilic bisphosphonates as dual farnesyl/geranylgeranyl diphosphate synthase inhibitors: An X-ray and NMR investigation. J. Am. Chem. Soc. 2009, 131, 5153–5162. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Cao, R.; Leon, A.; Guo, R.T.; Krysiak, K.; Yin, F.; Hudock, M.P.; Mukherjee, S.; Gao, Y.; Robinson, H.; et al. Bisphosphonates: Teaching old drugs with new tricks. Unpublished work. 2007; doi:10.2210/pdb2ogd/pdb. [Google Scholar]
- Gabelli, S.B.; McLellan, J.S.; Montalvetti, A.; Oldfield, E.; Docampo, R.; Amzel, L.M. Structure and mechanism of the farnesyl diphosphate synthase from Trypanosoma cruzi: Implications for drug design. Proteins 2006, 62, 80–88. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.H.; Gabelli, S.B.; Oldfield, E.; Amzel, L.M. Binding of nitrogen-containing bisphosphonates (N-BPs) to the Trypanosoma cruzi farnesyl diphosphate synthase homodimer. Proteins 2010, 78, 888–899. [Google Scholar] [CrossRef] [PubMed]
- Aripirala, S.; Szajnman, S.H.; Jakoncic, J.; Rodriguez, J.B.; Docampo, R.; Gabelli, S.B.; Amzel, L.M. Design, synthesis, calorimetry, and crystallographic analysis of 2-alkylaminoethyl-1,1-bisphosphonates as inhibitors of Trypanosoma cruzi farnesyl diphosphate synthase. J. Med. Chem. 2012, 55, 6445–6454. [Google Scholar] [CrossRef] [PubMed]
- Milani, M.; Balconi, E.; Aliverti, A.; Mastrangelo, E.; Seeber, F.; Bolognesi, M.; Zanetti, G. Ferredoxin-NADP+ reductase from Plasmodium falciparum undergoes NADP+-dependent dimerization and inactivation: Functional and crystallographic analysis. J. Mol. Biol. 2007, 367, 501–513. [Google Scholar] [CrossRef] [PubMed]
- Harikishore, A.; Niang, M.; Rajan, S.; Preiser, P.R.; Yoon, H.S. Small molecule Plasmodium FKBP35 inhibitor as a potential antimalaria agent. Sci. Rep. 2013, 3, 2501. [Google Scholar] [CrossRef] [PubMed]
- Chudzik, D.M.; Michels, P.A.; de Walque, S.; Hol, W.G. Structures of type 2 peroxisomal targeting signals in two trypanosomatid aldolases. J. Mol. Biol. 2000, 300, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Lafrance-Vanasse, J.; Sygusch, J. Carboxy-terminus recruitment induced by substrate binding in eukaryotic fructose bis-phosphate aldolases. Biochemistry 2007, 46, 9533–9540. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Certa, U.; Döbeli, H.; Jakob, P.; Hol, W.G. Crystal structure of fructose-1,6-bisphosphate aldolase from the human malaria parasite Plasmodium falciparum. Biochemistry 1998, 37, 4388–4396. [Google Scholar] [CrossRef] [PubMed]
- Zocher, K.; Fritz-Wolf, K.; Kehr, S.; Fischer, M.; Rahlfs, S.; Becker, K. Biochemical and structural characterization of Plasmodium falciparum glutamate dehydrogenase 2. Mol. Biochem. Parasitol. 2012, 183, 52–62. [Google Scholar] [CrossRef] [PubMed]
- Patel, S.; Hussain, S.; Harris, R.; Sardiwal, S.; Kelly, J.M.; Wilkinson, S.R.; Driscoll, P.C.; Djordjevic, S. Structural insights into the catalytic mechanism of Trypanosoma cruzi GPXI (glutathione peroxidase-like enzyme I). Biochem. J. 2010, 425, 513–522. [Google Scholar] [CrossRef] [PubMed]
- Sarma, G.N.; Savvides, S.N.; Becker, K.; Schirmer, M.; Schirmer, R.H.; Karplus, P.A. Glutathione reductase of the malarial parasite Plasmodium falciparum: Crystal structure and inhibitor development. J. Mol. Biol. 2003, 328, 893–907. [Google Scholar] [CrossRef]
- Fritz-Wolf, K.; Becker, A.; Rahlfs, S.; Harwaldt, P.; Schirmer, R.H.; Kabsch, W.; Becker, K. X-ray structure of glutathione S-transferase from the malarial parasite Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 2003, 100, 13821–13826. [Google Scholar] [CrossRef] [PubMed]
- Perbandt, M.; Burmeister, C.; Walter, R.D.; Betzel, C.; Liebau, E. Native and inhibited structure of a Mu class-related glutathione S-transferase from Plasmodium falciparum. J. Biol. Chem. 2004, 279, 1336–1342. [Google Scholar] [CrossRef] [PubMed]
- Hiller, N.; Fritz-Wolf, K.; Deponte, M.; Wende, W.; Zimmermann, H.; Becker, K. Plasmodium falciparum glutathione S-transferase—Structural and mechanistic studies on ligand binding and enzyme inhibition. Protein Sci. 2006, 15, 281–289. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Feil, I.K.; Verlinde, C.L.; Petra, P.H.; Hol, W.G. Crystal structure of glycosomal glyceraldehyde-3-phosphate dehydrogenase from Leishmania mexicana: Implications for structure-based drug design and a new position for the inorganic phosphate binding site. Biochemistry 1995, 34, 14975–14986. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Hol, W.G. Crystal structure of Leishmania mexicana glycosomal glyceraldehyde-3-phosphate dehydrogenase in a new crystal form confirms the putative physiological active site structure. J. Mol. Biol. 1998, 278, 5–11. [Google Scholar] [CrossRef] [PubMed]
- Aronov, A.M.; Suresh, S.; Buckner, F.S.; van Voorhis, W.C.; Verlinde, C.L.; Opperdoes, F.R.; Hol, W.G.; Gelb, M.H. Structure-based design of submicromolar, biologically active inhibitors of trypanosomatid glyceraldehyde-3-phosphate dehydrogenase. Proc. Natl. Acad. Sci. USA 1999, 96, 4273–4278. [Google Scholar] [CrossRef] [PubMed]
- Satchell, J.F.; Malby, R.L.; Luo, C.S.; Adisa, A.; Alpyurek, A.E.; Klonis, N.; Smith, B.J.; Tilley, L.; Colman, P.M. Structure of glyceraldehyde-3-phosphate dehydrogenase from Plasmodium falciparum. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 1213–1221. [Google Scholar] [CrossRef] [PubMed]
- Robien, M.A.; Bosch, J.; Buckner, F.S.; van Voorhis, W.C.; Worthey, E.A.; Myler, P.; Mehlin, C.; Boni, E.E.; Kalyuzhniy, O.; Anderson, L.; et al. Crystal structure of glyceraldehyde-3-phosphate dehydrogenase from Plasmodium falciparum at 2.25 Å resolution reveals intriguing extra electron density in the active site. Proteins 2006, 62, 570–577. [Google Scholar] [CrossRef] [PubMed]
- Vellieux, F.M.; Hajdu, J.; Verlinde, C.L.; Groendijk, H.; Read, R.J.; Greenhough, T.J.; Campbell, J.W.; Kalk, K.H.; Littlechild, J.A.; Watson, H.C.; et al. Structure of glycosomal glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma brucei determined from Laue data. Proc. Natl. Acad. Sci. USA 1993, 90, 2355–2359. [Google Scholar] [CrossRef] [PubMed]
- Seattle Structural Genomics Center for Infectious Disease; Abendroth, J.; Lorimer, D.; Edwards, T.E. Structure of a glycosomal glyceraldehyde 3-phosphate dehydrogenase from Trypanosoma brucei. Unpublished work. 2014; doi:10.2210/pdb4p8r/pdb. [Google Scholar]
- Pavão, F.; Castilho, M.S.; Pupo, M.T.; Dias, R.L.; Correa, A.G.; Fernandes, J.B.; da Silva, M.F.; Mafezoli, J.; Vieira, P.C.; Oliva, G. Structure of Trypanosoma cruzi glycosomal glyceraldehyde-3-phosphate dehydrogenase complexed with chalepin, a natural product inhibitor, at 1.95 Å resolution. FEBS Lett. 2002, 520, 13–17. [Google Scholar] [CrossRef]
- Castilho, M.S.; Pavão, F.; Oliva, G.; Ladame, S.; Willson, M.; Périé, J. Evidence for the two phosphate binding sites of an analogue of the thioacyl intermediate for the Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase-catalyzed reaction, from its crystal structure. Biochemistry 2003, 42, 7143–7151. [Google Scholar] [CrossRef] [PubMed]
- Ladame, S.; Castilho, M.S.; Silva, C.H.; Denier, C.; Hannaert, V.; Périé, J.; Oliva, G.; Willson, M. Crystal structure of Trypanosoma cruzi glyceraldehyde-3-phosphate dehydrogenase complexed with an analogue of 1,3-bisphospho-d-glyceric acid. Eur. J. Biochem. 2003, 270, 4574–4586. [Google Scholar] [CrossRef] [PubMed]
- Balliano, T.L.; Guido, R.V.C.; Andricopulo, A.D.; Oliva, G. Structure of glycosomal glyceraldehyde-3-phosphate dehydrogenase from Trypanosoma cruzi in complex with the irreversible iodoacetamide inhibitor. Unpublished work. 2009; doi:10.2210/pdb3ids/pdb. [Google Scholar]
- Suresh, S.; Turley, S.; Opperdoes, F.R.; Michels, P.A.; Hol, W.G. A potential target enzyme for trypanocidal drugs revealed by the crystal structure of NAD-dependent glycerol-3-phosphate dehydrogenase from Leishmania mexicana. Structure 2000, 8, 541–552. [Google Scholar] [CrossRef]
- Choe, J.; Suresh, S.; Wisedchaisri, G.; Kennedy, K.J.; Gelb, M.H.; Hol, W.G. Anomalous differences of light elements in determining precise binding modes of ligands to glycerol-3-phosphate dehydrogenase. Chem. Biol. 2002, 9, 1189–1197. [Google Scholar] [CrossRef]
- Choe, J.; Guerra, D.; Michels, P.A.; Hol, W.G. Leishmania mexicana glycerol-3-phosphate dehydrogenase showed conformational changes upon binding a bi-substrate adduct. J. Mol. Biol. 2003, 329, 335–349. [Google Scholar] [CrossRef]
- Ariza, A.; Vickers, T.J.; Greig, N.; Armour, K.A.; Dixon, M.J.; Eggleston, I.M.; Fairlamb, A.H.; Bond, C.S. Specificity of the trypanothione-dependent Leishmania major glyoxalase I: Structure and biochemical comparison with the human enzyme. Mol. Microbiol. 2006, 59, 1239–1248. [Google Scholar] [CrossRef] [PubMed]
- Silva, M.S.; Barata, L.; Ferreira, A.E.; Romão, S.; Tomás, A.M.; Freire, A.P.; Cordeiro, C. Catalysis and structural properties of Leishmania infantum glyoxalase II: Trypanothione specificity and phylogeny. Biochemistry 2008, 47, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.K.; Dong, A.; Hills, T.; Amani, M.; Perieteanu, A.; Lin, Y.H.; Loppnau, P.; Arrowsmith, C.H.; Edwards, A.M.; Bountra, C.; et al. Crystal Structure of PF10_0123, a GMP synthetase from Plasmodium falciparum. Unpublished work. 2011; doi:10.2210/pdb3uow/pdb. [Google Scholar]
- Vedadi, M.; Lew, J.; Artz, J.; Amani, M.; Zhao, Y.; Dong, A.; Wasney, G.A.; Gao, M.; Hills, T.; Brokx, S.; et al. Genome-scale protein expression and structural biology of Plasmodium falciparum and related Apicomplexan organisms. Mol. Biochem. Parasitol. 2007, 151, 100–110. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.K.; Tempel, W.; Lin, Y.H.; Hutchinson, A.; Mackenzie, F.; Fairlamb, A.; Kozieradzki, I.; Cossar, D.; Zhao, Y.; Schapira, M.; et al. Crystal structure of the amino-terminal domain of HSP90 from Leishmania major, LmjF33.0312:M1-K213. Unpublished work. 2009; doi:10.2210/pdb3h80/pdb. [Google Scholar]
- Wernimont, A.K.; Tempel, W.; Lin, Y.H.; Hutchinson, A.; MacKenzie, F.; Fairlamb, A.; Cossar, D.; Zhao, Y.; Schapira, M.; Arrowsmith, C.H.; et al. Crystal structure of the amino-terminal domain of HSP90 from Leishmania major, LMJF33.0312:M1-K213. Unpublished work. 2011; doi:10.2210/pdb3q5j/pdb, doi:10.2210/pdb3q5k/pdb, doi:10.2210/pdb3q5l/pdb. [Google Scholar]
- Hills, T.; Pizarro, J.C.; Wernimont, A.K.; Ferguson, M.A.J.; Hui, R. Crystal structure of the N-terminal domain of Hsp90 from Leishmania major (LmjF33.0312) in complex with ADP. Unpublished work. 2012; doi:10.2210/pdb3u67/pdb. [Google Scholar]
- Merritt, E.A.; Arakaki, T.L.; Gillespie, J.R.; Larson, E.T.; Kelley, A.; Mueller, N.; Napuli, A.J.; Kim, J.; Zhang, L.; Verlinde, C.L.; et al. Crystal structures of trypanosomal histidyl-tRNA synthetase illuminate differences between eukaryotic and prokaryotic homologs. J. Mol. Biol. 2010, 397, 481–494. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, P.; Xiao, H.; Parr, C.L.; Kiso, Y.; Gustchina, A.; Yada, R.Y.; Wlodawer, A. Crystal structures of the histo-aspartic protease (HAP) from Plasmodium falciparum. J. Mol. Biol. 2009, 388, 520–540. [Google Scholar] [CrossRef] [PubMed]
- Maity, K.; Venkata, B.S.; Kapoor, N.; Surolia, N.; Surolia, A.; Suguna, K. Structural basis for the functional and inhibitory mechanisms of β-hydroxyacyl-acyl carrier protein dehydratase (FabZ) of Plasmodium falciparum. J. Struct. Biol. 2011, 176, 238–249. [Google Scholar] [CrossRef] [PubMed]
- Rekittke, I.; Olkhova, E.; Wiesner, J.; Demmer, U.; Warkentin, E.; Jomaa, H.; Ermler, U. Structure of the (E)-4-hydroxy-3-methyl-but-2-enyl-diphosphate reductase from Plasmodium falciparum. FEBS Lett. 2013, 587, 3968–3972. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Li, C.M.; Tyler, P.C.; Furneaux, R.H.; Cahill, S.M.; Girvin, M.E.; Grubmeyer, C.; Schramm, V.L.; Almo, S.C. The 2.0 Å structure of malarial purine phosphoribosyltransferase in complex with a transition-state analogue inhibitor. Biochemistry 1999, 38, 9872–9880. [Google Scholar] [CrossRef] [PubMed]
- Focia, P.J.; Craig, S.P.; Eakin, A.E. Approaching the transition state in the crystal structure of a phosphoribosyltransferase. Biochemistry 1998, 37, 17120–17127. [Google Scholar] [CrossRef] [PubMed]
- Canyuk, B.; Medrano, F.J.; Wenck, M.A.; Focia, P.J.; Eakin, A.E.; Craig, S.P. Interactions at the dimer interface influence the relative efficiencies for purine nucleotide synthesis and pyrophosphorolysis in a phosphoribosyltransferase. J. Mol. Biol. 2004, 335, 905–921. [Google Scholar] [CrossRef] [PubMed]
- Dunn, C.R.; Banfield, M.J.; Barker, J.J.; Higham, C.W.; Moreton, K.M.; Turgut-Balik, D.; Brady, R.L.; Holbrook, J.J. The structure of lactate dehydrogenase from Plasmodium falciparum reveals a new target for anti-malarial design. Nat. Struct. Biol. 1996, 3, 912–915. [Google Scholar] [CrossRef] [PubMed]
- Read, J.A.; Wilkinson, K.W.; Tranter, R.; Sessions, R.B.; Brady, R.L. Chloroquine binds in the cofactor binding site of Plasmodium falciparum lactate dehydrogenase. J. Biol. Chem. 1999, 274, 10213–10218. [Google Scholar] [CrossRef] [PubMed]
