
Evaluation and Comparison of the Inhibition Effect of Astragaloside IV and Aglycone Cycloastragenol on Various UDP-Glucuronosyltransferase (UGT) Isoforms


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