- Cameron, A.; Read, J.; Tranter, R.; Winter, V.J.; Sessions, R.B.; Brady, R.L.; Vivas, L.; Easton, A.; Kendrick, H.; Croft, S.L.; et al. Identification and activity of a series of azole-based compounds with lactate dehydrogenase-directed anti-malarial activity. J. Biol. Chem. 2004, 279, 31429–31439. [Google Scholar] [CrossRef] [PubMed]
- Conners, R.; Schambach, F.; Read, J.; Cameron, A.; Sessions, R.B.; Vivas, L.; Easton, A.; Croft, S.L.; Brady, R.L. Mapping the binding site for gossypol-like inhibitors of Plasmodium falciparum lactate dehydrogenase. Mol. Biochem. Parasitol. 2005, 142, 137–148. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Fairweather, V.; Conners, R.; Joseph-Horne, T.; Turgut-Balik, D.; Brady, R.L. Structure of lactate dehydrogenase from Plasmodium vivax: Complexes with NADH and APADH. Biochemistry 2005, 44, 16221–16228. [Google Scholar] [CrossRef] [PubMed]
- Birkinshaw, R.W.; Brady, R.L. The crystal structure of Plasmodium falciparum L-lactate dehydrogenase in complex with a novel bicine ligand. Unpublished work. 2012; doi:10.2210/pdb4b7u/pdb. [Google Scholar]
- Werner, C.; Krauth-Siegel, R.L.; Stubbs, M.T.; Klebe, G. Crystal structure of lipoamide dehydrogenase from Trypanosoma cruzi: A putative target for the design of new drugs against Chagas disease. Unpublished work. 2008; doi:10.2210/pdb2qae/pdb. [Google Scholar]
- Khan, S.; Garg, A.; Camacho, N.; van Rooyen, J.; Kumar Pole, A.; Belrhali, H.; Ribas de Pouplana, L.; Sharma, V.; Sharma, A. Structural analysis of malaria-parasite lysyl-tRNA synthetase provides a platform for drug development. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Kannan Sivaraman, K.; Paiardini, A.; Sieńczyk, M.; Ruggeri, C.; Oellig, C.A.; Dalton, J.P.; Scammells, P.J.; Drag, M.; McGowan, S. Synthesis and structure-activity relationships of phosphonic arginine mimetics as inhibitors of the M1 and M17 aminopeptidases from Plasmodium falciparum. J. Med. Chem. 2013, 56, 5213–5217. [Google Scholar] [CrossRef] [PubMed]
- McGowan, S.; Oellig, C.A.; Birru, W.A.; Caradoc-Davies, T.T.; Stack, C.M.; Lowther, J.; Skinner-Adams, T.; Mucha, A.; Kafarski, P.; Grembecka, J.; et al. Structure of the Plasmodium falciparum M17 aminopeptidase and significance for the design of drugs targeting the neutral exopeptidases. Proc. Natl. Acad. Sci. USA 2010, 107, 2449–2454. [Google Scholar] [CrossRef] [PubMed]
- Harbut, M.B.; Velmourougane, G.; Dalal, S.; Reiss, G.; Whisstock, J.C.; Onder, O.; Brisson, D.; McGowan, S.; Klemba, M.; Greenbaum, D.C. Bestatin-based chemical biology strategy reveals distinct roles for malaria M1- and M17-family aminopeptidases. Proc. Natl. Acad. Sci. USA 2011, 108, E526–E334. [Google Scholar] [CrossRef] [PubMed]
- Sivaraman, K.K.; Oellig, C.A.; Huynh, K.; Atkinson, S.C.; Poreba, M.; Perugini, M.A.; Trenholme, K.R.; Gardiner, D.L.; Salvesen, G.; Drag, M.; et al. X-ray crystal structure and specificity of the Plasmodium falciparum malaria aminopeptidase PfM18AAP. J. Mol. Biol. 2012, 422, 495–507. [Google Scholar] [CrossRef] [PubMed]
- Pereira, P.J.; Vega, M.C.; González-Rey, E.; Fernández-Carazo, R.; Macedo-Ribeiro, S.; Gomis-Rüth, F.X.; González, A.; Coll, M. Trypanosoma cruzi macrophage infectivity potentiator has a rotamase core and a highly exposed alpha-helix. EMBO Rep. 2002, 3, 88–94. [Google Scholar] [CrossRef] [PubMed]
- McLuskey, K.; Rudolf, J.; Proto, W.R.; Isaacs, N.W.; Coombs, G.H.; Moss, C.X.; Mottram, J.C. Crystal structure of a Trypanosoma brucei metacaspase. Proc. Natl. Acad. Sci. USA 2012, 109, 7469–7474. [Google Scholar] [CrossRef] [PubMed]
- Niemirowicz, G.; Fernández, D.; Solà, M.; Cazzulo, J.J.; Avilés, F.X.; Gomis-Rüth, F.X. The molecular analysis of Trypanosoma cruzi metallocarboxypeptidase 1 provides insight into fold and substrate specificity. Mol. Microbiol. 2008, 70, 853–866. [Google Scholar] [PubMed]
- Wernimont, A.K.; Artz, J.D.; Crombet, L.; Lew, J.; Weadge, J.; Arrowsmith, C.H.; Edwards, A.M.; Weigelt, J.; Bountra, C.; Hui, R.; et al. Crystal structure of methionine aminopeptidase 1b from Plasmodium falciparum, PF10_0150. Unpublished work. 2011; doi:10.2210/pdb3s6b/pdb. [Google Scholar]
- Larson, E.T.; Kim, J.E.; Zucker, F.H.; Kelley, A.; Mueller, N.; Napuli, A.J.; Verlinde, C.L.; Fan, E.; Buckner, F.S.; van Voorhis, W.C.; et al. Structure of Leishmania major methionyl-tRNA synthetase in complex with intermediate products methionyladenylate and pyrophosphate. Biochimie 2011, 93, 570–582. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kim, J.E.; Shibata, S.; Ranade, R.M.; Yu, M.; Liu, J.; Gillespie, J.R.; Buckner, F.S.; Verlinde, C.L.; Fan, E.; et al. Distinct states of methionyl-tRNA synthetase indicate inhibitor binding by conformational selection. Structure 2012, 20, 1681–1691. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kim, J.E.; Wetzel, A.B.; de van der Schueren, W.J.; Shibata, S.; Ranade, R.M.; Liu, J.; Zhang, Z.; Gillespie, J.R.; Buckner, F.S.; et al. Structures of Trypanosoma brucei methionyl-tRNA synthetase with urea-based inhibitors provide guidance for drug design against sleeping sickness. PLoS Negl. Trop. Dis. 2014, 8, e2775. [Google Scholar] [CrossRef] [PubMed]
- Horjales, S.; Schmidt-Arras, D.; Limardo, R.R.; Leclercq, O.; Obal, G.; Prina, E.; Turjanski, A.G.; Späth, G.F.; Buschiazzo, A. The crystal structure of the MAP kinase LmaMPK10 from Leishmania major reveals parasite-specific features and regulatory mechanisms. Structure 2012, 20, 1649–1660. [Google Scholar] [CrossRef] [PubMed]
- Eadsforth, T.C.; Cameron, S.; Hunter, W.N. The crystal structure of Leishmania major N5,N10-methylenetetrahydrofolate dehydrogenase/cyclohydrolase and assessment of a potential drug target. Mol. Biochem. Parasitol. 2012, 181, 178–185. [Google Scholar] [CrossRef] [PubMed]
- Gazanion, E.; Garcia, D.; Silvestre, R.; Gérard, C.; Guichou, J.F.; Labesse, G.; Seveno, M.; Cordeiro-Da-Silva, A.; Ouaissi, A.; Sereno, D.; et al. The Leishmania nicotinamidase is essential for NAD+ production and parasite proliferation. Mol. Microbiol. 2011, 82, 21–38. [Google Scholar] [CrossRef] [PubMed]
- Brannigan, J.A.; Smith, B.A.; Yu, Z.; Brzozowski, A.M.; Hodgkinson, M.R.; Maroof, A.; Price, H.P.; Meier, F.; Leatherbarrow, R.J.; Tate, E.W.; et al. N-myristoyltransferase from Leishmania donovani: Structural and functional characterisation of a potential drug target for visceral leishmaniasis. J. Mol. Biol. 2010, 396, 985–999. [Google Scholar] [CrossRef] [PubMed]
- Frearson, J.A.; Brand, S.; McElroy, S.P.; Cleghorn, L.A.; Smid, O.; Stojanovski, L.; Price, H.P.; Guther, M.L.; Torrie, L.S.; Robinson, D.A.; et al. N-myristoyltransferase inhibitors as new leads to treat sleeping sickness. Nature 2010, 464, 728–732. [Google Scholar] [CrossRef] [PubMed]
- Brand, S.; Cleghorn, L.A.; McElroy, S.P.; Robinson, D.A.; Smith, V.C.; Hallyburton, I.; Harrison, J.R.; Norcross, N.R.; Spinks, D.; Bayliss, T.; et al. Discovery of a novel class of orally active trypanocidal N-myristoyltransferase inhibitors. J. Med. Chem. 2012, 55, 140–152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosch, J.; Robien, M.A.; Mehlin, C.; Boni, E.; Riechers, A.; Buckner, F.S.; van Voorhis, W.C.; Myler, P.J.; Worthey, E.A.; DeTitta, G.; et al. Using fragment cocktail crystallography to assist inhibitor design of Trypanosoma brucei nucleoside 2-deoxyribosyltransferase. J. Med. Chem. 2006, 49, 5939–5946. [Google Scholar] [CrossRef] [PubMed]
- Souza, T.A.; Trindade, D.M.; Tonoli, C.C.; Santos, C.R.; Ward, R.J.; Arni, R.K.; Oliveira, A.H.; Murakami, M.T. Molecular adaptability of nucleoside diphosphate kinase b from trypanosomatid parasites: Stability, oligomerization and structural determinants of nucleotide binding. Mol. Biosyst. 2011, 7, 2189–2195. [Google Scholar] [CrossRef] [PubMed]
- Gardberg, A.S.; Edwards, T.E.; Seattle Structural Genomics Center for Infectious Disease. Crystal structure of nucleoside diphosphate kinase B from Trypanosoma brucei. Unpublished work. 2012; doi:10.2210/pdb4fkx/pdb, doi:10.2210/pdb4fky/pdb. [Google Scholar]
- Seattle Structural Genomics Center for Infectious Disease; Gardberg, A.S.; Edwards, T.E.; Staker, B.; Stewart, L. Crystal structure of nucleoside diphosphate kinase B from Trypanosoma brucei. Unpublished work. 2012; doi:10.2210/pdb4f4a/pdb, doi:10.2210/pdb4f36/pdb. [Google Scholar]
- Shi, W.; Schramm, V.L.; Almo, S.C. Nucleoside hydrolase from Leishmania major. Cloning, expression, catalytic properties, transition state inhibitors, and the 2.5-Ǻ crystal structure. J. Biol. Chem. 1999, 274, 21114–21120. [Google Scholar] [CrossRef] [PubMed]
- Giannese, F.; Berg, M.; van der Veken, P.; Castagna, V.; Tornaghi, P.; Augustyns, K.; Degano, M. Structures of purine nucleosidase from Trypanosoma brucei bound to isozyme-specific trypanocidals and a novel metalorganic inhibitor. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1553–1566. [Google Scholar] [CrossRef] [PubMed]
- Vandemeulebrouke, A.; Minici, C.; Bruno, I.; Muzzolini, L.; Tornaghi, P.; Parkin, D.W.; Versées, W.; Steyaert, J.; Degano, M. Structure and mechanism of the 6-oxopurine nucleosidase from Trypanosoma brucei brucei. Biochemistry 2010, 49, 8999–9010. [Google Scholar] [CrossRef] [PubMed]
- Gill, J.; Yogavel, M.; Kumar, A.; Belrhali, H.; Jain, S.K.; Rug, M.; Brown, M.; Maier, A.G.; Sharma, A. Crystal structure of malaria parasite nucleosome assembly protein: Distinct modes of protein localization and histone recognition. J. Biol. Chem. 2009, 284, 10076–10087. [Google Scholar] [CrossRef] [PubMed]
- Yogavel, M.; Gill, J.; Sharma, A. Iodide-SAD, SIR and SIRAS phasing for structure solution of a nucleosome assembly protein. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 618–622. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, N.; Yamaguchi, K.; Mizohata, E.; Tokuoka, K.; Uchiyama, N.; Sugiyama, S.; Matsumura, H.; Inaka, K.; Urade, Y.; Inoue, T. Structural insight into the stereoselective production of PGF2α by Old Yellow Enzyme from Trypanosoma cruzi. J. Biochem. 2011, 150, 563–568. [Google Scholar] [CrossRef] [PubMed]
- Murakami, M.T.; Rodrigues, N.C.; Gava, L.M.; Canduri, F.; Oliva, G.; Barbosa, L.R.S.; Borgers, J.C. High resolution crystal structure and in solution studies of the old yellow enzyme from Trypanosoma cruzi: Insights into oligomerization, enzyme dynamics and specificity. Unpublished work. 2013; doi:10.2210/pdb4e2b/pdb, doi:10.2210/pdb4e2d/pdb. [Google Scholar]
- McLuskey, K.; Paterson, N.G.; Bland, N.D.; Isaacs, N.W.; Mottram, J.C. Crystal structure of Leishmania major oligopeptidase B gives insight into the enzymatic properties of a trypanosomatid virulence factor. J. Biol. Chem. 2010, 285, 39249–39259. [Google Scholar] [CrossRef] [PubMed]
- Canning, P.; Rea, D.; Morty, R.E.; Fülöp, V. Crystal structures of Trypanosoma brucei oligopeptidase B broaden the paradigm of catalytic regulation in prolyl oligopeptidase family enzymes. PLoS ONE 2013, 8, e79349. [Google Scholar] [CrossRef] [PubMed]
- Grishin, N.V.; Osterman, A.L.; Brooks, H.B.; Phillips, M.A.; Goldsmith, E.J. X-ray structure of ornithine decarboxylase from Trypanosoma brucei: The native structure and the structure in complex with alpha-difluoromethylornithine. Biochemistry 1999, 38, 15174–15184. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.K.; Brooks, H.B.; Osterman, A.L.; Goldsmith, E.J.; Phillips, M.A. Altering the reaction specificity of eukaryotic ornithine decarboxylase. Biochemistry 2000, 39, 11247–11257. [Google Scholar] [CrossRef] [PubMed]
- Jackson, L.K.; Goldsmith, E.J.; Phillips, M.A. X-ray structure determination of Trypanosoma brucei ornithine decarboxylase bound to d-ornithine and to G418: Insights into substrate binding and ODC conformational flexibility. J. Biol. Chem. 2003, 278, 22037–22043. [Google Scholar] [CrossRef] [PubMed]
- Jortzik, E.; Fritz-Wolf, K.; Sturm, N.; Hipp, M.; Rahlfs, S.; Becker, K. Redox regulation of Plasmodium falciparum ornithine δ-aminotransferase. J. Mol. Biol. 2010, 402, 445–459. [Google Scholar] [CrossRef] [PubMed]
- French, J.B.; Yates, P.A.; Soysa, D.R.; Boitz, J.M.; Carter, N.S.; Chang, B.; Ullman, B.; Ealick, S.E. The Leishmania donovani UMP synthase is essential for promastigote viability and has an unusual tetrameric structure that exhibits substrate-controlled oligomerization. J. Biol. Chem. 2011, 286, 20930–20941. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.M.; Poduch, E.; Liu, Y.; Wei, L.; Crandall, I.; Wang, X.; Dyanand, C.; Kain, K.C.; Pai, E.F.; Kotra, L.P. Structure-activity relationships of C6-uridine derivatives targeting plasmodia orotidine monophosphate decarboxylase. J. Med. Chem. 2008, 51, 439–448. [Google Scholar] [CrossRef] [PubMed]
- Langley, D.B.; Shojaei, M.; Chan, C.; Lok, H.C.; Mackay, J.P.; Traut, T.W.; Guss, J.M.; Christopherson, R.I. Structure and inhibition of orotidine 5′-monophosphate decarboxylase from Plasmodium falciparum. Biochemistry 2008, 47, 3842–3854. [Google Scholar] [CrossRef] [PubMed]
- Tokuoka, K.; Kusakari, Y.; Krungkrai, S.R.; Matsumura, H.; Kai, Y.; Krungkrai, J.; Horii, T.; Inoue, T. Structural basis for the decarboxylation of orotidine 5′-monophosphate (OMP) by Plasmodium falciparum OMP decarboxylase. J. Biochem. 2008, 143, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Bello, A.M.; Konforte, D.; Poduch, E.; Furlonger, C.; Wei, L.; Liu, Y.; Lewis, M.; Pai, E.F.; Paige, C.J.; Kotra, L.P. Structure-activity relationships of orotidine-5′-monophosphate decarboxylase inhibitors as anticancer agents. J. Med. Chem. 2009, 52, 1648–1658. [Google Scholar] [CrossRef] [PubMed]
- Takashima, Y.; Mizohata, E.; Krungkrai, S.R.; Fukunishi, Y.; Kinoshita, T.; Sakata, T.; Matsumura, H.; Krungkrai, J.; Horii, T.; Inoue, T. The in silico screening and X-ray structure analysis of the inhibitor complex of Plasmodium falciparum orotidine 5′-monophosphate decarboxylase. J. Biochem. 2012, 152, 133–138. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Lau, W.; Lew, J.; Amani, M.; Hui, R.; Pai, E.F. Crystal structure of orotidine 5′-phosphate decarboxylase from Plasmodium falciparum. Unpublished work. 2005; doi:10.2210/pdb2f84/pdb. [Google Scholar]
- Caruthers, J.M.; Robein, M.; Zucker, F.; Mehlin, C.; Luft, J.; Boni, E.; Lauricella, A.; Merritt, E.A.; Hol, W.G.J. Crystal structure of an orotidine-5′-monophosphate decarboxylase homolog from P. falciparum. Unpublished work. 2005; doi:10.2210/pdb2f84/pdb. [Google Scholar]
- Liu, Y.; Kotra, L.P.; Pai, E.F. Crystal structure of Plasmodium falciparum orotidine 5′-monophosphate decarboxylase. Unpublished work. 2011; doi:10.2210/pdb3mwa/pdb, doi:10.2210/pdb3n2m/pdb, doi:10.2210/pdb3n34/pdb, doi:10.2210/pdb3n3m/pdb. [Google Scholar]
- Wickramasinghe, S.R.; Inglis, K.A.; Urch, J.E.; Müller, S.; van Aalten, D.M.; Fairlamb, A.H. Kinetic, inhibition and structural studies on 3-oxoacyl-ACP reductase from Plasmodium falciparum, a key enzyme in fatty acid biosynthesis. Biochem. J. 2006, 393, 447–557. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Nguyen, K.T.; Srivathsan, S.; Ornstein, B.; Turley, S.; Hirsh, I.; Pei, D.; Hol, W.G. Crystals of peptide deformylase from Plasmodium falciparum reveal critical characteristics of the active site for drug design. Structure 2002, 10, 357–367. [Google Scholar] [CrossRef]
- Robien, M.A.; Nguyen, K.T.; Kumar, A.; Hirsh, I.; Turley, S.; Pei, D.; Hol, W.G. An improved crystal form of Plasmodium falciparum peptide deformylase. Protein Sci. 2004, 13, 1155–1163. [Google Scholar] [CrossRef] [PubMed]
- Sampathkumar, P.; Roach, C.; Michels, P.A.; Hol, W.G. Structural insights into the recognition of peroxisomal targeting signal 1 by Trypanosoma brucei peroxin 5. J. Mol. Biol. 2008, 381, 867–880. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.G.; Kim, Y.; Alpert, T.D.; Nagata, A.; Jez, J.M. Structure and reaction mechanism of phosphoethanolamine methyltransferase from the malaria parasite Plasmodium falciparum: An antiparasitic drug target. J. Biol. Chem. 2012, 287, 1426–1434. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yan, Z.; Geng, J.; Kunz, S.; Seebeck, T.; Ke, H. Crystal structure of the Leishmania major phosphodiesterase LmjPDEB1 and insight into the design of the parasite-selective inhibitors. Mol. Microbiol. 2007, 66, 1029–1038. [Google Scholar] [CrossRef] [PubMed]
- Jansen, C.; Wang, H.; Kooistra, A.J.; de Graaf, C.; Orrling, K.M.; Tenor, H.; Seebeck, T.; Bailey, D.; de Esch, I.J.; Ke, H.; et al. Discovery of novel Trypanosoma brucei phosphodiesterase B1 inhibitors by virtual screening against the unliganded TbrPDEB1 crystal structure. J. Med. Chem. 2013, 56, 2087–2096. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kunz, S.; Chen, G.; Seebeck, T.; Wan, Y.; Robinson, H.; Martinelli, S.; Ke, H. Biological and structural characterization of Trypanosoma cruzi phosphodiesterase C and implications for design of parasite selective inhibitors. J. Biol. Chem. 2012, 287, 11788–11797. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Kunz, S.; Chen, G.; Seebeck, T.; Wan, Y.; Robinson, H.; Martinelli, S.; Ke, H. TcrPDEC1 catalytic domain in complex with inhibitor wyq16. Unpublished work. 2012; doi:10.2210/pdb3v94/pdb. [Google Scholar]
- Trapani, S.; Linss, J.; Goldenberg, S.; Fischer, H.; Craievich, A.F.; Oliva, G. Crystal structure of the dimeric phosphoenolpyruvate carboxykinase (PEPCK) from Trypanosoma cruzi at 2 Å resolution. J. Mol. Biol. 2001, 313, 1059–1072. [Google Scholar] [CrossRef] [PubMed]
- McNae, I.W.; Martinez-Oyanedel, J.; Keillor, J.W.; Michels, P.A.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. The crystal structure of ATP-bound phosphofructokinase from Trypanosoma brucei reveals conformational transitions different from those of other phosphofructokinases. J. Mol. Biol. 2009, 385, 1519–1533. [Google Scholar] [CrossRef] [PubMed]
- Delarue, M.; Duclert-Savatier, N.; Miclet, E.; Haouz, A.; Giganti, D.; Ouazzani, J.; Lopez, P.; Nilges, M.; Stoven, V. Three dimensional structure and implications for the catalytic mechanism of 6-phosphogluconolactonase from Trypanosoma brucei. J. Mol. Biol. 2007, 366, 868–881. [Google Scholar] [CrossRef] [PubMed]
- Duclert-Savatier, N.; Poggi, L.; Miclet, E.; Lopes, P.; Ouazzani, J.; Chevalier, N.; Nilges, M.; Delarue, M.; Stoven, V. Insights into the enzymatic mechanism of 6-phosphogluconolactonase from Trypanosoma brucei using structural data and molecular dynamics simulation. J. Mol. Biol. 2009, 388, 1009–1021. [Google Scholar] [CrossRef] [PubMed]
- Phillips, C.; Dohnalek, J.; Gover, S.; Barrett, M.P.; Adams, M.J. A 2.8 Å resolution structure of 6-phosphogluconate dehydrogenase from the protozoan parasite Trypanosoma brucei: Comparison with the sheep enzyme accounts for differences in activity with coenzyme and substrate analogues. J. Mol. Biol. 1998, 282, 667–681. [Google Scholar] [CrossRef] [PubMed]
- Cordeiro, A.T.; Michels, P.A.; Delboni, L.F.; Thiemann, O.H. The crystal structure of glucose-6-phosphate isomerase from Leishmania mexicana reveals novel active site features. Eur. J. Biochem. 2004, 271, 2765–2772. [Google Scholar] [CrossRef] [PubMed]
- Arsenieva, D.; Appavu, B.L.; Mazock, G.H.; Jeffery, C.J. Crystal structure of phosphoglucose isomerase from Trypanosoma brucei complexed with glucose-6-phosphate at 1.6 Å resolution. Proteins 2009, 74, 72–80. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.D.; Chattopadhyay, D.; Pal, B. Crystal structure of Plasmodium falciparum phosphoglycerate kinase: Evidence for anion binding in the basic patch. Biochem. Biophys. Res. Commun. 2011, 412, 203–206. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Michels, P.A.; Hol, W.G. Synergistic effects of substrate-induced conformational changes in phosphoglycerate kinase activation. Nature 1997, 385, 275–278. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, B.E.; Williams, D.M.; Bressi, J.C.; Kuhn, P.; Gelb, M.H.; Blackburn, G.M.; Hol, W.G. A bisubstrate analog induces unexpected conformational changes in phosphoglycerate kinase from Trypanosoma brucei. J. Mol. Biol. 1998, 279, 1137–1148. [Google Scholar] [CrossRef] [PubMed]
- Nowicki, M.W.; Kuaprasert, B.; McNae, I.W.; Morgan, H.P.; Harding, M.M.; Michels, P.A.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. Crystal structures of Leishmania mexicana phosphoglycerate mutase suggest a one-metal mechanism and a new enzyme subclass. J. Mol. Biol. 2009, 394, 535–543. [Google Scholar] [CrossRef] [PubMed]
- Hills, T.; Srivastava, A.; Ayi, K.; Wernimont, A.K.; Kain, K.; Waters, A.P.; Hui, R.; Pizarro, J.C. Characterization of a new phosphatase from Plasmodium. Mol. Biochem. Parasitol. 2011, 179, 69–79. [Google Scholar] [CrossRef] [PubMed]
- Mercaldi, G.F.; Pereira, H.M.; Cordeiro, A.T.; Michels, P.A.; Thiemann, O.H. Structural role of the active-site metal in the conformation of Trypanosoma brucei phosphoglycerate mutase. FEBS J. 2012, 279, 2012–2021. [Google Scholar] [CrossRef] [PubMed]
- Kedzierski, L.; Malby, R.L.; Smith, B.J.; Perugini, M.A.; Hodder, A.N.; Ilg, T.; Colman, P.M.; Handman, E. Structure of Leishmania mexicana phosphomannomutase highlights similarities with human isoforms. J. Mol. Biol. 2006, 363, 215–227. [Google Scholar] [CrossRef] [PubMed]
- Wernimont, A.K.; Lam, A.; Ali, A.; Lin, Y.H.; Guther, L.; Shamshad, A.; Bandini, G.; MacKenzie, F.; Kozieradzki, I.; Cossar, D.; et al. Crystal structure of Trypanosoma brucei phosphomannosemutase, TB.10.700.370. Unpublished work. 2009; doi:10.2210/pdb3f9r/pdb. [Google Scholar]
- Liu, P.; Marzahn, M.R.; Robbins, A.H.; Gutiérrez-de-Terán, H.; Rodríguez, D.; McClung, S.H.; Stevens, S.M.; Yowell, C.A.; Dame, J.B.; McKenna, R.; et al. Recombinant plasmepsin 1 from the human malaria parasite Plasmodium falciparum: Enzymatic characterization, active site inhibitor design, and structural analysis. Biochemistry 2009, 48, 4086–4099. [Google Scholar] [CrossRef] [PubMed]
- Bhaumik, P.; Horimoto, Y.; Xiao, H.; Miura, T.; Hidaka, K.; Kiso, Y.; Wlodawer, A.; Yada, R.Y.; Gustchina, A. Crystal structures of the free and inhibited forms of plasmepsin I (PMI) from Plasmodium falciparum. J. Struct. Biol. 2011, 175, 73–84. [Google Scholar] [CrossRef] [PubMed]
- Silva, A.M.; Lee, A.Y.; Gulnik, S.V.; Maier, P.; Collins, J.; Bhat, T.N.; Collins, P.J.; Cachau, R.E.; Luker, K.E.; Gluzman, I.Y.; et al. Structure and inhibition of plasmepsin II, a hemoglobin-degrading enzyme from Plasmodium falciparum. Proc. Natl. Acad. Sci. USA 1996, 93, 10034–10039. [Google Scholar] [CrossRef] [PubMed]
- Asojo, O.A.; Afonina, E.; Gulnik, S.V.; Yu, B.; Erickson, J.W.; Randad, R.; Medjahed, D.; Silva, A.M. Structures of Ser205 mutant plasmepsin II from Plasmodium falciparum at 1.8 Å in complex with the inhibitors rs367 and rs370. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 2001–2008. [Google Scholar] [CrossRef] [PubMed]
- Asojo, O.A.; Gulnik, S.V.; Afonina, E.; Yu, B.; Ellman, J.A.; Haque, T.S.; Silva, A.M. Novel uncomplexed and complexed structures of plasmepsin II, an aspartic protease from Plasmodium falciparum. J. Mol. Biol. 2003, 327, 173–181. [Google Scholar] [CrossRef]
- Prade, L.; Jones, A.F.; Boss, C.; Richard-Bildstein, S.; Meyer, S.; Binkert, C.; Bur, D. X-ray structure of plasmepsin II complexed with a potent achiral inhibitor. J. Biol. Chem. 2005, 280, 23837–23843. [Google Scholar] [CrossRef] [PubMed]
- Boss, C.; Corminboeuf, O.; Grisostomi, C.; Meyer, S.; Jones, A.F.; Prade, L.; Binkert, C.; Fischli, W.; Weller, T.; Bur, D. Achiral, cheap, and potent inhibitors of Plasmepsins I, II, and IV. ChemMedChem 2006, 1, 1341–1345. [Google Scholar] [CrossRef] [PubMed]
- Robbins, A.H.; Dunn, B.M.; Agbandje-McKenna, M.; McKenna, R. Crystallographic evidence for noncoplanar catalytic aspartic acids in plasmepsin II resides in the Protein Data Bank. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 294–296. [Google Scholar] [CrossRef] [PubMed]
- Asojo, O.A.; Silva, A.M.; Gulnik, S. Novel uncomplexed and complex structures of PM II, an aspartic protease from P. falciparum. Unpublished work. 2005; doi:10.2210/pdb1m43/pdb. [Google Scholar]
- Freire, E.; Nezami, A.G.; Amzel, L.M. Crystal structure of plasmepsin II, an aspartyl protease from Plasmodium falciparum, in complex with a statine-based inhibitor. Unpublished work. 2004; doi:10.2210/pdb1me6/pdb. [Google Scholar]
- Lindberg, J.; Johansson, P.-O.; Rosenquist, A.; Kvarnstroem, I.; Vrang, L.; Samuelsson, B.; Unge, T. Structural study of a novel inhibitor with bulky P1 side chain in complex with Plasmepsin II—Implications for drug design. Unpublished work. 2006; doi:10.2210/pdb1w6h/pdb, doi:10.2210/pdb1w6i/pdb. [Google Scholar]
- Prade, L. Structure of plasmepsin II. Unpublished work. 2005; doi:10.2210/pdb1xdh/pdb, doi:10.2210/pdb1xe5/pdb, doi:10.2210/pdb1xe6/pdb. [Google Scholar]
- Buschiazzo, A.; Goytia, M.; Schaeffer, F.; Degrave, W.; Shepard, W.; Grégoire, C.; Chamond, N.; Cosson, A.; Berneman, A.; Coatnoan, N.; et al. Crystal structure, catalytic mechanism, and mitogenic properties of Trypanosoma cruzi proline racemase. Proc. Natl. Acad. Sci. USA 2006, 103, 1705–1710. [Google Scholar] [CrossRef] [PubMed]
- Holton, S.; Merckx, A.; Burgess, D.; Doerig, C.; Noble, M.; Endicott, J. Structures of P. falciparum PfPK5 test the CDK regulation paradigm and suggest mechanisms of small molecule inhibition. Structure 2003, 11, 1329–1337. [Google Scholar] [CrossRef] [PubMed]
- Chou, S.; Jensen, B.C.; Parsons, M.; Alber, T.; Grundner, C. The Trypanosoma brucei life cycle switch TbPTP1 is structurally conserved and dephosphorylates the nucleolar protein NOPP44/46. J. Biol. Chem. 2010, 285, 22075–22081. [Google Scholar] [CrossRef] [PubMed]
- Lountos, G.T.; Tropea, J.E.; Waugh, D.S. Structure of the Trypanosoma cruzi protein tyrosine phosphatase TcPTP1, a potential therapeutic target for Chagas’ disease. Mol. Biochem. Parasitol. 2013, 187, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Barrack, K.L.; Tulloch, L.B.; Burke, L.A.; Fyfe, P.K.; Hunter, W.N. Structure of recombinant Leishmania donovani pteridine reductase reveals a disordered active site. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2011, 67, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Gourley, D.G.; Schüttelkopf, A.W.; Leonard, G.A.; Luba, J.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Pteridine reductase mechanism correlates pterin metabolism with drug resistance in trypanosomatid parasites. Nat. Struct. Biol. 2001, 8, 521–525. [Google Scholar] [CrossRef] [PubMed]
- McLuskey, K.; Gibellini, F.; Carvalho, P.; Avery, M.A.; Hunter, W.N. Inhibition of Leishmania major pteridine reductase by 2,4,6-triaminoquinazoline: Structure of the NADPH ternary complex. Acta Crystallogr. D Biol. Crystallogr. 2004, 60, 1780–1785. [Google Scholar] [CrossRef] [PubMed]
- Schüttelkopf, A.W.; Hardy, L.W.; Beverley, S.M.; Hunter, W.N. Structures of Leishmania major pteridine reductase complexes reveal the active site features important for ligand binding and to guide inhibitor design. J. Mol. Biol. 2005, 352, 105–116. [Google Scholar] [CrossRef] [PubMed]
- Cavazzuti, A.; Paglietti, G.; Hunter, W.N.; Gamarro, F.; Piras, S.; Loriga, M.; Allecca, S.; Corona, P.; McLuskey, K.; Tulloch, L.; et al. Discovery of potent pteridine reductase inhibitors to guide antiparasite drug development. Proc. Natl. Acad. Sci. USA 2008, 105, 1448–1453. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, A.; Gibellini, F.; Sienkiewicz, N.; Tulloch, L.B.; Fyfe, P.K.; McLuskey, K.; Fairlamb, A.H.; Hunter, W.N. Structure and reactivity of Trypanosoma brucei pteridine reductase: Inhibition by the archetypal antifolate methotrexate. Mol. Microbiol. 2006, 61, 1457–1468. [Google Scholar] [CrossRef] [PubMed]
- Mpamhanga, C.P.; Spinks, D.; Tulloch, L.B.; Shanks, E.J.; Robinson, D.A.; Collie, I.T.; Fairlamb, A.H.; Wyatt, P.G.; Frearson, J.A.; Hunter, W.N.; et al. One scaffold, three binding modes: Novel and selective pteridine reductase 1 inhibitors derived from fragment hits discovered by virtual screening. J. Med. Chem. 2009, 52, 4454–4465. [Google Scholar] [CrossRef] [PubMed]
- Shanks, E.J.; Ong, H.B.; Robinson, D.A.; Thompson, S.; Sienkiewicz, N.; Fairlamb, A.H.; Frearson, J.A. Development and validation of a cytochrome c-coupled assay for pteridine reductase 1 and dihydrofolate reductase. Anal. Biochem. 2010, 396, 194–203. [Google Scholar] [CrossRef] [PubMed]
- Tulloch, L.B.; Martini, V.P.; Iulek, J.; Huggan, J.K.; Lee, J.H.; Gibson, C.L.; Smith, T.K.; Suckling, C.J.; Hunter, W.N. Structure-based design of pteridine reductase inhibitors targeting African sleeping sickness and the leishmaniases. J. Med. Chem. 2010, 53, 221–229. [Google Scholar] [CrossRef] [PubMed]
- Dawson, A.; Tulloch, L.B.; Barrack, K.L.; Hunter, W.N. High-resolution structures of Trypanosoma brucei pteridine reductase ligand complexes inform on the placement of new molecular entities in the active site of a potential drug target. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 1334–1340. [Google Scholar] [CrossRef] [PubMed]
- Nerini, E.; Dawson, A.; Hannaert, V.; Michels, P.A.; Hunter, W.N.; Costi, M.P. Structural studies of thiadiazole derivatives that inhibit Trypanosoma brucei growth. Unpublished work. 2012; doi:10.2210/pdb2yhi/pdb. [Google Scholar]
- Schormann, N.; Pal, B.; Senkovich, O.; Carson, M.; Howard, A.; Smith, C.; Delucas, L.; Chattopadhyay, D. Crystal structure of Trypanosoma cruzi pteridine reductase 2 in complex with a substrate and an inhibitor. J. Struct. Biol. 2005, 152, 64–75. [Google Scholar] [CrossRef] [PubMed]
- Shi, W.; Ting, L.M.; Kicska, G.A.; Lewandowicz, A.; Tyler, P.C.; Evans, G.B.; Furneaux, R.H.; Kim, K.; Almo, S.C.; Schramm, V.L. Plasmodium falciparum purine nucleoside phosphorylase: Crystal structures, immucillin inhibitors, and dual catalytic function. J. Biol. Chem. 2004, 279, 18103–18106. [Google Scholar] [CrossRef] [PubMed]
- Schnick, C.; Robien, M.A.; Brzozowski, A.M.; Dodson, E.J.; Murshudov, G.N.; Anderson, L.; Luft, J.R.; Mehlin, C.; Hol, W.G.; Brannigan, J.A.; et al. Structures of Plasmodium falciparum purine nucleoside phosphorylase complexed with sulfate and its natural substrate inosine. Acta Crystallogr. D Biol. Crystallogr. 2005, 61, 1245–1254. [Google Scholar] [CrossRef] [PubMed]
- Chaikuad, A.; Brady, R.L. Conservation of structure and activity in Plasmodium purine nucleoside phosphorylases. BMC Struct. Biol. 2009, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.C.; Alphey, M.S.; Wyllie, S.; Fairlamb, A.H. Chemical, genetic and structural assessment of pyridoxal kinase as a drug target in the African trypanosome. Mol. Microbiol. 2012, 86, 51–64. [Google Scholar] [CrossRef] [PubMed]
- Rigden, D.J.; Phillips, S.E.; Michels, P.A.; Fothergill-Gilmore, L.A. The structure of pyruvate kinase from Leishmania mexicana reveals details of the allosteric transition and unusual effector specificity. J. Mol. Biol. 1999, 291, 615–635. [Google Scholar] [CrossRef] [PubMed]
- Tulloch, L.B.; Morgan, H.P.; Hannaert, V.; Michels, P.A.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. Sulphate removal induces a major conformational change in Leishmania mexicana pyruvate kinase in the crystalline state. J. Mol. Biol. 2008, 383, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.P.; McNae, I.W.; Hsin, K.Y.; Michels, P.A.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. An improved strategy for the crystallization of Leishmania mexicana pyruvate kinase. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 2010, 66, 215–218. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.P.; McNae, I.W.; Nowicki, M.W.; Hannaert, V.; Michels, P.A.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. Allosteric mechanism of pyruvate kinase from Leishmania mexicana uses a rock and lock model. J. Biol. Chem. 2010, 285, 12892–12898. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.P.; McNae, I.W.; Nowicki, M.W.; Zhong, W.; Michels, P.A.; Auld, D.S.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. The trypanocidal drug suramin and other trypan blue mimetics are inhibitors of pyruvate kinases and bind to the adenosine site. J. Biol. Chem. 2011, 286, 31232–31240. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.P.; Walsh, M.; Blackburn, E.A.; Wear, M.A.; Boxer, M.; Shen, M.; McNae, I.W.; Michels, P.A.M.; Auld, D.S.; Fothergill-Gilmore, L.A.; et al. A new class of suicide inhibitor blocks nucleotide binding to pyruvate kinase. Unpublished work. 2012; doi:10.2210/pdb3srk/pdb. [Google Scholar]
- Wernimont, A.K.; Hutchinson, A.; Hassanali, A.; Mackenzie, F.; Cossar, D.; Bochkarev, A.; Arrowsmith, C.H.; Bountra, C.; Weigelt, J.; Edwards, A.M.; et al. Crystal structure of PFF1300w. Unpublished work. 2005; doi:10.2210/pdb3khd/pdb. [Google Scholar]
- Zhong, W.; Morgan, H.P.; McNae, I.W.; Michels, P.A.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. ‘In crystallo’ substrate binding triggers major domain movements and reveals magnesium as a co-activator of Trypanosoma brucei pyruvate kinase. Acta Crystallogr. D Biol. Crystallogr. 2013, 69, 1768–1779. [Google Scholar] [CrossRef] [PubMed]
- Zhong, W.; Morgan, H.P.; Nowicki, M.W.; McNae, I.W.; Yuan, M.; Bella, J.; Michels, P.A.; Fothergill-Gilmore, L.A.; Walkinshaw, M.D. Pyruvate kinases have an intrinsic and conserved decarboxylase activity. Biochem. J. 2014, 458, 301–311. [Google Scholar] [CrossRef] [PubMed]
- Marion, R.; Hansell, E.; Caffrey, C.; Roush, W.R.; Brinen, L.S. The high resolution structure of rhodesain, the major cathepsin L protease from Trypanosoma brucei rhodesiense, illustrates the basis for differences in inhibition profiles from other papain family cysteine proteases. Unpublished work. 2008; doi:10.2210/pdb2p86/pdb. [Google Scholar]
- Stern, A.L.; Naworyta, A.; Cazzulo, J.J.; Mowbray, S.L. Structures of type B ribose 5-phosphate isomerase from Trypanosoma cruzi shed light on the determinants of sugar specificity in the structural family. FEBS J. 2011, 278, 793–808. [Google Scholar] [CrossRef] [PubMed]
- Caruthers, J.; Bosch, J.; Buckner, F.; van Voorhis, W.; Myler, P.; Worthey, E.; Mehlin, C.; Boni, E.; DeTitta, G.; Luft, J.; et al. Structure of a ribulose 5-phosphate 3-epimerase from Plasmodium falciparum. Proteins 2006, 62, 338–342. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Schnaufer, A.; Salavati, R.; Stuart, K.D.; Hol, W.G. High resolution crystal structure of a key editosome enzyme from Trypanosoma brucei: RNA editing ligase 1. J. Mol. Biol. 2004, 343, 601–613. [Google Scholar] [CrossRef] [PubMed]
- Siponen, M.I.; Welin, M.; Arrowsmith, C.H.; Berglund, H.; Bountra, C.; Collins, R.; Dahlgren, L.G.; Edwards, A.M.; Flodin, S.; Flores, A.; et al. Crystal structure of Leishmania major S-adenosylhomocysteine hydrolase. Unpublished work. 2009; doi:10.2210/pdb3g1u/pdb. [Google Scholar]
- Tanaka, N.; Nakanishi, M.; Kusakabe, Y.; Shiraiwa, K.; Yabe, S.; Ito, Y.; Kitade, Y.; Nakamura, K.T. Crystal structure of S-adenosyl-l-homocysteine hydrolase from the human malaria parasite Plasmodium falciparum. J. Mol. Biol. 2004, 343, 1007–1017. [Google Scholar] [CrossRef] [PubMed]
- Siponen, M.I.; Schutz, P.; Arrowsmith, C.H. Crystal structure of S-adenosyl homocysteine hydrolase (SAHH) from Trypanosoma brucei. Unpublished work. 2009; doi:10.2210/pdb3h9u/pdb. [Google Scholar]
- Larson, E.T.; Zhang, L.; Napuli, A.; Mueller, N.; Verlinde, C.L.M.J.; van Voorhis, W.C.; Buckner, F.S.; Fan, E.; Hol, W.G.J.; Merritt, E.A. X-ray crystal structure of Seryl-tRNA synthetase from the eukaryotic parasite Trypanosoma brucei. Unpublished work. 2010; doi:10.2210/pdb3lsq/pdb, doi:10.2210/pdb3lss/pdb. [Google Scholar]
- Zhu, A.Y.; Zhou, Y.; Khan, S.; Deitsch, K.W.; Hao, Q.; Lin, H. Plasmodium falciparum Sir2A preferentially hydrolyzes medium and long chain fatty acyl lysine. ACS Chem. Biol. 2012, 7, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Dufe, V.T.; Qiu, W.; Müller, I.B.; Hui, R.; Walter, R.D.; Al-Karadaghi, S. Crystal structure of Plasmodium falciparum spermidine synthase in complex with the substrate decarboxylated S-adenosylmethionine and the potent inhibitors 4MCHA and AdoDATO. J. Mol. Biol. 2007, 373, 167–177. [Google Scholar] [CrossRef] [PubMed]
- Qiu, W.; Dong, A.; Ren, H.; Wu, H.; Zhao, Y.; Schapira, M.; Wasney, G.; Vedadi, M.; Lew, J.; Kozieradzki, I.; Edwards, A.M.; et al. Crystal structure of spermidine synthase from Plasmodium falciparum. Unpublished work. 2007; doi:10.2210/pdb2pwp/pdb. [Google Scholar]
- Burger, P.B.; Williams, M.; Reeksting, S.B.; Muller, I.B.; Al-Karadaghi, S.; Briggs, J.M.; Joubert, F.; Birkholtz, L.; Louw, A.I. Insights into the design of novel inhibitors against Plasmodium falciparum spermidine synthase using structurally derived binding descriptors. Unpublished work. 2012; doi:10.2210/pdb3rie/pdb. [Google Scholar]
- Bosch, J.; Arakaki, T.L.; Le Trong, I.; Merritt, E.A.; Hol, W.G.J. Crystal structure of spermidine synthase from Trypanosoma cruzi. Unpublished work. 2008; doi:10.2210/pdb3bwc/pdb. [Google Scholar]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Nes, W.D.; Waterman, M.R.; Lepesheva, G.I. Substrate preferences and catalytic parameters determined by structural characteristics of sterol 14α-demethylase (CYP51) from Leishmania infantum. J. Biol. Chem. 2011, 286, 26838–26848. [Google Scholar] [CrossRef] [PubMed]
- Lepesheva, G.I.; Park, H.W.; Hargrove, T.Y.; Vanhollebeke, B.; Wawrzak, Z.; Harp, J.M.; Sundaramoorthy, M.; Nes, W.D.; Pays, E.; Chaudhuri, M.; et al. Crystal structures of Trypanosoma brucei sterol 14α-demethylase and implications for selective treatment of human infections. J. Biol. Chem. 2010, 285, 1773–1780. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.K.; Leung, S.S.; Guilbert, C.; Jacobson, M.P.; McKerrow, J.H.; Podust, L.M. Structural characterization of CYP51 from Trypanosoma cruzi and Trypanosoma brucei bound to the antifungal drugs posaconazole and fluconazole. PLoS Negl. Trop. Dis. 2010, 4, e651. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Liu, J.; Waterman, M.R.; Nes, W.D.; Lepesheva, G.I. Structural complex of sterol 14α-demethylase (CYP51) with 14α-methylenecyclopropyl-Δ7-24,25-dihydrolanosterol. J. Lipid Res. 2012, 53, 311–320. [Google Scholar] [CrossRef] [PubMed]
- Buckner, F.S.; Bahia, M.T.; Suryadevara, P.K.; White, K.L.; Shackleford, D.M.; Chennamaneni, N.K.; Hulverson, M.A.; Laydbak, J.U.; Chatelain, E.; Scandale, I.; et al. Pharmacological characterization, structural studies, and in vivo activities of anti-Chagas disease lead compounds derived from tipifarnib. Antimicrob. Agents Chemother. 2012, 56, 4914–4921. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Calvet, C.M.; Gunatilleke, S.S.; Ruiz, C.; Cameron, M.D.; McKerrow, J.H.; Podust, L.M.; Roush, W.R. Rational development of 4-aminopyridyl-based inhibitors targeting Trypanosoma cruzi CYP51 as anti-Chagas agents. J. Med. Chem. 2013, 56, 7651–7668. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Waterman, M.R.; Lepesheva, G.I. CYP51 structure-based VNI scaffold development. Unpublished work. 2005; doi:10.2210/pdb4g7g/pdb. [Google Scholar]
- Lepesheva, G.I.; Hargrove, T.Y.; Anderson, S.; Kleshchenko, Y.; Furtak, V.; Wawrzak, Z.; Villalta, F.; Waterman, M.R. Structural insights into inhibition of sterol 14α-demethylase in the human pathogen Trypanosoma cruzi. J. Biol. Chem. 2010, 285, 25582–25590. [Google Scholar] [CrossRef] [PubMed]
- Andriani, G.; Amata, E.; Beatty, J.; Clements, Z.; Coffey, B.J.; Courtemanche, G.; Devine, W.; Erath, J.; Juda, C.E.; Wawrzak, Z.; et al. Antitrypanosomal lead discovery: Identification of a ligand-efficient inhibitor of Trypanosoma cruzi CYP51 and parasite growth. J. Med. Chem. 2013, 56, 2556–2567. [Google Scholar] [CrossRef] [PubMed]
- Hargrove, T.Y.; Wawrzak, Z.; Alexander, P.W.; Chaplin, J.H.; Keenan, M.; Charman, S.A.; Perez, C.J.; Waterman, M.R.; Chatelain, E.; Lepesheva, G.I. Complexes of Trypanosoma cruzi sterol 14α-demethylase (CYP51) with two pyridine-based drug candidates for Chagas disease: Structural basis for pathogen selectivity. J. Biol. Chem. 2013, 288, 31602–31615. [Google Scholar] [CrossRef] [PubMed]
- Vieira, D.F.; Choi, J.Y.; Roush, W.R.; Podust, L.M. Expanding the binding envelope of CYP51 inhibitors targeting Trypanosoma cruzi with 4-aminopyridyl-based sulfonamide derivatives. ChemBiochem 2014, 15, 1111–1120. [Google Scholar] [CrossRef] [PubMed]
- Choi, J.Y.; Calvet, C.M.; Vieira, D.F.; Gunatilleke, S.S.; Cameron, M.D.; Mckerrow, J.H.; Podust, L.M.; Roush, W.R. R-Configuration of 4-aminopyridyl-based inhibitors of CYP51 confers superior efficacy against Trypanosoma cruzi. ACS Med. Chem. Lett. 2014, 5, 434–439. [Google Scholar] [CrossRef] [PubMed]
- Calvet, C.M.; Vieira, D.F.; Choi, J.Y.; Kellar, D.; Cameron, M.D.; Siqueira-Neto, J.L.; Gut, J.; Johnston, J.B.; Lin, L.; et al. 4-Aminopyridyl-based CYP51 inhibitors as anti-Trypanosoma cruzi drug leads with improved pharmacokinetic profile and in vivo potency. J. Med. Chem. 2014, 57, 6989–7005. [Google Scholar] [CrossRef] [PubMed]
- Harijan, R.K.; Kiema, T.R.; Karjalainen, M.P.; Janardan, N.; Murthy, M.R.; Weiss, M.S.; Michels, P.A.; Wierenga, R.K. Crystal structures of SCP2-thiolases of Trypanosomatidae, human pathogens causing widespread tropical diseases: The importance for catalysis of the cysteine of the unique HDCF loop. Biochem. J. 2013, 455, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Boucher, I.W.; Brzozowski, A.M.; Brannigan, J.A.; Schnick, C.; Smith, D.J.; Kyes, S.A.; Wilkinson, A.J. The crystal structure of superoxide dismutase from Plasmodium falciparum. BMC Struct. Biol. 2006, 6, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bachega, J.F.; Navarro, M.V.; Bleicher, L.; Bortoleto-Bugs, R.K.; Dive, D.; Hoffmann, P.; Viscogliosi, E.; Garratt, R.C. Systematic structural studies of iron superoxide dismutases from human parasites and a statistical coupling analysis of metal binding specificity. Proteins 2009, 77, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Ernst, N.L.; Turley, S.; Stuart, K.D.; Hol, W.G. Structural basis for UTP specificity of RNA editing TUTases from Trypanosoma brucei. EMBO J. 2005, 24, 4007–4017. [Google Scholar] [CrossRef] [PubMed]
- Stagno, J.; Aphasizheva, I.; Rosengarth, A.; Luecke, H.; Aphasizhev, R. UTP-bound and Apo structures of a minimal RNA uridylyltransferase. J. Mol. Biol. 2007, 366, 882–899. [Google Scholar] [CrossRef] [PubMed]
- Stagno, J.; Aphasizheva, I.; Aphasizhev, R.; Luecke, H. Dual role of the RNA substrate in selectivity and catalysis by terminal uridylyl transferases. Proc. Natl. Acad. Sci. USA 2007, 104, 14634–14639. [Google Scholar] [CrossRef] [PubMed]
- Saab-Rincón, G.; Olvera, L.; Olvera, M.; Rudiño-Piñera, E.; Benites, E.; Soberón, X.; Morett, E. Evolutionary walk between (β/α)(8) barrels: Catalytic migration from triosephosphate isomerase to thiamin phosphate synthase. J. Mol. Biol. 2012, 416, 255–270. [Google Scholar] [CrossRef] [PubMed]
- Fyfe, P.K.; Westrop, G.D.; Silva, A.M.; Coombs, G.H.; Hunter, W.N. Leishmania TDR1 structure, a unique trimeric glutathione transferase capable of deglutathionylation and antimonial prodrug activation. Proc. Natl. Acad. Sci. USA 2012, 109, 11693–11698. [Google Scholar] [CrossRef] [PubMed]
- Fritz-Wolf, K.; Jortzik, E.; Stumpf, M.; Preuss, J.; Iozef, R.; Rahlfs, S.; Becker, K. Crystal structure of the Plasmodium falciparum thioredoxin reductase-thioredoxin complex. J. Mol. Biol. 2013, 425, 3446–3460. [Google Scholar] [CrossRef] [PubMed]
- Whittingham, J.L.; Carrero-Lerida, J.; Brannigan, J.A.; Ruiz-Perez, L.M.; Silva, A.P.; Fogg, M.J.; Wilkinson, A.J.; Gilbert, I.H.; Wilson, K.S.; González-Pacanowska, D. Structural basis for the efficient phosphorylation of AZT-MP (3′-azido-3′-deoxythymidine monophosphate) and dGMP by Plasmodium falciparum type I thymidylate kinase. Biochem. J. 2010, 428, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Cui, H.; Carrero-Lérida, J.; Silva, A.P.; Whittingham, J.L.; Brannigan, J.A.; Ruiz-Pérez, L.M.; Read, K.D.; Wilson, K.S.; González-Pacanowska, D.; Gilbert, I.H. Synthesis and evaluation of α-thymidine analogues as novel antimalarials. J. Med. Chem. 2012, 55, 10948–10957. [Google Scholar] [CrossRef] [PubMed]
- Veitch, N.J.; Maugeri, D.A.; Cazzulo, J.J.; Lindqvist, Y.; Barrett, M.P. Transketolase from Leishmania mexicana has a dual subcellular localization. Biochem. J. 2004, 382, 759–767. [Google Scholar] [CrossRef] [PubMed]
- Eichhorn, T.; Winter, D.; Büchele, B.; Dirdjaja, N.; Frank, M.; Lehmann, W.D.; Mertens, R.; Krauth-Siegel, R.L.; Simmet, T.; Granzin, J.; et al. Molecular interaction of artemisinin with translationally controlled tumor protein (TCTP) of Plasmodium falciparum. Biochem. Pharmacol. 2013, 85, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, A.; Amaya, M.F.; Cremona, M.L.; Frasch, A.C.; Alzari, P.M. The crystal structure and mode of action of trans-sialidase, a key enzyme in Trypanosoma cruzi pathogenesis. Mol. Cell 2002, 10, 757–768. [Google Scholar] [CrossRef]
- Amaya, M.F.; Watts, A.G.; Damager, I.; Wehenkel, A.; Nguyen, T.; Buschiazzo, A.; Paris, G.; Frasch, A.C.; Withers, S.G.; Alzari, P.M. Structural insights into the catalytic mechanism of Trypanosoma cruzi trans-sialidase. Structure 2004, 12, 775–784. [Google Scholar] [CrossRef] [PubMed]
- Buchini, S.; Buschiazzo, A.; Withers, S.G. A new generation of specific Trypanosoma cruzi trans-sialidase inhibitors. Angew. Chem. Int. Ed. Engl. 2008, 47, 2700–2703. [Google Scholar] [CrossRef] [PubMed]
- Buschiazzo, A.; Muiá, R.; Larrieux, N.; Pitcovsky, T.; Mucci, J.; Campetella, O. Trypanosoma cruzi trans-sialidase in complex with a neutralizing antibody: Structure/function studies towards the rational design of inhibitors. PLoS Pathog. 2012, 8, e1002474. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.C.; Zeelen, J.P.; Neubauer, G.; Vriend, G.; Backmann, J.; Michels, P.A.; Lambeir, A.M.; Wierenga, R.K. Structural and mutagenesis studies of Leishmania triosephosphate isomerase: A point mutation can convert a mesophilic enzyme into a superstable enzyme without losing catalytic power. Protein Eng. 1999, 12, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Kursula, I.; Partanen, S.; Lambeir, A.M.; Antonov, D.M.; Augustyns, K.; Wierenga, R.K. Structural determinants for ligand binding and catalysis of triosephosphate isomerase. Eur. J. Biochem. 2001, 268, 5189–5196. [Google Scholar] [CrossRef] [PubMed]
- Kursula, I.; Wierenga, R.K. Crystal structure of triosephosphate isomerase complexed with 2-phosphoglycolate at 0.83-Å resolution. J. Biol. Chem. 2003, 278, 9544–9551. [Google Scholar] [CrossRef] [PubMed]
- Alahuhta, M.; Wierenga, R.K. Atomic resolution crystallography of a complex of triosephosphate isomerase with a reaction-intermediate analog: New insight in the proton transfer reaction mechanism. Proteins 2010, 78, 1878–1888. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, R.; Alahuhta, M.; Pihko, P.M.; Wierenga, R.K. High resolution crystal structures of triosephosphate isomerase complexed with its suicide inhibitors: The conformational flexibility of the catalytic glutamate in its closed, liganded active site. Protein Sci. 2011, 20, 1387–1397. [Google Scholar] [CrossRef] [PubMed]
- Velanker, S.S.; Ray, S.S.; Gokhale, R.S.; Suma, S.; Balaram, H.; Balaram, P.; Murthy, M.R. Triosephosphate isomerase from Plasmodium falciparum: The crystal structure provides insights into antimalarial drug design. Structure 1997, 5, 751–761. [Google Scholar] [CrossRef]
- Parthasarathy, S.; Ravindra, G.; Balaram, H.; Balaram, P.; Murthy, M.R. Structure of the Plasmodium falciparum triosephosphate isomerase-phosphoglycolate complex in two crystal forms: Characterization of catalytic loop open and closed conformations in the ligand-bound state. Biochemistry 2002, 41, 13178–13188. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Balaram, H.; Balaram, P.; Murthy, M.R. Structures of Plasmodium falciparum triosephosphate isomerase complexed to substrate analogues: Observation of the catalytic loop in the open conformation in the ligand-bound state. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 1992–2000. [Google Scholar] [CrossRef] [PubMed]
- Parthasarathy, S.; Eaazhisai, K.; Balaram, H.; Balaram, P.; Murthy, M.R. Structure of Plasmodium falciparum triose-phosphate isomerase-2-phosphoglycerate complex at 1.1-Å resolution. J. Biol. Chem. 2003, 278, 52461–52470. [Google Scholar] [CrossRef] [PubMed]
- Gayathri, P.; Banerjee, M.; Vijayalakshmi, A.; Balaram, H.; Balaram, P.; Murthy, M.R. Biochemical and structural characterization of residue 96 mutants of Plasmodium falciparum triosephosphate isomerase: Active-site loop conformation, hydration and identification of a dimer-interface ligand-binding site. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 847–857. [Google Scholar] [CrossRef] [PubMed]
- Verlinde, C.L.; Noble, M.E.; Kalk, K.H.; Groendijk, H.; Wierenga, R.K.; Hol, W.G. Anion binding at the active site of trypanosomal triosephosphate isomerase. Monohydrogen phosphate does not mimic sulphate. Eur. J. Biochem. 1991, 198, 53–57. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, R.K.; Noble, M.E.; Postma, J.P.; Groendijk, H.; Kalk, K.H.; Hol, W.G.; Opperdoes, F.R. The crystal structure of the “open” and the “closed” conformation of the flexible loop of trypanosomal triosephosphate isomerase. Proteins 1991, 10, 33–49. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.E.; Wierenga, R.K.; Lambeir, A.M.; Opperdoes, F.R.; Thunnissen, A.M.; Kalk, K.H.; Groendijk, H.; Hol, W.G. The adaptability of the active site of trypanosomal triosephosphate isomerase as observed in the crystal structures of three different complexes. Proteins 1991, 10, 50–69. [Google Scholar] [CrossRef] [PubMed]
- Wierenga, R.K.; Noble, M.E.; Vriend, G.; Nauche, S.; Hol, W.G. Refined 1.83 Å structure of trypanosomal triosephosphate isomerase crystallized in the presence of 2.4 M-ammonium sulphate. A comparison with the structure of the trypanosomal triosephosphate isomerase-glycerol-3-phosphate complex. J. Mol. Biol. 1991, 220, 995–1015. [Google Scholar] [CrossRef]
- Noble, M.E.; Verlinde, C.L.; Groendijk, H.; Kalk, K.H.; Wierenga, R.K.; Hol, W.G. Crystallographic and molecular modeling studies on trypanosomal triosephosphate isomerase: A critical assessment of the predicted and observed structures of the complex with 2-phosphoglycerate. J. Med. Chem. 1991, 34, 2709–2718. [Google Scholar] [CrossRef] [PubMed]
- Noble, M.E.; Zeelen, J.P.; Wierenga, R.K. Structures of the “open” and “closed” state of trypanosomal triosephosphate isomerase, as observed in a new crystal form: Implications for the reaction mechanism. Proteins 1993, 16, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Kishan, K.V.; Zeelen, J.P.; Noble, M.E.; Borchert, T.V.; Wierenga, R.K. Comparison of the structures and the crystal contacts of trypanosomal triosephosphate isomerase in four different crystal forms. Protein Sci. 1994, 3, 779–787. [Google Scholar] [PubMed]
- Thanki, N.; Zeelen, J.P.; Mathieu, M.; Jaenicke, R.; Abagyan, R.A.; Wierenga, R.K.; Schliebs, W. Protein engineering with monomeric triosephosphate isomerase (monoTIM): The modelling and structure verification of a seven-residue loop. Protein Eng. 1997, 10, 159–167. [Google Scholar] [CrossRef] [PubMed]
- Norledge, B.V.; Lambeir, A.M.; Abagyan, R.A.; Rottmann, A.; Fernandez, A.M.; Filimonov, V.V.; Peter, M.G.; Wierenga, R.K. Modeling, mutagenesis, and structural studies on the fully conserved phosphate-binding loop (loop 8) of triosephosphate isomerase: Toward a new substrate specificity. Proteins 2001, 42, 383–389. [Google Scholar] [CrossRef]
- Casteleijn, M.G.; Alahuhta, M.; Groebel, K.; El-Sayed, I.; Augustyns, K.; Lambeir, A.M.; Neubauer, P.; Wierenga, R.K. Functional role of the conserved active site proline of triosephosphate isomerase. Biochemistry 2006, 45, 15483–15494. [Google Scholar] [CrossRef] [PubMed]
- Salin, M.; Kapetaniou, E.G.; Vaismaa, M.; Lajunen, M.; Casteleijn, M.G.; Neubauer, P.; Salmon, L.; Wierenga, R.K. Crystallographic binding studies with an engineered monomeric variant of triosephosphate isomerase. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 934–944. [Google Scholar] [CrossRef] [PubMed]
- Maldonado, E.; Soriano-García, M.; Moreno, A.; Cabrera, N.; Garza-Ramos, G.; de Gómez-Puyou, M.; Gómez-Puyou, A.; Perez-Montfort, R. Differences in the intersubunit contacts in triosephosphate isomerase from two closely related pathogenic trypanosomes. J. Mol. Biol. 1998, 283, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.G.; Maldonado, E.; Pérez-Montfort, R.; Garza-Ramos, G.; de Gómez-Puyou, M.T.; Gómez-Puyou, A.; Rodríguez-Romero, A. Crystal structure of triosephosphate isomerase from Trypanosoma cruzi in hexane. Proc. Natl. Acad. Sci. USA 1999, 96, 10062–10067. [Google Scholar] [CrossRef] [PubMed]
- Téllez-Valencia, A.; Olivares-Illana, V.; Hernández-Santoyo, A.; Pérez-Montfort, R.; Costas, M.; Rodríguez-Romero, A.; López-Calahorra, F.; Tuena De Gómez-Puyou, M.; Gómez-Puyou, A. Inactivation of triosephosphate isomerase from Trypanosoma cruzi by an agent that perturbs its dimer interface. J. Mol. Biol. 2004, 341, 1355–1365. [Google Scholar] [CrossRef] [PubMed]
- Olivares-Illana, V.; Rodríguez-Romero, A.; Becker, I.; Berzunza, M.; García, J.; Pérez-Montfort, R.; Cabrera, N.; López-Calahorra, F.; de Gómez-Puyou, M.T.; Gómez-Puyou, A. Perturbation of the dimer interface of triosephosphate isomerase and its effect on Trypanosoma cruzi. PLoS Negl. Trop. Dis. 2007, 1, e1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zárate-Pérez, F.; Chánez-Cárdenas, M.E.; Vázquez-Contreras, E. The folding pathway of triosephosphate isomerase. Prog. Mol. Biol. Transl. Sci. 2008, 84, 251–267. [Google Scholar] [PubMed]
- García-Torres, I.; Cabrera, N.; Torres-Larios, A.; Rodríguez-Bolaños, M.; Díaz-Mazariegos, S.; Gómez-Puyou, A.; Perez-Montfort, R. Identification of amino acids that account for long-range interactions in two triosephosphate isomerases from pathogenic trypanosomes. PLoS ONE 2011, 6, e18791. [Google Scholar] [CrossRef] [PubMed]
- Aguirre, Y.; Cabrera, N.; Aguirre, B.; Pérez-Montfort, R.; Hernandez-Santoyo, A.; Reyes-Vivas, H.; Enríquez-Flores, S.; de Gómez-Puyou, M.T.; Gómez-Puyou, A.; Sanchez-Ruiz, J.M.; et al. Different contribution of conserved amino acids to the global properties of triosephosphate isomerases. Proteins 2014, 82, 323–335. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Colotti, G.; Franceschini, S.; Ilari, A. Molecular basis of antimony treatment in leishmaniasis. J. Med. Chem. 2009, 52, 2603–2612. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Ilari, A.; Ceci, P.; Orsini, S.; Gramiccia, M.; di Muccio, T.; Colotti, G. Inhibitory effect of silver nanoparticles on trypanothione reductase activity and Leishmania infantum proliferation. ACS Med. Chem. Lett. 2011, 2, 230–233. [Google Scholar] [CrossRef] [PubMed]
- Ilari, A.; Baiocco, P.; Messori, L.; Fiorillo, A.; Boffi, A.; Gramiccia, M.; di Muccio, T.; Colotti, G. A gold-containing drug against parasitic polyamine metabolism: The X-ray structure of trypanothione reductase from Leishmania infantum in complex with auranofin reveals a dual mechanism of enzyme inhibition. Amino Acids 2012, 42, 803–811. [Google Scholar] [CrossRef] [PubMed]
- Baiocco, P.; Poce, G.; Alfonso, S.; Cocozza, M.; Porretta, G.C.; Colotti, G.; Biava, M.; Moraca, F.; Botta, M.; Yardley, V.; et al. Inhibition of Leishmania infantum trypanothione reductase by azole-based compounds: A comparative analysis with its physiological substrate by X-ray crystallography. ChemMedChem 2013, 8, 1175–1183. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.C.; Ariza, A.; Chow, W.H.; Oza, S.L.; Fairlamb, A.H. Comparative structural, kinetic and inhibitor studies of Trypanosoma brucei trypanothione reductase with T. cruzi. Mol. Biochem. Parasitol. 2010, 169, 12–19. [Google Scholar] [CrossRef] [PubMed]
- Patterson, S.; Alphey, M.S.; Jones, D.C.; Shanks, E.J.; Street, I.P.; Frearson, J.A.; Wyatt, P.G.; Gilbert, I.H.; Fairlamb, A.H. Dihydroquinazolines as a novel class of Trypanosoma brucei trypanothione reductase inhibitors: Discovery, synthesis, and characterization of their binding mode by protein crystallography. J. Med. Chem. 2011, 54, 6514–6530. [Google Scholar] [CrossRef] [PubMed]
- Persch, E.; Bryson, S.; Todoroff, N.K.; Eberle, C.; Thelemann, J.; Dirdjaja, N.; Kaiser, M.; Weber, M.; Derbani, H.; Brun, R.; et al. Binding to large enzyme pockets: Small-molecule inhibitors of trypanothione reductase. ChemMedChem 2014, 9, 1880–1891. [Google Scholar] [CrossRef] [PubMed]
- Lantwin, C.B.; Schlichting, I.; Kabsch, W.; Pai, E.F.; Krauth-Siegel, R.L. The structure of Trypanosoma cruzi trypanothione reductase in the oxidized and NADPH reduced state. Proteins 1994, 18, 161–173. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Bond, C.S.; Bailey, S.; Cunningham, M.L.; Fairlamb, A.H.; Hunter, W.N. The crystal structure of trypanothione reductase from the human pathogen Trypanosoma cruzi at 2.3 Å resolution. Protein Sci. 1996, 5, 52–61. [Google Scholar] [CrossRef] [PubMed]
- Bond, C.S.; Zhang, Y.; Berriman, M.; Cunningham, M.L.; Fairlamb, A.H.; Hunter, W.N. Crystal structure of Trypanosoma cruzi trypanothione reductase in complex with trypanothione, and the structure-based discovery of new natural product inhibitors. Structure 1999, 7, 81–89. [Google Scholar] [CrossRef]
- Saravanamuthu, A.; Vickers, T.J.; Bond, C.S.; Peterson, M.R.; Hunter, W.N.; Fairlamb, A.H. Two interacting binding sites for quinacrine derivatives in the active site of trypanothione reductase: A template for drug design. J. Biol. Chem. 2004, 279, 29493–29500. [Google Scholar] [CrossRef] [PubMed]
- Alphey, M.S.; König, J.; Fairlamb, A.H. Structural and mechanistic insights into type II trypanosomatid tryparedoxin-dependent peroxidases. Biochem. J. 2008, 414, 375–381. [Google Scholar] [CrossRef] [PubMed]
- Piñeyro, M.D.; Pizarro, J.C.; Lema, F.; Pritsch, O.; Cayota, A.; Bentley, G.A.; Robello, C. Crystal structure of the tryparedoxin peroxidase from the human parasite Trypanosoma cruzi. J. Struct. Biol. 2005, 150, 11–22. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.Y.; Kim, J.E.; Napoli, A.J.; Verlinde, C.L.; Fan, E.; Buckner, F.S.; van Voorhis, W.C.; Hol, W.G. Crystal structures of Plasmodium falciparum cytosolic tryptophanyl-tRNA synthetase and its potential as a target for structure-guided drug design. Mol. Biochem. Parasitol. 2013, 189, 26–32. [Google Scholar] [CrossRef] [PubMed]
- Merritt, E.A.; Arakaki, T.L.; Gillespie, R.; Napuli, A.J.; Kim, J.E.; Buckner, F.S.; van Voorhis, W.C.; Verlinde, C.L.; Fan, E.; Zucker, F.; et al. Crystal structures of three protozoan homologs of tryptophanyl-tRNA synthetase. Mol. Biochem. Parasitol. 2011, 177, 20–28. [Google Scholar] [CrossRef] [PubMed]
- Larson, E.T.; Kim, J.E.; Castaneda, L.J.; Napuli, A.J.; Zhang, Z.; Fan, E.; Zucker, F.H.; Verlinde, C.L.; Buckner, F.S.; van Voorhis, W.C.; et al. The double-length tyrosyl-tRNA synthetase from the eukaryote Leishmania major forms an intrinsically asymmetric pseudo-dimer. J. Mol. Biol. 2011, 409, 159–176. [Google Scholar] [CrossRef] [PubMed]
- Bhatt, T.K.; Khan, S.; Dwivedi, V.P.; Banday, M.M.; Sharma, A.; Chandele, A.; Camacho, N.; de Pouplana, L.R.; Wu, Y.; Craig, A.G.; et al. Malaria parasite tyrosyl-tRNA synthetase secretion triggers pro-inflammatory responses. Nat. Commun. 2011, 2, 530. [Google Scholar] [CrossRef] [PubMed]
- Artavanis-Tsakonas, K.; Weihofen, W.A.; Antos, J.M.; Coleman, B.I.; Comeaux, C.A.; Duraisingh, M.T.; Gaudet, R.; Ploegh, H.L. Characterization and structural studies of the Plasmodium falciparum ubiquitin and Nedd8 hydrolase UCHL3. J. Biol. Chem. 2010, 285, 6857–6866. [Google Scholar] [CrossRef] [PubMed]
- Shaw, M.P.; Bond, C.S.; Roper, J.R.; Gourley, D.G.; Ferguson, M.A.; Hunter, W.N. High-resolution crystal structure of Trypanosoma brucei UDP-galactose 4′-epimerase: A potential target for structure-based development of novel trypanocides. Mol. Biochem. Parasitol. 2003, 126, 173–180. [Google Scholar] [CrossRef]
- Dhatwalia, R.; Singh, H.; Oppenheimer, M.; Sobrado, P.; Tanner, J.J. Crystal structures of Trypanosoma cruzi UDP-galactopyranose mutase implicate flexibility of the histidine loop in enzyme activation. Biochemistry 2012, 51, 4968–4979. [Google Scholar] [CrossRef] [PubMed]
- Führing, J.; Cramer, J.T.; Routier, F.H.; Lamerz, A.C.; Baruch, P.; Gerardy-Schahn, R.; Fedorov, R. Catalytic mechanism and allosteric regulation of UDP-glucose pyrophosphorylase from Leishmania major. ACS Catal. 2013, 3, 2976–2985. [Google Scholar] [CrossRef]
- Urbaniak, M.D.; Collie, I.T.; Fang, W.; Aristotelous, T.; Eskilsson, S.; Raimi, O.G.; Harrison, J.; Navratilova, I.H.; Frearson, J.A.; van Aalten, D.M.; et al. A novel allosteric inhibitor of the uridine diphosphate N-acetylglucosamine pyrophosphorylase from Trypanosoma brucei. ACS Chem. Biol. 2013, 8, 1981–1987. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larson, E.T.; Mudeppa, D.G.; Gillespie, J.R.; Mueller, N.; Napuli, A.J.; Arif, J.A.; Ross, J.; Arakaki, T.L.; Lauricella, A.; Detitta, G.; et al. The crystal structure and activity of a putative trypanosomal nucleoside phosphorylase reveal it to be a homodimeric uridine phosphorylase. J. Mol. Biol. 2010, 396, 1244–1259. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Holien, J.; Ramsland, P.A. Improvements, trands, and new ideas in molecular docking: 2012–2013 in review. J. Mol. Recognit. 2015, 28, 581–604. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, L.G.; dos Santos, R.N.; Oliva, G.; Andricopulo, A.D. Molecular docking and structure-based drug design strategies. Molecules 2015, 20, 13384–13421. [Google Scholar] [CrossRef] [PubMed]
- Berry, M.; Fielding, B.; Gamieldien, J. Practical considerations in virtual screening and molecular docking. In Emerging Trends in Computational Biology, Bioinformatics, and Systems Biology; Tran, Q.N., Hamid, A.R., Eds.; Elsevier: Amsterdam, The Netherlands, 2015; pp. 487–502. [Google Scholar]
- De Ruyck, J.; Brysbaert, G.; Blossey, R.; Lensink, M.F. Molecular docking as a popular tool in drug design, an in silico travel. Adv. Appl. Bioinform. Chem. 2016, 9, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Agostino, M.; Ramsland, P.A. Challenges and advances in computational docking: 2009 in review. J. Mol. Recognit. 2011, 24, 149–164. [Google Scholar] [CrossRef] [PubMed]
- Yuriev, E.; Ramsland, P.A. Latest developments in molecular docking: 2010–2011 in review. J. Mol. Recognit. 2013, 26, 215–239. [Google Scholar] [CrossRef] [PubMed]
- Irwin, J.J.; Shoichet, B.K. Docking screens for novel ligands conferring new biology. J. Med. Chem. 2016, 59, 4103–4120. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.K.; Saudagar, P.; Shukla, A.K.; Dubey, V.K. Screening natural products database for identification of potential antileishmanial chemotherapeutic agents. Interdiscip. Sci. 2011, 3, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, F.C.; Schmidt, T.J. In silico screening of natural product databases reveals new potential leads against neglected diseases. Planta Med. 2013, 79. [Google Scholar] [CrossRef]
- Buckingham, J. (Ed.); Dictionary of Natural Products on DVD; CRC Press: Boca Raton, FL, USA, 2016. [Google Scholar]
- Napralert. Natural Products Alert. Available online: http://napralert.org (accessed on 16 August 2016).
- ZINC12. Bioinformatics and Chemical Informatics Research Center, Department of Pharmaceutical Chemistry, University of California: San Francisco, CA, USA. Available online: http://zinc.docking.org/browse/catalogs/natural-products (accessed on 16 August 2016).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Autodock4 and AutoDockTools4: Automated docking with selective receptor flexiblity. J. Comput. Chem. 2009, 16, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Thomsen, R.; Christensen, M.H. MolDock: A new technique for high-accuracy molecular docking. J. Med. Chem. 2006, 49, 3315–3321. [Google Scholar] [CrossRef] [PubMed]
- Friesner, R.A.; Banks, J.L.; Murphy, R.B.; Halgren, T.A.; Klicic, J.J.; Mainz, D.T.; Repasky, M.P.; Knoll, E.H.; Shaw, D.E.; Shelley, M.; et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]
- Jain, A.N. Surflex-Dock 2.1: Robust performance from ligand energetic modeling, ring, flexibility, and knowledge-based search. J. Comput. Aided Mol. Des. 2007, 21, 281–306. [Google Scholar] [CrossRef] [PubMed]
- Ogungbe, I.V.; Ng, J.D.; Setzer, W.N. Interactions of antiparasitic alkaloids with Leishmania protein targets: A molecular docking analysis. Future Med. Chem. 2013, 5, 1777–1799. [Google Scholar] [CrossRef] [PubMed]
- Ogungbe, I.V.; Setzer, W.N. In-silico Leishmania target selectivity of antiparasitic terpenoids. Molecules 2013, 18, 7761–7847. [Google Scholar] [CrossRef] [PubMed]
- Kumar, M.; Dagar, A.; Gupta, V.K.; Sharma, A. In silico docking studies of bioactive natural plant products as putative DHFR antagonists. Med. Chem. Res. 2014, 23, 810–817. [Google Scholar] [CrossRef]
- Ogungbe, I.V.; Erwin, W.R.; Setzer, W.N. Antileishmanial phytochemical phenolics: Molecular docking to potential protein targets. J. Mol. Graph. Model. 2014, 48, 105–117. [Google Scholar] [CrossRef] [PubMed]
- Bernal, F.A.; Coy-Barrera, E. In-silico analyses of sesquiterpene-related compounds on selected Leishmania enzyme-based targets. Molecules 2014, 19, 5550–5569. [Google Scholar] [CrossRef] [PubMed]
- Setzer, W.N.; Ogungbe, I.V. In-silico investigation of antitrypanosomal phytochemicals from Nigerian medicinal plants. PLoS Negl. Trop. Dis. 2012, 6, e1727. [Google Scholar] [CrossRef] [PubMed]
- McCulley, S.F.; Setzer, W.N. An in-silico investigation of anti-Chagas phytochemicals. Curr. Clin. Pharmacol. 2014, 9, 205–257. [Google Scholar] [CrossRef] [PubMed]
- Melo, T.S.; Gattass, C.R.; Soares, D.C.; Cunha, M.R.; Ferreira, C.; Tavares, M.T.; Saraiva, E.; Parise-Filho, R.; Braden, H.; Delorenzi, J.C. Oleanolic acid (OA) as an antileishmanial agent: Biological evaluation and in silico mechanistic insights. Parasitol. Int. 2016, 65, 227–237. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, M.B.G.; Manjolin, L.C.; Maquiaveli, C.D.C.; Santos-Filho, O.A.; da Silva, E.R. Inhibition of Leishmania (Leishmania) amazonensis and rat arginases by green tea EGCG, (+)-catechin and (2)-epicatechin: A comparative structural analysis of enzyme-inhibitor interactions. PLoS ONE 2013, 8, e78387. [Google Scholar] [CrossRef] [PubMed]
- Venkatesan, S.K.; Saudagar, P.; Dubey, V.K. Identification of novel inhibitor of trypanothione synthase from two Leishmania species: Comparative in silico analysis. J. Protein Proteom. 2011, 2, 41–48. [Google Scholar]
- Krauth-Siegel, R.L.; Inhoff, O. Parasite-specific trypanothione reductase as a drug target molecule. Parasitol. Res. 2003, 90 (Suppl. 2), S77–S85. [Google Scholar] [CrossRef] [PubMed]
- Gundampati, R.K.; Jagannadham, M.V. Molecular docking based inhibition of trypanothione reductase activity of taxifolin novel target for antileishmanial activity. J. Appl. Pharmaceut. Sci. 2012, 2, 133–136. [Google Scholar] [CrossRef]
- Ribeiro, F.F.; Junior, F.J.B.M.; da Silva, M.S.; Scotti, M.T.; Scotti, L. Computational and investigative study of flavonoids against Trypanosoma cruzi and Leishmania spp. Nat. Prod. Commun. 2015, 10, 917–920. [Google Scholar] [PubMed]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Deliv. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Gundampati, R.K.; Chandrasekaran, S.; Jagannadham, M.V. Molecular docking study on the interaction between trypanothione reductase and mangiferin for antileishmanial activity. Bangladesh J. Pharmacol. 2013, 8, 40–43. [Google Scholar]
- Chauhan, R. In Silico Approach towards Finding Inhibitory Effect of Phytochemicals on Trypanothione Reductase in Leishmania donovani. Master’s Thesis, National Institute of Technology, Orissa, India, 2014. [Google Scholar]
- Venkatesan, S.K.; Dubey, V.K. Footprinting of inhibitor interactions of in silico identified inhibitors of trypanothione reductase of Leishmania parasite. Sci. World J. 2012, 2012, 963658. [Google Scholar] [CrossRef] [PubMed]
- Lavie, Y.; Harel-Orbital, T.; Gaffield, W.; Liscovitch, M. Inhibitory effect of steroidal alkaloids on drug transport and multidrug resistance in human cancer cells. Anticancer Res. 2001, 21, 1189–1194. [Google Scholar] [PubMed]
- Medina, J.M.; Rodrigues, J.C.; de Souza, W.; Atella, G.C.; Barrabin, H. Tomatidine promotes the inhibition of 24-alkylated sterol biosynthesis and mitochondrial dysfunction in Leishmania amazonensis promastigotes. Parasitology 2012, 139, 1253–1265. [Google Scholar] [CrossRef] [PubMed]
- Ogungbe, I.V.; Setzer, W.N. Comparative molecular docking of antitrypanosomal natural products into multiple Trypanosoma brucei drug targets. Molecules 2009, 14, 1513–1536. [Google Scholar] [CrossRef] [PubMed]
- Izumi, E.; Ueda-Nakamura, T.; Dias Milho, B.P.; Veiga Júnior, V.F.; Vataru Nakamura, C. Natural products and Chagas’ disease: A review of plant compounds studied for activity against Trypanosoma cruzi. Nat. Prod. Rep. 2011, 28, 809–823. [Google Scholar] [CrossRef] [PubMed]
- Argüelles, A.J.; Cordell, G.A.; Maruenda, H. Molecular docking and binding mode analysis of plant alkaloids as in vitro and in silico inhibitors of trypanothione reductase from Trypanosoma cruzi. Nat. Prod. Commun. 2016, 11, 57–62. [Google Scholar] [PubMed]
- Asthana, S.; Agarwal, T.; Banerjee, I.; Ray, S.S. In silico screening to elucidate the therapeutic potentials of asparagamine A. Int. J. Pharm. Pharmaceut. Sci. 2014, 6, 247–253. [Google Scholar]
- Saha, D.; Sharma, A. Docking-based screening of natural product database in quest for dual site inhibitors of Trypanosoma cruzi trypanothione reductase (TcTR). Med. Chem. Res. 2015, 24, 316–333. [Google Scholar] [CrossRef]
- Sahi, S.; Tewatia, P.; Shosal, S. Leishmania donovani pteridine reductase 1: Comparative protein modeling and protein-ligand interaction studies of the leishmanicidal constituents isolated from the fruits of Piper longum. J. Mol. Model. 2012, 18, 5065–5073. [Google Scholar] [CrossRef] [PubMed]
- Herrmann, F.C.; Lenz, M.; Jose, J.; Kaiser, M.; Brun, R.; Schmidt, T.J. In silico identification and in vitro activity of novel natural inhibitors of Trypanosoma brucei glyceraldehyde-3-phosphate dehydrogenase. Molecules 2015, 20, 16154–16169. [Google Scholar] [CrossRef] [PubMed]
- Scotti, L.; Ishiki, H.; Mondonça Júnior, F.J.B.; da Silva, M.S.; Scotti, M.T. In-silico analyses of natural products on Leishmania enzyme targets. Mini Rev. Med. Chem. 2015, 15, 253–269. [Google Scholar] [CrossRef] [PubMed]
- Rakesh, N.R.; Pradeep, S. Comparative genomic studies and in-silico strategies on Leishmania braziliensis, Leishmania infantum and Leishmania major: Conserved features, putative functions and potential drug target. Int. J. Appl. Sci. Biotechnol. 2013, 1, 62–66. [Google Scholar] [CrossRef]
- Soares, M.B.P.; Silva, C.V.; Bastos, T.M.; Guimarães, E.T.; Figueira, C.P.; Smirlis, D.; Azevedo, W.F., Jr. Anti-Trypanosoma cruzi activity of nicotinamide. Acta Trop. 2012, 122, 224–229. [Google Scholar] [CrossRef] [PubMed]
- Sacconnay, L.; Angleviel, M.; Randazzo, G.M.; Queiroz, M.M.F.; Queiroz, E.F.; Wolfender, J.L.; Carrupt, P.A.; Nurisso, A. Computational studies on sirtuins from Trypansomoma cruzi: Structures, conformations and interactions with phytochemicals. PLoS Negl. Trop. Dis. 2014, 8, e2689. [Google Scholar] [CrossRef] [PubMed]
- Looker, D.L.; Berens, R.L.; Marr, J.J. Purine metabolism in Leishmania donovani amastigotes and promastigotes. Mol. Biochem. Parasitol. 1983, 9, 15–28. [Google Scholar] [CrossRef]
- Kar, R.K.; Ansari, M.Y.; Suryadevara, P.; Sahoo, B.R.; Sahoo, G.C.; Dikhit, M.R.; Das, P. Computational elucidation of structural basis for ligand binding with Leishmania donovani adenosine kinase. BioMed Res. Int. 2013, 2013, 609289. [Google Scholar] [CrossRef] [PubMed]
- Chakrabarti, R.; Rawat, P.S.; Cooke, B.M.; Coppel, R.L.; Patankar, S. Cellular effects of curcumin on Plasmodium falciparum include disruption of microtubules. PLoS ONE 2013, 8, e57302. [Google Scholar] [CrossRef] [PubMed]
- Dahlström, S.; Veiga, M.I.; Ferreira, P.; Mårtensson, A.; Kaneko, A.; Andersson, B.; Björkman, A.; Gil, J.P. Diversity of the sarco/endoplasmic reticulum Ca2+-ATPase orthologue of Plasmodium falciparum (PfATP6). Infect. Genet. Evol. 2008, 8, 340–345. [Google Scholar] [CrossRef] [PubMed]
- Jung, M.; Kim, H.; Nam, K.Y.; No, K.T. Three-dimensional structure of Plasmodium falciparum Ca2+-ATPase (PfATP6) and docking of artemisinin derivatives to PfATP6. Bioorg. Med. Chem. Lett. 2005, 15, 2994–2997. [Google Scholar] [CrossRef] [PubMed]
- Ji, H.F.; Shen, L. Interactions of curcumin with the PfATP6 model and the implications for its antimalarial mechanism. Bioorg. Med. Chem. Lett. 2009, 19, 2453–2455. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Singh, A.; Singh, A.; Pathak, L.P.; Shrivastava, N.; Tripathi, P.K.; Singh, M.P.; Singh, K. (Inhibition of P. falciparum PfATP6 by curcumin and its derivatives: A bioinformatics study. Cell. Mol. Biol. 2012, 58, 182–186. [Google Scholar] [PubMed]
- Bousejra-El Garah, F.; Stigliani, J.L.; Coslédan, F.; Meunier, B.; Robert, A. Docking studies of structurally diverse antimalarial drugs targeting PfATP6: No correlation between in silico binding affinity and in vitro antimalarial activity. ChemMedChem 2009, 4, 1469–1479. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.M.; Yowell, C.A.; Hoard, A.; Vander Jagt, T.A.; Hunsaker, L.A.; Deck, L.M.; Royer, R.E.; Piper, R.C.; Dame, J.B.; Makler, M.T.; et al. Comparative structural analysis and kinetic properties of lactate dehydrogenases from the four species of human malarial parasites. Biochemistry 2004, 43, 6219–6229. [Google Scholar] [CrossRef] [PubMed]
- Tegar, M.; Purnomo, H. Tea leaves extracted as anti-malaria based on molecular docking PLANTS. Proc. Environ. Sci. 2013, 17, 188–194. [Google Scholar] [CrossRef]
- Kalani, K.; Agarwal, J.; Alam, S.; Khan, F.; Pal, A.; Srivastava, S.K. In silico and in vivo anti-malarial studies of 18β-glycyrrhetinic acid from Glychrrhiza glabra. PLoS ONE 2013, 8, e74761. [Google Scholar] [CrossRef] [PubMed]
- Lauinger, I.L.; Vivas, L.; Perozzo, R.; Stairiker, C.; Tarun, A.; Zloh, M.; Zhang, X.; Xu, H.; Tonge, P.J.; Franzblau, S.G.; et al. Potential of lichen secondary metabolites against Plasmodium liver stage parasites with FAS-II as the potential target. J. Nat. Prod. 2013, 76, 1064–1070. [Google Scholar] [CrossRef] [PubMed]
- Tallorin, L.; Durrant, J.D.; Nguyen, Q.G.; McCammon, J.A.; Burkart, M.D. Celastrol inhibits Plasmodium falciparum enoyl-acyl carrier protein reductase. Bioorg. Med. Chem. 2014, 22, 6053–6061. [Google Scholar] [CrossRef] [PubMed]
- Wadhwa, P.; Saha, D.; Sharma, A. Combined 3D-QSAR and molecular docking study for identification of diverse natural products as potent Pf ENR inhibitors. Curr. Comput. Aided Drug Des. 2015, 11, 245–257. [Google Scholar] [CrossRef] [PubMed]
- Gupta, C.L.; Akhtar, S.; Kumar, N.; Ali, J.; Pathak, N.; Bajpai, P. In silico elucidation and inhibition studies of selected phytoligands against mitogen-activated protein kinases of protozoan parasites. Inderdiscip. Sci. Comput. Life Sci. 2016, 8, 41–52. [Google Scholar] [CrossRef] [PubMed]
- García-Sosa, A.T.; Maran, U.; Hetényi, C. Molecular property filters describing pharmacokinetics and drug binding. Curr. Med. Chem. 2012, 19, 1646–1662. [Google Scholar] [CrossRef] [PubMed]
- Tian, S.; Wang, J.; Li, Y.; Li, D.; Xu, L.; Hou, T. The application of in silico drug-likeness predictions in pharmaceutical research. Adv. Drug Deliv. Rev. 2015, 86, 2–10. [Google Scholar] [CrossRef] [PubMed]
- Hou, T.; Wang, J. Structure-ADME relationship: Still a long way to go? Expert. Opin. Drug Metab. Toxicol. 2008, 4, 759–770. [Google Scholar] [CrossRef] [PubMed]
- Valerio, L.G. In silico toxicology for the pharmaceutical sciences. Toxicol. Appl. Pharmacol. 2009, 241, 356–370. [Google Scholar] [CrossRef] [PubMed]
























{kind=link}
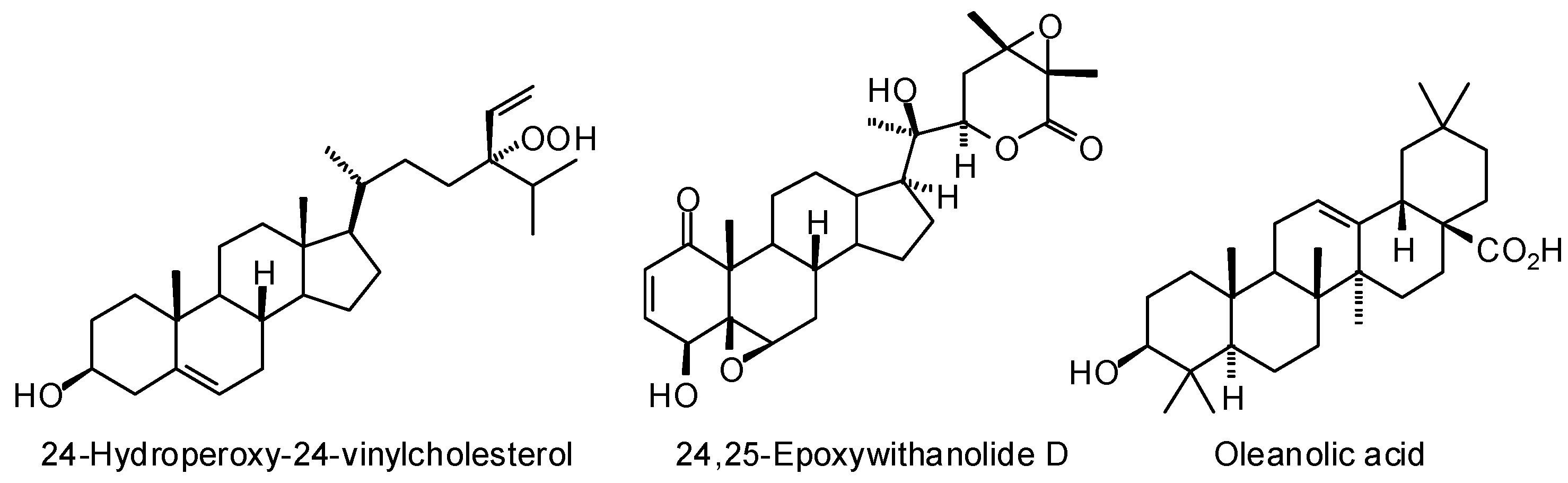{kind=link}
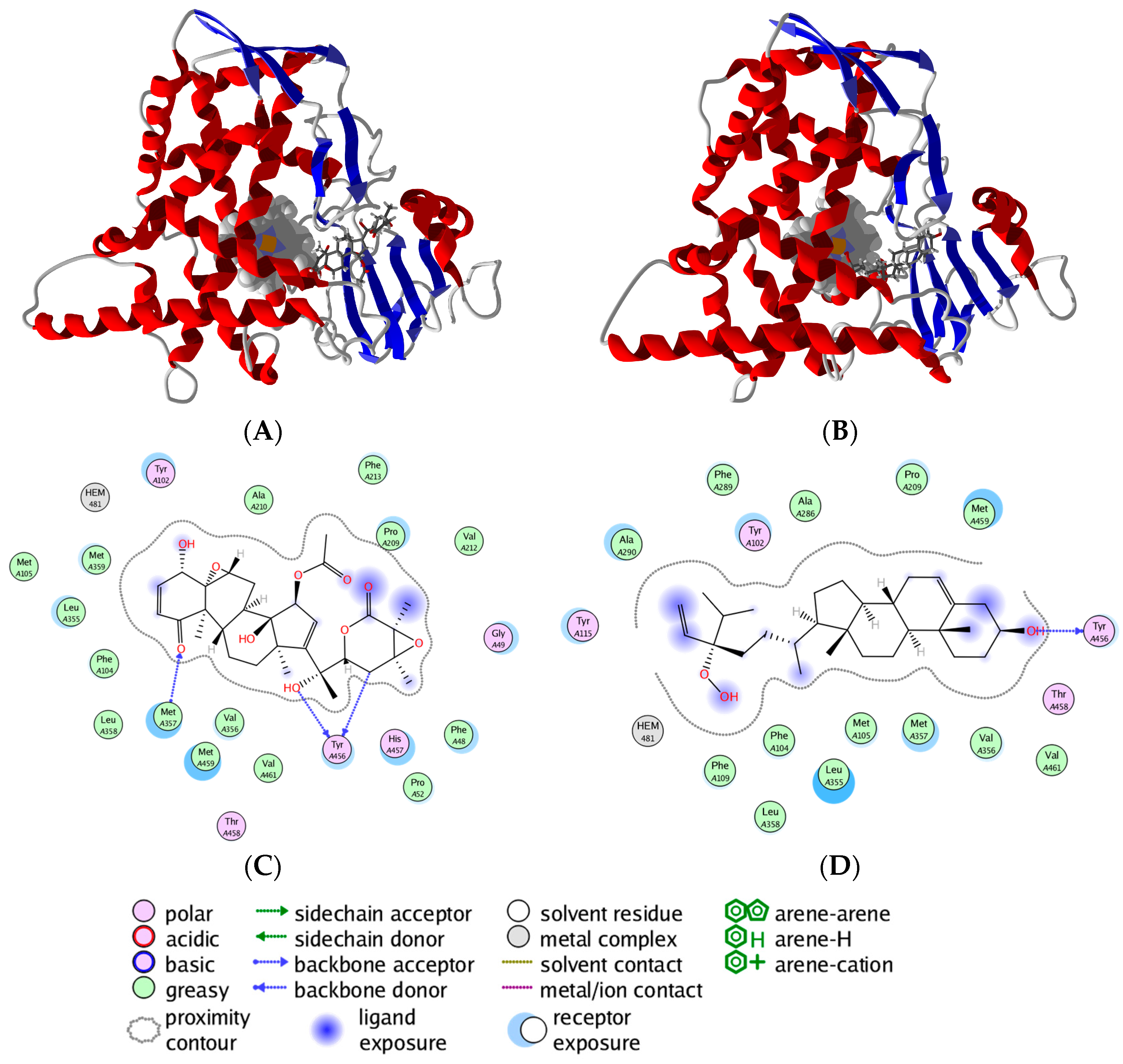{kind=link}
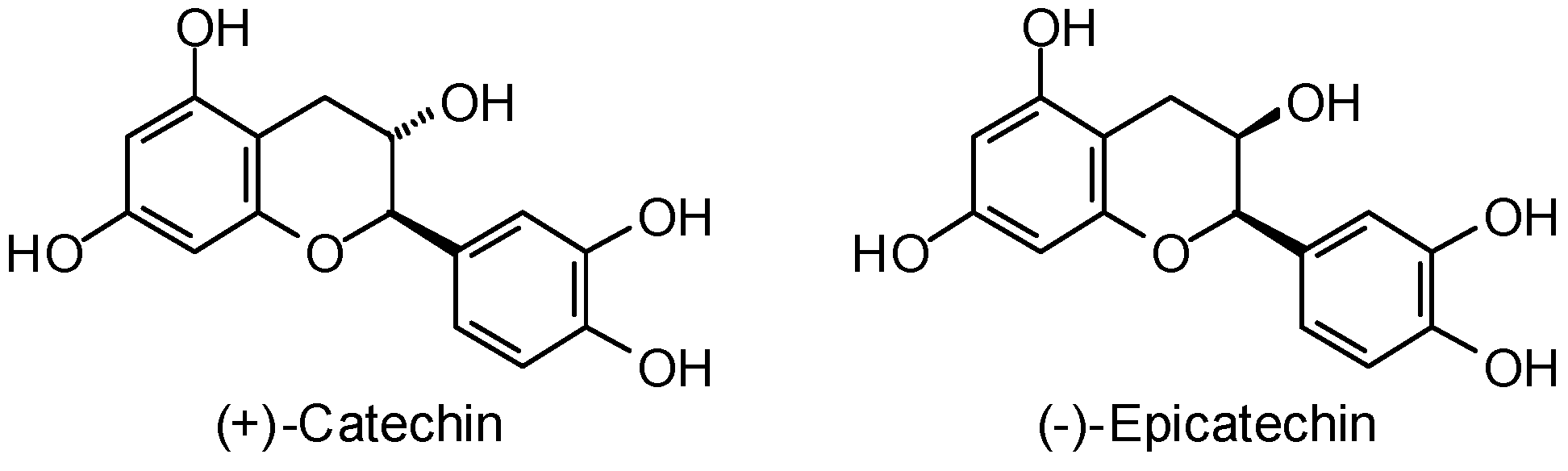{kind=link}
{kind=link}
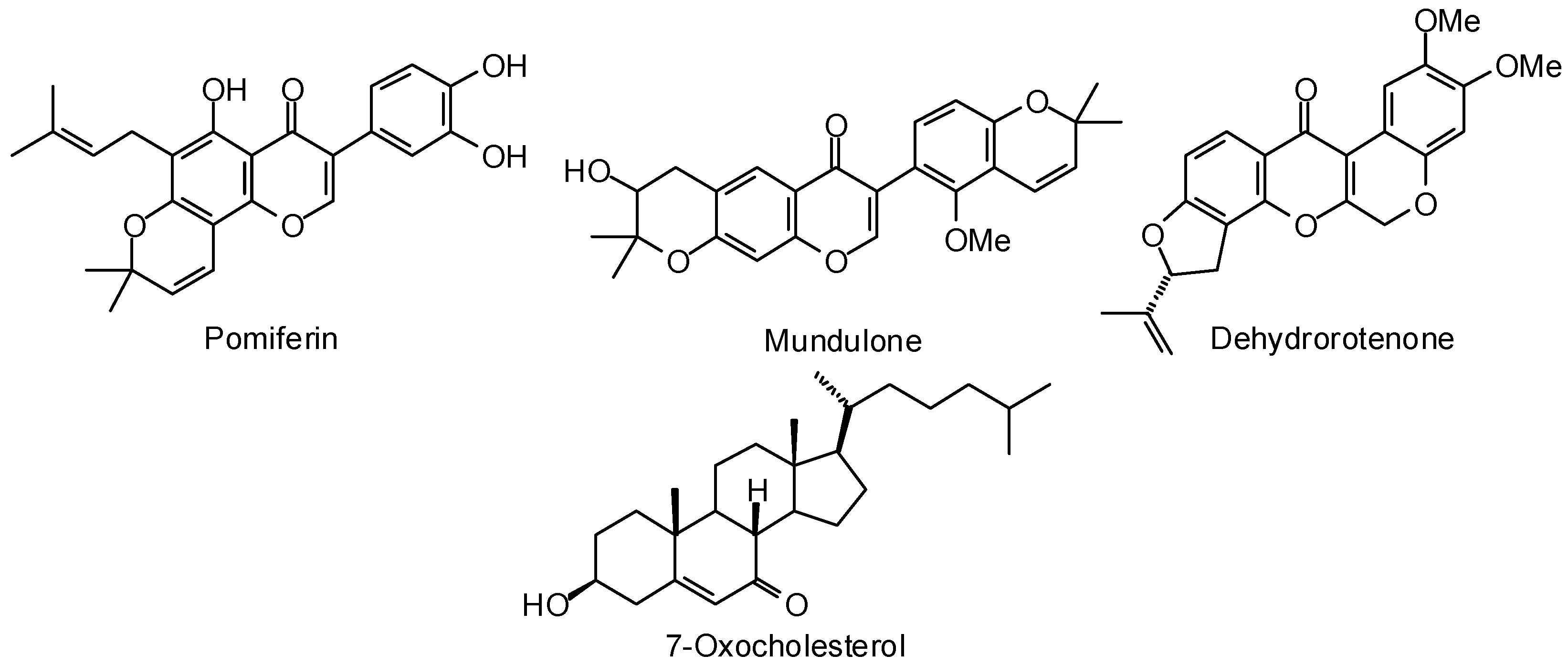{kind=link}
{kind=link}
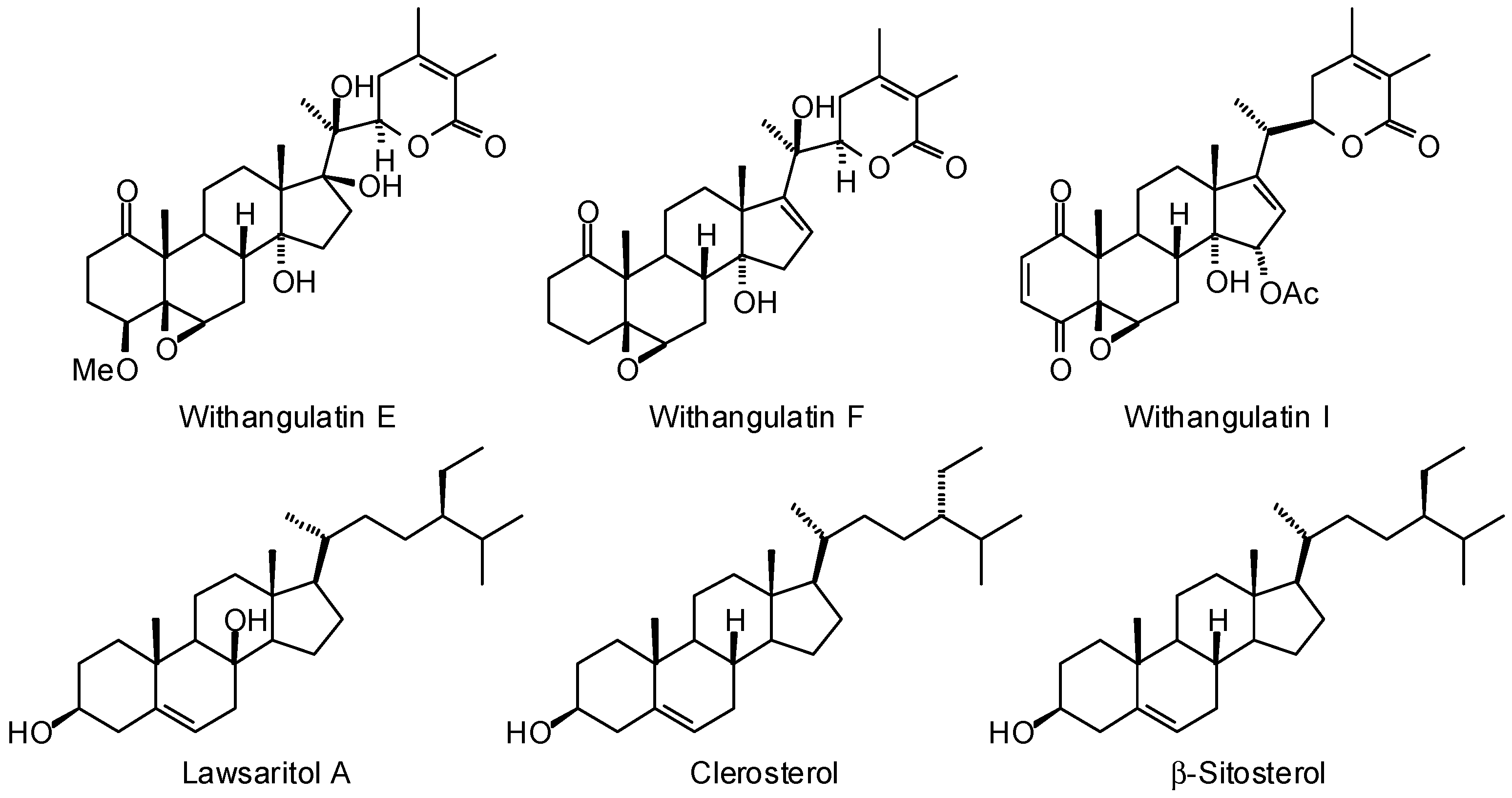{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
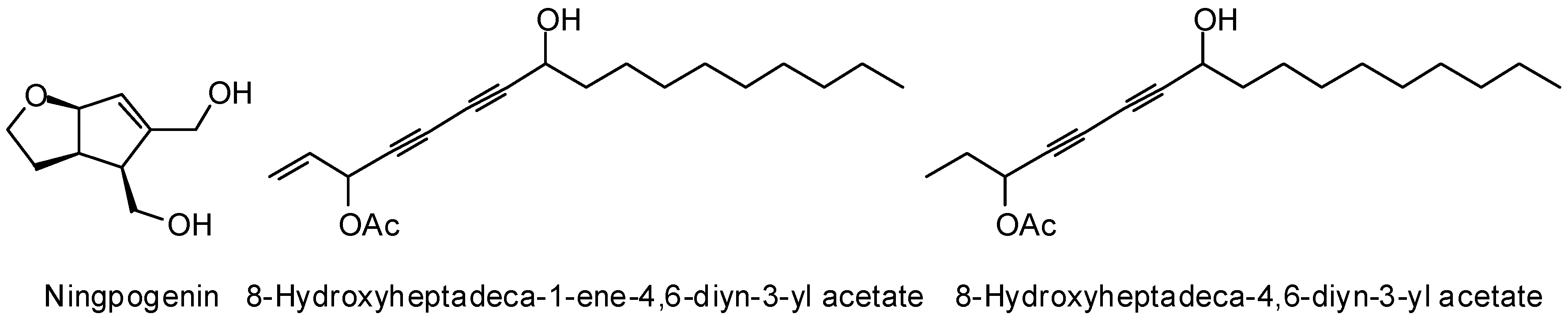{kind=link}
{kind=link}
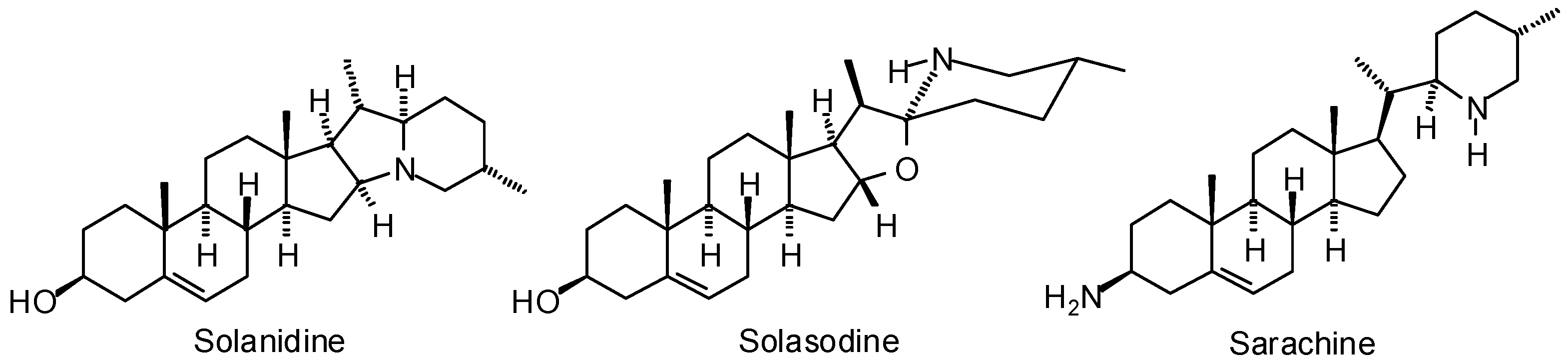{kind=link}
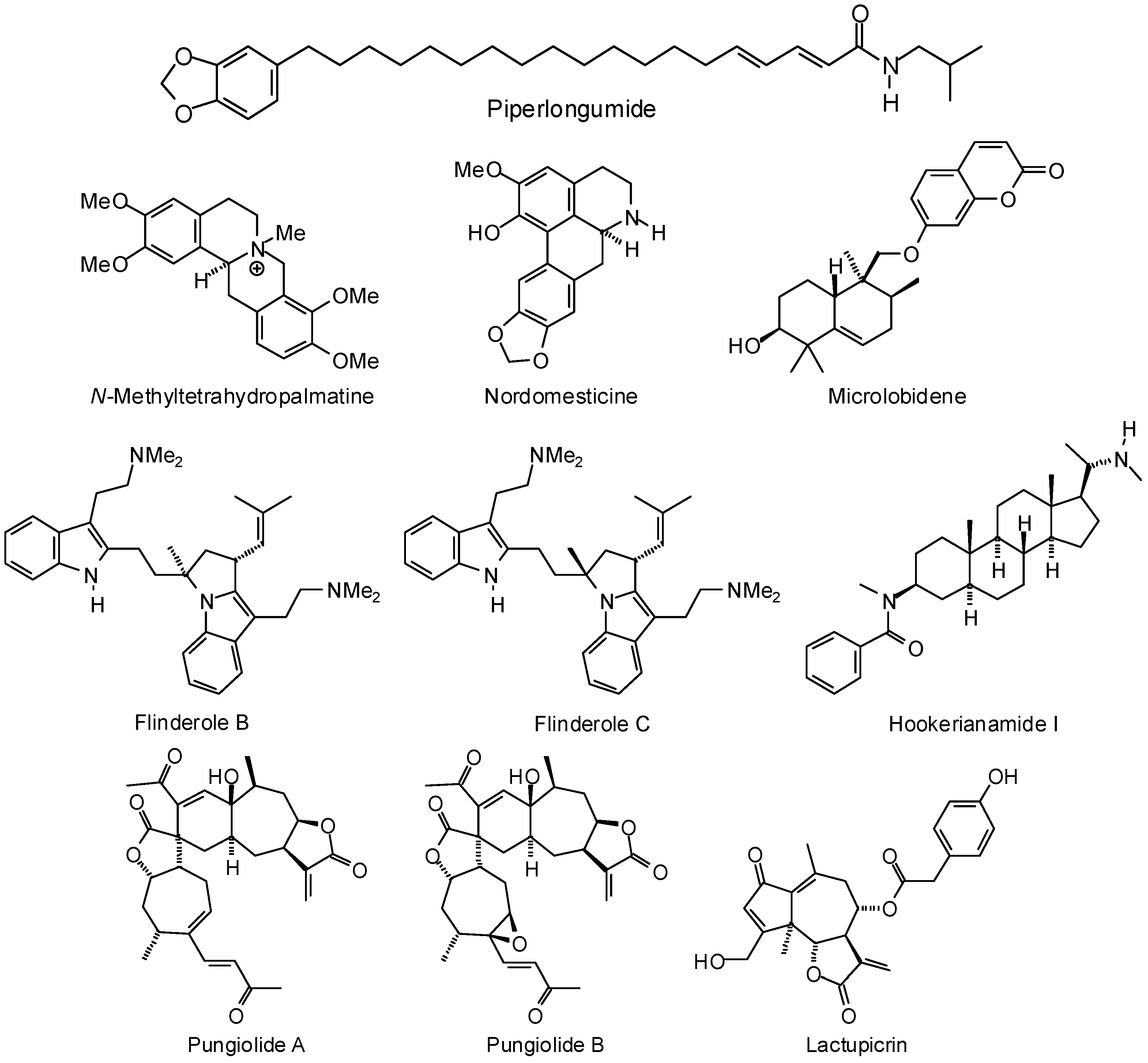{kind=link}
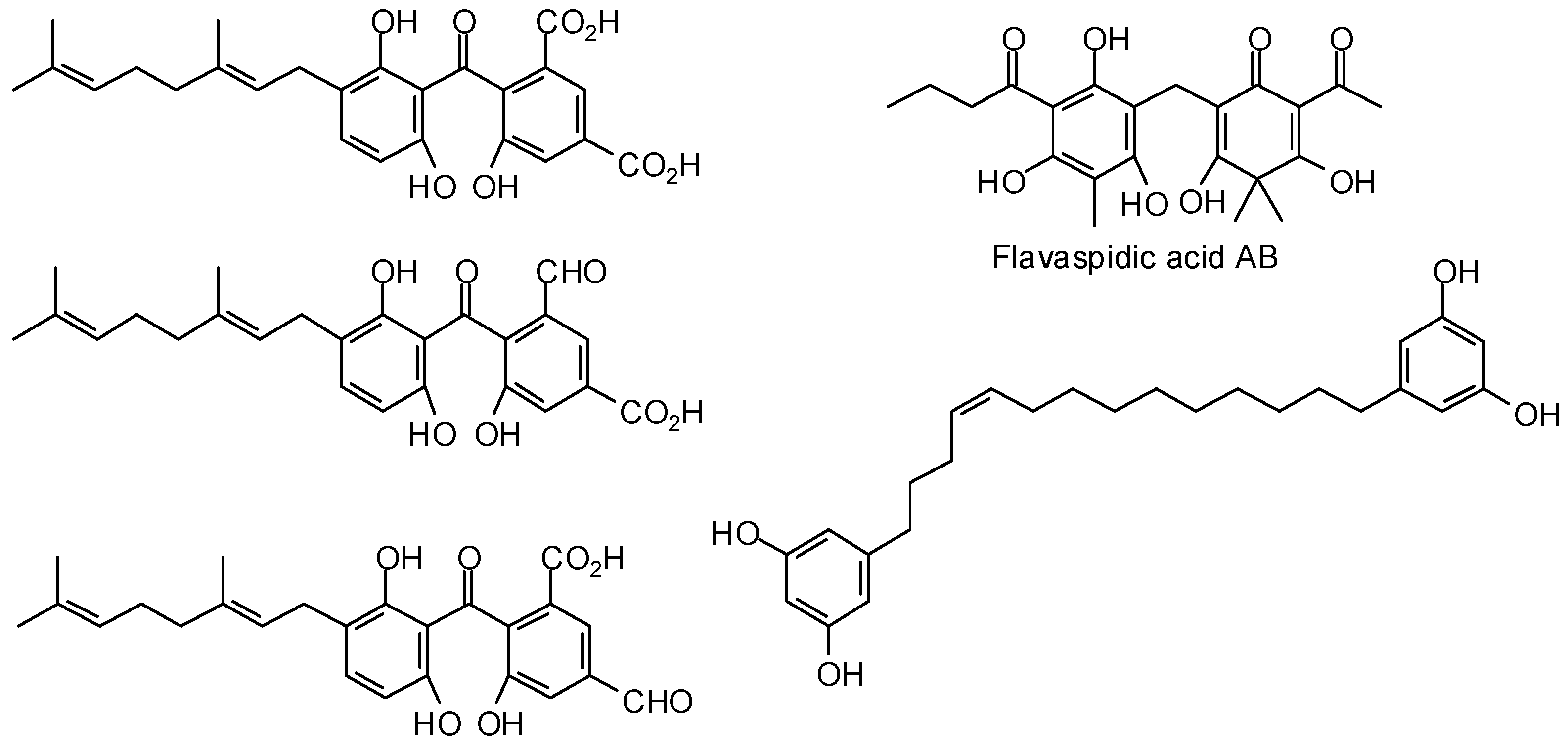{kind=link}
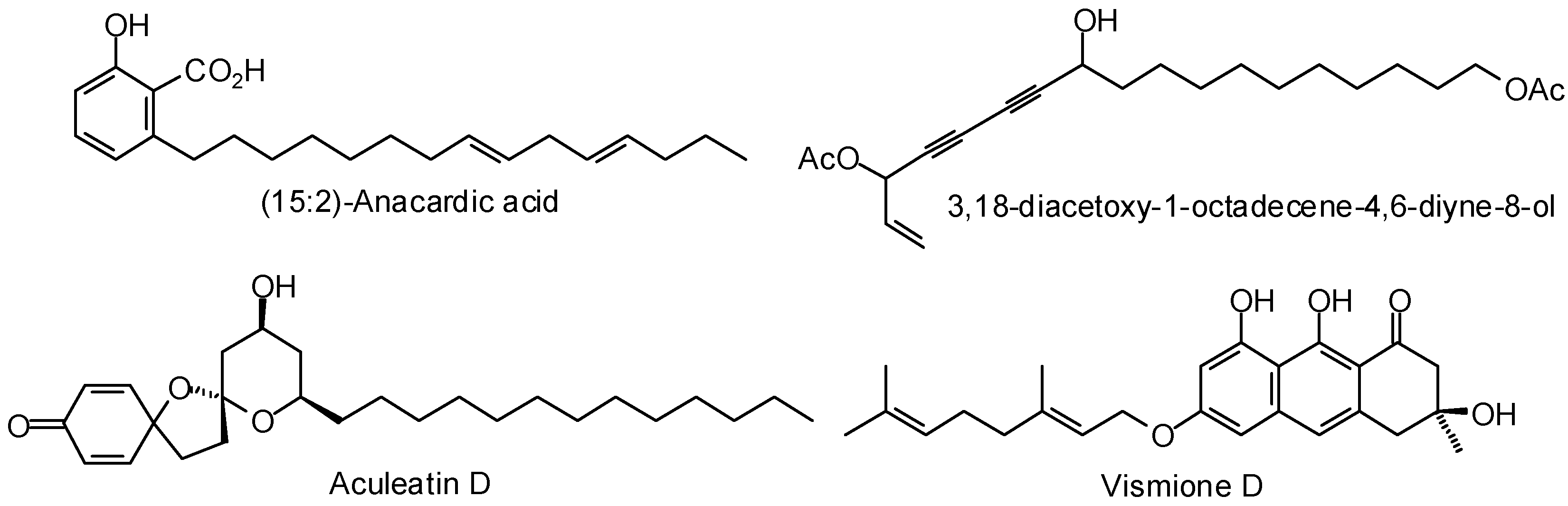{kind=link}
{kind=link}
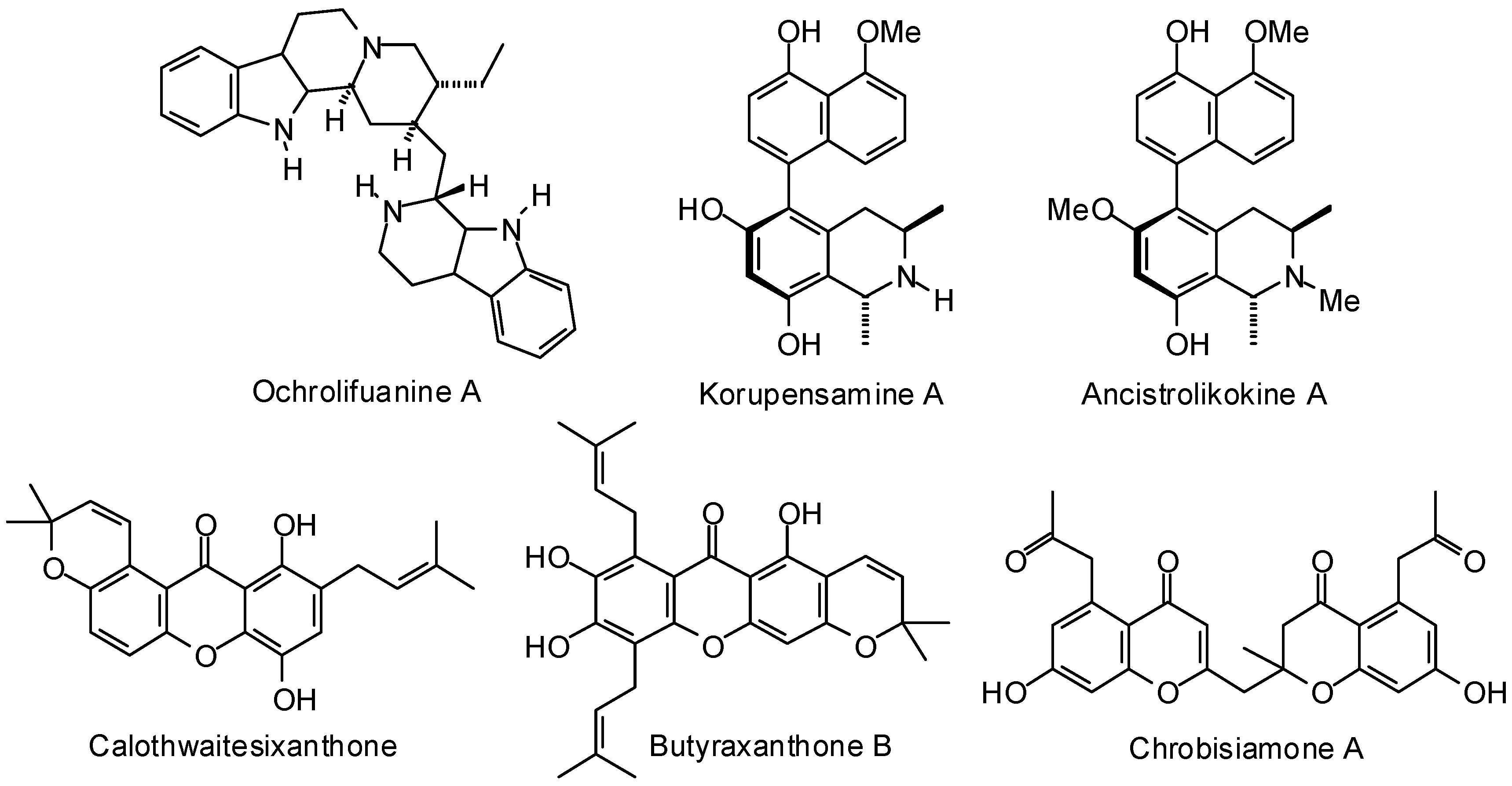{kind=link}
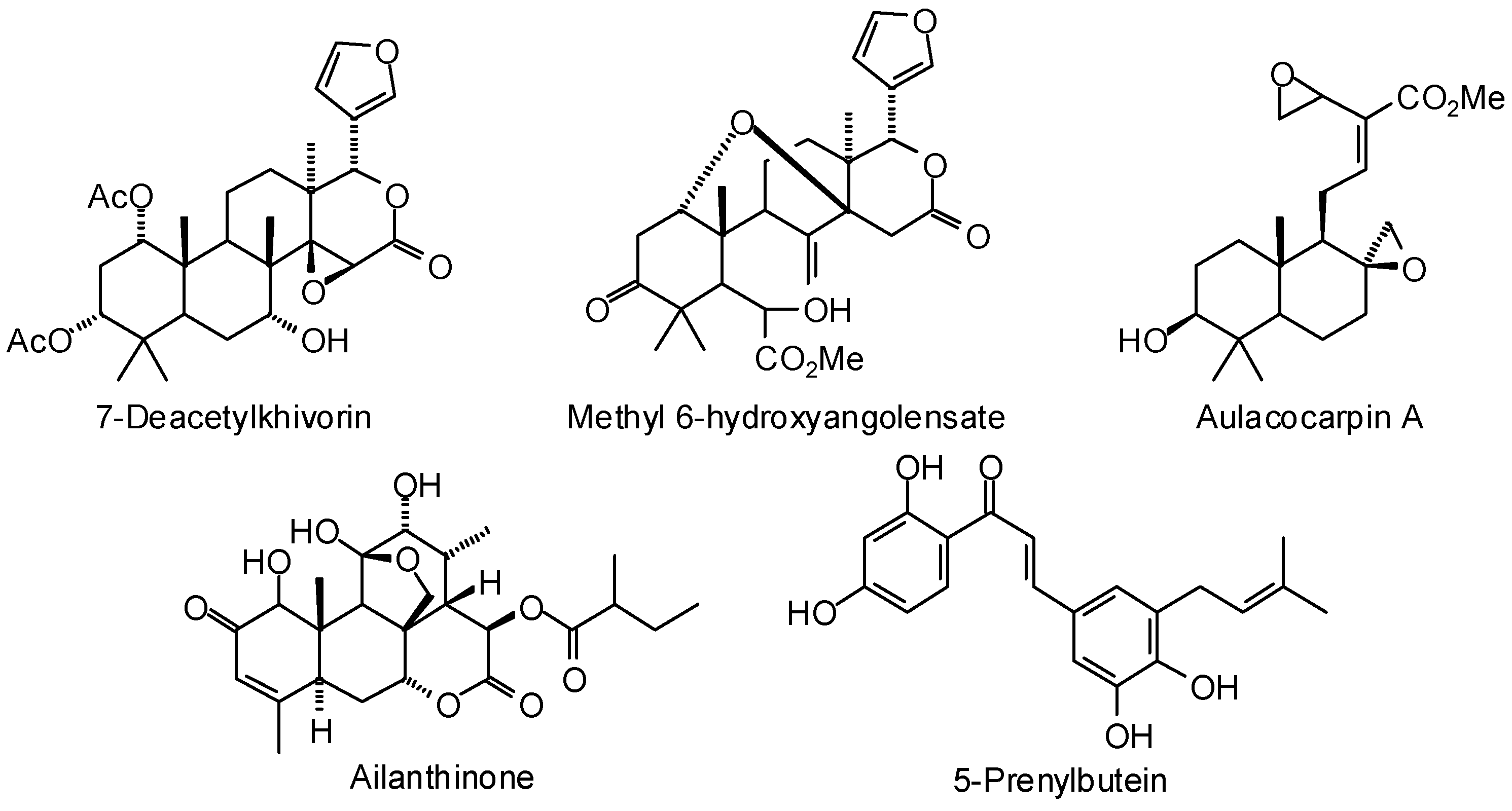{kind=link}
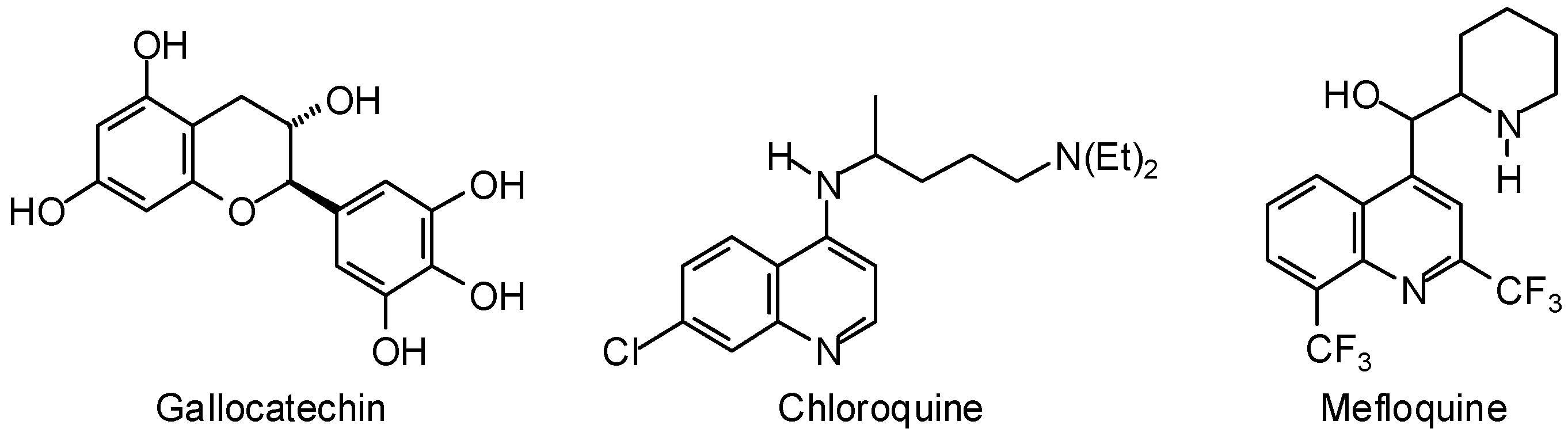{kind=link}
{kind=link}
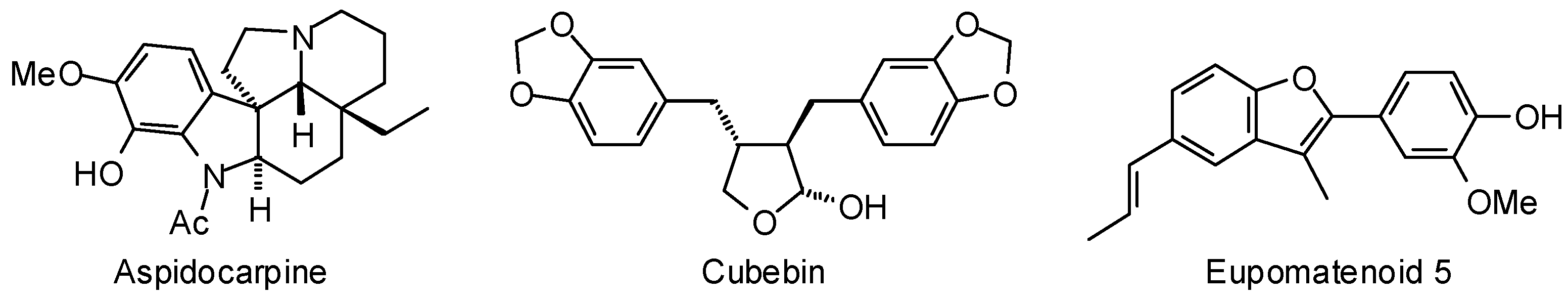{kind=link}
| Protein Target | PDB Protein Structure | ||||||
|---|---|---|---|---|---|---|---|
| L. donovani | L. infantum | L. major | L. mexicana | P. falciparum | T. brucei | T. cruzi | |
| Adenine phosphoribosyl transferase (APRT) | 1QB7, 1QB8, 1QCC, 1QCD [18] | ||||||
| Adenosine kinase (AK) | 2XTB, 3OTX [19], 4N09 [20] | ||||||
| Adenoylsuccinate synthetase (AdSS) | 1P9B [21] | ||||||
| Aminopeptidase (Apase) | 4EFD; 4FUK [22] | ||||||
| Apical membrane antigen I (AMA1) | 3SRI, 3SRJ, 3ZWZ [23] | ||||||
| Arginase (ARG) | 4ITU, 4IU0, 4IU1, 4IU4, 4IU5 [24] | 3MMR [25], 3SL0, 3SL1 [26] | |||||
| Arginine kinase (ArgK) | 2J1Q [27] | ||||||
| Aspartate aminotransferase (AspAT) | 3K7Y [28] | ||||||
| Autophagy protein 8 (Afg8) | 4EOY [29] | ||||||
| Cathepsin B (CatB) | 3HHI [30], 3MOR [31], 4HWY [32] | ||||||
| Choline kinase (CK) | 3F18 [33] | ||||||
| Cruzain | 2AIM [34], 1F29, 1F2A, 1F2B, 1F2C [35], 1ME3, 1ME4 [36], 1U9Q [37], 2OZ2 [38], 3HD3 [39], 3I06 [40], 3IUT [41], 3LXS [42], 4BKL [43], 1EWL; 1EWM; 1EWO; 1AIM [44] | ||||||
| Cyclophilin (Cyp) | 2HAQ, 3EOV [45] | 2HQJ [46] | 1QNG [47] | ||||
| Cysteine synthase (CS) | 4AIR [48] | ||||||
| Deoxyuridine triphosphate nucleotidohydrolase (dUTPase) | 2CJE, 2YAY, 2YAZ, 2YB0 [49] | 1VYQ [50], 2Y8C [51], 3T60, 3T64, 3T6Y, 3T70 [52] | 4DK2, 4DK4, 4DKB, 4DL8, 4DLC [53] | 1OGK, 1OGL [54] | |||
| Diadenosine tetraphosphatase (DATP) | 1QJC [55] | ||||||
| Dihydrofolate reductase-thymidylate synthase (HDFR-TS) | 1J3I [56]; 3DGA [57]; 3QGT [58]; 3UM8 [59]; 4DDR, 4DP3, 4DPD, 4DPH [60] | 3QFX, 3QGT, 3RG9 [58] | 2H2Q, 3CL9, 3CLB [61]; 3HBB [62]; 3KJS [63] | ||||
| Dihydroorate dehydrogenase (DHODH) | 3C61 [64] | 3MHU, 3MJY [65]; 3TQ0 [66]; 4EF8, 4EF9 [67]; 3GYE, 3GZ3 [68] | 1TV5 [69]; 3I65, 3I68; 3I6R [70]; 3O8A [71]; 3SFK [72]; 4CQ8, 4CQ9, 4CQA [73] | 2B4G [74] | 3C3N [75]; 2E6D [76]; 2E68, 2E6A, 2E6F, 2DJL, 2DJX [77]; 3W1A , 3W1L, 3W1M, 3W1N, 3W1P, 3W1Q, 3W1R, 3W1T, 3W1U, 3W1X, 3W22, 3W23, 3W2J, 3W2K, 3W2L, 3W2M, 3W2N, 3W2U [78]; 3W3O [79]; 3W6Y, 3W70, 3W71, 3W72, 3W73, 3W74, 3W75, 3W76, 3W7C, 3W7D, 3W7E, 3W7G, 3W7H, 3W7I, 3W7J, 3W7K, 3W7L, 3W7M, 3W7N, 3W7O, 3W7P, 3W7Q, 4JD4, 4JDB [80]; 3W83, 3W84, 3W85 [81]; 3W86, 3W87, 3W88 [82] | ||
| d-Tyrosyl-tRNATyr deacylase (DTD) | 3KNP, 3KNF, 3KO3, 3KO4, 3KO5, 3KO7, 3KO9, 3KOB, 3KOC [83]; 3LMT, 3LMU, 3LMV [84]; 4NBI, 4NBJ [85] | ||||||
| Enolase | 1OEP [86]; 2PTW, 2PTX, 2PTY, 2PTZ, 2PU0, 2PU1 [87] | ||||||
| Enoyl acyl-carrier-protein reductase (FabI = ENR) | 1NHD, 1NHG, 1NHW, 1NNU, 1VRW [88]; 1UH5, 1V35 [89]; 1ZSN, 1ZW1, 1ZXB, 1ZXL [90]; 2O2S, 2O2Y [91]; 2FOI, 2NQ8, 2OL4, 2OOS, 2OP0, 2OP1 [92]; 3LSY, 3LT0, 3LT1, 3LT2, 3LT4 [93]; 4IGE, 4IGF [94] | ||||||
| Falcipain 2 (FP-2) | 1YVB [95]; 2GHU [96]; 2OUL [97]; 3BPF [98]; 3PNR [99] | ||||||
| Falcipain 3 (FP-3) | 3BPM [98]; 3BWK [38] | ||||||
| Farnesyl diphosphate synthase (FPPS) | 4K10, 4JZX, 4JZB [100] | 2EWG, 2I19 [101]; 2P1C [102]; 3DYF, 3DYG, 3DYH, 3EFQ, 3EGT [103]; 2OGD [104] | 1YHL, 1YHM [105]; 3IBA, 3ICK, 3ICM, 3ICN, 3ICZ, 3ID0 [106]; 4DWB, 4DWG, 4DXJ, 4DZW, 4E1E [107] | ||||
| Ferredoxin-NADP+ reductase (FNR) | 2OK7, 2OK8 [108] | ||||||
| FK506 binding protein (FKBP35) | 4J4N [109] | ||||||
| Fructose-1,6-bisphosphate aldolase (ALDO) | 1EPX [110]; 2QAP, 2QDG, 2QDH [111] | 1A5C [112] | |||||
| Glutamate dehydrogenase 2 (GDH2) | 3R3J [113] | ||||||
| Glutathione peroxidase-like enzyme 1 (GPX1) | 3E0U [114] | ||||||
| Glutathione reductase (GR) | 1ONF [115] | ||||||
| Glutathione S-transferase (GST) | 1OKT [116]; 1PA3, 1Q4J [117]; 2AAW [118] | ||||||
| Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) | 1GYP [119]; 1A7K [120]; 1GYQ [121] | 1YWG [122]; 2B4R, 2B4T [123] | 2X0N [124]; 4P8R [125] | 1K3T [126]; 1ML3 [127]; 1QXS [128]; 3IDS [129] | |||
| Glycerol-3-phosphate dehydrogenase (GPDH) | 1EVY, 1EVZ [130];1JDJ, 1M66, 1M67, 1N1G [131]; 1N1E [132] | ||||||
| Glyoxalase I (GLO1) | 2C21 [133] | ||||||
| Glyoxalase II (GLO2) | 2P18, 2P1E [134] | ||||||
| GMP synthetase (GMPS) | 3UOW [135] | ||||||
| Guanylate kinase (GK) | 1Z6G [136] | ||||||
| Heat shock protein 90 (HSP90) | 3H80 [137], 3Q5J, 3Q5K, 3Q5L, [138]; 3U67 [139] | ||||||
| Histidyl-tRNA synthetase (HisRS) | 3HRI [140] | 3HRK, 3LC0 [140] | |||||
| Histo-aspartic protease (HAP) | 3FNS, 3FNT, 3FNU [141] | ||||||
| β-Hydroxyacyl-acyl carrier protein Dehydratase (FabZ) | 3AZ8, 3AZ9, 3AZA, 3AZB [142] | ||||||
| (E)-4-hydroxy-3-methyl-but-2-enyl-diphosphate reductase (LytB) | 4N7B [143] | ||||||
| Hypoxanthine-guanine phosphoribosyl transferase (HGPRT) | 1CJB [144] | 1TC1, 1TC2 [145]; 1P19 [146] | |||||
| Lactate dehydrogenase (LDH) | 1LDG [147]; 1CEQ, 1CET [148]; 1T24, 1T25, 1T26, 1T2C, 1T2D [149]; 1U4O, 1U4S, 1U5A, 1U5C, 1XIV [150]; 2A94 [151]; 4B7U [152] | ||||||
| Lipoamide dehydrogenase (LADH) | 2QAE [153] | ||||||
| Lysyl-tRNA synthetase (Lys-RS) | 4H02 [154] | ||||||
| M1 amino peptidase (A-M1) | 4K5L, 4K5M, 4K5N, 4K5O, 4K5P [155] | ||||||
| M17 amino peptidase (A-M17) | 4K3N [155]; 3KQX, 3KQZ, 3KR4, 3KR5 [156]; 3T8V, 3T8W [157] | ||||||
| M18 aspartyl aminopeptidase (M18AAP) | 4EME [158] | ||||||
| Macrophage infectivity potentiator (MIP) | 1JVW [159] | ||||||
| Metacaspase-2 (MCA2) | 4AF8, 4AFP, 4AFV [160] | ||||||
| Metallocarboxypeptidase 1 (MCP-1) | 3DWC [161] | ||||||
| Methionine aminopeptidase 1b (MAP1b) | 3S6B [162] | ||||||
| Methionyl-tRNA synthetase (MetRS) | 3KFL [163] | 4EG1, 4EG3, 4EG4, 4EG5, 4EG6, 4EG7, 4EG8, 4EGA [164]; 4MVW, 2MVX, 4MVY, 4MW0, 2MW1, 4MW2, 4MW4, 4MW5, 4MW6, 4MW7, 4MW9, 4MWB, 4MWC, 4MWD, 4MWE [165] | |||||
| Mitogen-activated protein kinase (MAPK) | 3PGI, 3UIB [166] | ||||||
| N5,N10-Methylenetetrahydrofolate dehydrogenase/cyclohydrolase (DHCH) | 4A26 [167] | ||||||
| Nicotinamidase (PnC1) | 3R2J [168] | ||||||
| N-Myristoyl transferase (NMT) | 2WUU [169] | 3H5Z, 2WSA [170]; 4A2Z, 4A30, 4A31, 4A32, 4A33 [171] | |||||
| Nucleoside 2-deoxyribosyltransferase (NDRT) | 2A0K, 2F2T, 2F62, 2F64, 2F67 [172] | ||||||
| Nucleoside diphosphate kinase B (NDKB) | 3NGR, 3NGS, 3NGT, 3NGU [173] | 4FKX, 4FKY [174]; 4F4A, 4F36 [175] | 3NGR, 3NGS, NGT, 3NGU, 3PRV [173] | ||||
| Nucleoside hydrolase (NH) | 1EZR [176] | ||||||
| Inosine-Adenosine-Guanosine nucleoside hydrolase (IAGNH) | 4I70, 4I71, 4I72, 4I73, 4I74, 4I75 [177] | ||||||
| Inosine-Guanosine nucleoside hydrolase (IG-NH) | 3FZ0, 4I70, 4I71, 4I72, 4I73, 4I74, 4I75 [178] | ||||||
| Nucleosome assembly protein (NapL) | 3FS3 [179]; 3GYV, 3GYW [180] | ||||||
| Old yellow enzyme (OYE) | 3ATY, 3ATZ [181]; 4E2B, 4E2D [182] | ||||||
| Oligopeptidase B (OPB) | 2XE4 [183] | 4BP8, 4BP9 [184] | |||||
| Ornithine decarboxylase (ODC) | 1QU4 [185]; 1F3T [186]; 1NJJ [187] | ||||||
| Ornithine δ-aminotransferase (OAT) | 3NTJ, 3LG0 [188] | ||||||
| Orotidine 5′-monophophate decarboxylase (OMPDC) | 3QW3 [189] | 2QAF, 2Q8Z, 3BAR [190]; 2ZCG [191]; 2ZA1, 2ZA2, 2ZA3 [192]; 3S9Y [193]; 3VI2 [194]; 2Q8L [195]; 2F84 [196]; 3MWA, 3N2M, 3N34, 3N3M [197]; | |||||
| Oxoacyl acyl-carrier-protein reductase (OAR) | 2C07 [198] | ||||||
| Peptide deformylase (PDF) | 1JYM [199]; 1RL4, 1RQC [200] | ||||||
| Peroxisomal targeting signal 1 (PTS1) | 3CV0, 3CVL, CVN, 3CVP, 3CVQ [201] | ||||||
| Peroxisomal targeting signal 2 (PTS2) | 2F2J [110] | ||||||
| Phosphethanolamine methyltransferase (PMT) | 3UJ6, 3UJ7, 3UJ8, 3UJ9, 3UJA, 3UJB [202] | ||||||
| Phosphodiesterase B1 (PDEB1) | 2R8Q [203] | 4I15 [204] | |||||
| Phosphodiesterase C (PDEC) | 3V93 [205]; 3V94 [206] | ||||||
| Phosphoenolpyruvate carboxykinase (PEPCH) | 1II2 [207] | ||||||
| Phosphofructokinase (PFK) | 3F5M [208] | ||||||
| 6-Phosphoglucolactonase (6PGL) | 2J0E [209]; 3E7F, 3EB9 [210] | ||||||
| 6-Phosphogluconate dehydrogenase (6PGDH) | 1PGJ [211] | ||||||
| Phosphoglucose isomerase (PGI) | 1Q50, 1T10 [212] | 2O2C, 2O2D [213] | |||||
| Phosphoglycerate kinase (PGK) | 3OZA, 3OZ7 [214] | 13PK [215]; 16PK [216] | |||||
| Phosphoglycerate mutase (PGAM) | 3IGY, 3IGZ [217] | 3EOZ [218] | 3NVL [219] | ||||
| Phosphomannomutase (PMM) | 2I54, 2I55 [220] | 3F9R [221] | |||||
| Plasmepsin I (PMI) | 2R9B [222]; 3QRV, 3QS1 [223]; | ||||||
| Plasmepsin II (PMII) | 1SME [224]; 1LEE, 1LF2 [225]; 1LF3, 1LF4 [226]; 2BJU [227]; 2IGX, 2IGY [228]; 3F9Q [229]; 1M43 [230]; 1ME6 [231]; 1W6H, 1W6I [232]; 1XDH, 1XE5, 1XE6 [233] | ||||||
| Plasmepsin IV (PMIV) | 1LS5 [225] | ||||||
| Proline racemase (PRACA) | 1W61, 1W62 [234] | ||||||
| Protein Kinase 5 (PK5) | 1OB3, 1V0O, 1V0P [235] | ||||||
| Protein tyrosine phosphatase 1 (PTP1) | 3M4U [236] | 4AZ1 [237] | |||||
| Pteridine reductase 1 (PTR1) | 2XOX [238] | 1E7W, 1E92 [239]; 1W0C [240]; 2BF7, 2BFA,2BFM, 2BFO, 2BFP [241]; 2QHX, 3H4V [242] | 2C7V [243]; 2WD7, 2WD8, 3GN1, 3GN2 [244]; 2VZ0 [245]; 3BMC, 3BMN, 3BMO, 3BMQ, 3JQ6, 3JQ7, 3JQ8, 3JQ9, 3JQA, 3JQB, 3JQC, 3JQD, 3JQE, 3JQF, 3JQG [246]; 2X9N, 2X9G, 2X9V, 3MCV [247]; 2YHI [248] | ||||
| Pteridine reductase 2 (PTR2) | 1MXF, 1MXH [249] | ||||||
| Purine nucleoside phosphorylase (PNP) | 1NW4, 1Q1G [250]; 2BSX, 1SQ6 [251]; 3ENZ [252] | ||||||
| Pyridoxal kinase (PdxK) | 3ZS7 [253] | ||||||
| Pyruvate kinase (PYK) | 1PKL [254]; 3E0V, 3E0W [255]; 3IS4, 3KTX [256]; 3HQN, 3HQO, 3HQP, 3HQQ [257]; 3PP7, 3QV6, 3QV7, 3QV8, 3QV9 [258]; 3SRK [259] | 3KHD [260] | 4HYV, 4HYW [261]; 4KCT, 4KCU, 4KCV, 4KCW [262] | 3PP7, 3QV6, 3QV7, 3QV8, 3QV9 [258] | |||
| Rhodesain | 2P7U [38], 2P86 [263] | ||||||
| Ribose 5-phosphate isomerase type B (RPIB) | 3K7O, 3K7S, 3K8C, 3M1P [264] | ||||||
| Ribulose 5-phosphate 3-epimerase (a1RPE) | 1TQX [265] | ||||||
| RNA Editing ligase 1 (REL1) | 1XDN [266] | ||||||
| S-Adenosylhomocysteine hydrolase (SAHH) | 3G1U [267] | 1V8B [268] | 3H9U [269] | ||||
| Seryl-tRNA synthetase (SerRS) | 3LSQ, 3LSS [270] | ||||||
| Sirtuin 2A (Sir2A) | 3U31, 3U3D [271] | ||||||
| Spermidine synthase (SpdSyn) | 2HTE [136] ; 2I7C, 2PSS, 2PT6, 2PT9 [272]; 3B7P; 2PWP [273]; 3RIE [274] | 3BWC [275] | |||||
| Sterol 14-α Demethylase (CYP51) | 3L4D [276] | 3G1Q, 3GW9 [277]; 2WV2, 2X2N [278]; 3P99 [279]; 3TIK [280]; 4BJK [281]; 4G7G, 4G3J [282]; | 2WUZ, 2WX2 [278]; 3K1O, 3KHM, 3KSW [283]; 4H6O [284]; 3ZG2, 3ZG3 [285]; 4COH [286]; 4BY0 [287] ; 4BMM [288] | ||||
| Sterol carrier protein, type 2 thiolase (SCP2-thiolase) | 3ZBG, 4B19 [289] | 4BI9 [289] | |||||
| Superoxide dismutase (SOD) | 2BPI [290] | 3ESF [291] | 2GPC [291] | ||||
| Terminal RNA uridyltransferase (TUTase) | 2B4V, 2B51, 2B56 [292]; 2IKF, 2NOM [293]; 2Q0C, 2Q0D, 2Q0E, 2Q0F, 2Q0G [294] | ||||||
| Thiamine phosphate synthase (TPS) | 2Y6Z [295] | ||||||
| Thiol-dependent reductase 1 (TDR1) | 4AGS [296] | ||||||
| Thioredoxin reductase (TrxR) | 4J56, 4J57 [297] | ||||||
| Thymidylate kinase (TMPK) | 2WWF, 2WWG, 2WWH, 2WWI [298]; 2YOF, 2YOG, 2YOH [299] | ||||||
| Transkelolase (Tk) | 1R9J [300] | ||||||
| Translationally controlled tumor protein (TCTP) | 3P3K [301] | ||||||
| trans-Sialidase (TS) | 1MS0, 1MS1, 1MS3, 1MS4, 1MS5, 1MS8, 1MS9, 1MR5 [302]; 1S0I, 1S0J, 2AH2 [303]; 3B69 [304]; 3OPZ [305] | ||||||
| Triosephosphate isomerase (TIM) | 1AMK [306]; 1IF2; [307]; 1N55 [308]; 2VXN [309]; 2Y61, 2Y62, 2Y63 [310] | 1YDV [311]; 1LYX, 1LZO [312]; 1M7O, 1M7P [313]; 1O5X [314]; 2VFI [315] | 1AG1 [316]; 3TIM [317]; 1IIG, 1IIH; 6TIM [318]; 5TIM [319]; 4TIM [320]; 1TPD, 1TRD, 2V5L [321]; 1TPE, 1TPF [322]; 1ML1 [323]; 1DKW [324]; 2J24, 2J27 [325]; 2X1U [326] | 1TCD [327]; 1CI1 [328]; 1SUX [329]; 2OMA [330]; 2V5B [331]; 3Q37 [332]; 4JEQ [333] | |||
| Trypanothione reductase (TR) | 2JK6, 2W0H [334]; 2X50 [335]; 2YAU [336]; 4ADW, 4APN [337] | 2WBA [338]; 2WOI, 2WOV, 2WOW, 2WP5, 2WP6, 2 WPC, 2WPE, 2 WPF [339]; 4NEV [340]; | 1NDA [341]; 1AOG [342]; 1BZL [343]; 1GXF [344]; 4NEW [340] | ||||
| Tryparedoxin-dependent peroxidase (TDPX) | 2VUP [345] | 4LLR [346] | |||||
| Tryptophanyl-tRNA synthetase (TrpRS) | 4J75, 4J76 [347] | 3I05 [348] | |||||
| Tyrosyl-tRNA synthase (TyrRS) | 3P0H, 3P0I, 3P0J [349] | 3VGJ [350] | |||||
| Ubiquitin and Nedd8 Hydrolase (UCHL3) | 2WDT, 2WE6 [351] | ||||||
| UDP-Galactose 4′-epimerase (UGE) | 1GY8 [352] | ||||||
| UDP-Galactopyranose mutase (UGM) | 4DSG, 4DSH [353] | ||||||
| UDP-glucose pyrophosphorylase (UGP) | 4M28, 4M2A, 2OEF, 2OEG [354] | ||||||
| UDP-N-acetylglucosamine pyrophosphorylase (UAP) | 4BQH [355] | ||||||
| UMP synthase (UMPS) | 3QW4 [189] | ||||||
| Uridine phophorylase (UP) | 3BJE [356] | ||||||
| Docking Program | Source |
|---|---|
| AutoDock | Scripps Research Institute, http://autodock.scripps.edu/ [369] |
| Molegro Virtual Docker | Molegr ApS (no longer available) [370] |
| GLIDE | Schrödinger, https://www.schrodinger.com/Glide/ [371] |
| AutoDock Vina | Scripps Research Institute, http://vina.scripps.edu/ [372] |
| Molecular Operating Environment (MOE) | Chemical Computing Group, http://www.chemcomp.com/MOE-Molecular_Operating_Environment.htm |
| CDOCKER (Discovery Studio) | Dassault Systèmes BIOVIA, http://accelrys.com/products/collaborative-science/biovia-discovery-studio/ |
| ArgusLab | http://www.arguslab.com/arguslab.com/ArgusLab.html |
| iGemDock | National Chiao Tung University, http://gemdock.life.nctu.edu.tw/dock/download.php |
| Surflex-Dock | Certara USA, Inc., https://www.certara.com/ [373] |
| GOLD | Cambridge Crystallographic Data Centre (CCDC), http://www.ccdc.cam.ac.uk/solutions/csd-discovery/components/gold/ |
| FlexX | BioSolveIT, http://www.biosolveit.de/FlexX/ |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ogungbe, I.V.; Setzer, W.N. The Potential of Secondary Metabolites from Plants as Drugs or Leads against Protozoan Neglected Diseases—Part III: In-Silico Molecular Docking Investigations. Molecules 2016, 21, 1389. https://doi.org/10.3390/molecules21101389
Ogungbe IV, Setzer WN. The Potential of Secondary Metabolites from Plants as Drugs or Leads against Protozoan Neglected Diseases—Part III: In-Silico Molecular Docking Investigations. Molecules. 2016; 21(10):1389. https://doi.org/10.3390/molecules21101389
Chicago/Turabian StyleOgungbe, Ifedayo Victor, and William N. Setzer. 2016. "The Potential of Secondary Metabolites from Plants as Drugs or Leads against Protozoan Neglected Diseases—Part III: In-Silico Molecular Docking Investigations" Molecules 21, no. 10: 1389. https://doi.org/10.3390/molecules21101389
APA StyleOgungbe, I. V., & Setzer, W. N. (2016). The Potential of Secondary Metabolites from Plants as Drugs or Leads against Protozoan Neglected Diseases—Part III: In-Silico Molecular Docking Investigations. Molecules, 21(10), 1389. https://doi.org/10.3390/molecules21101389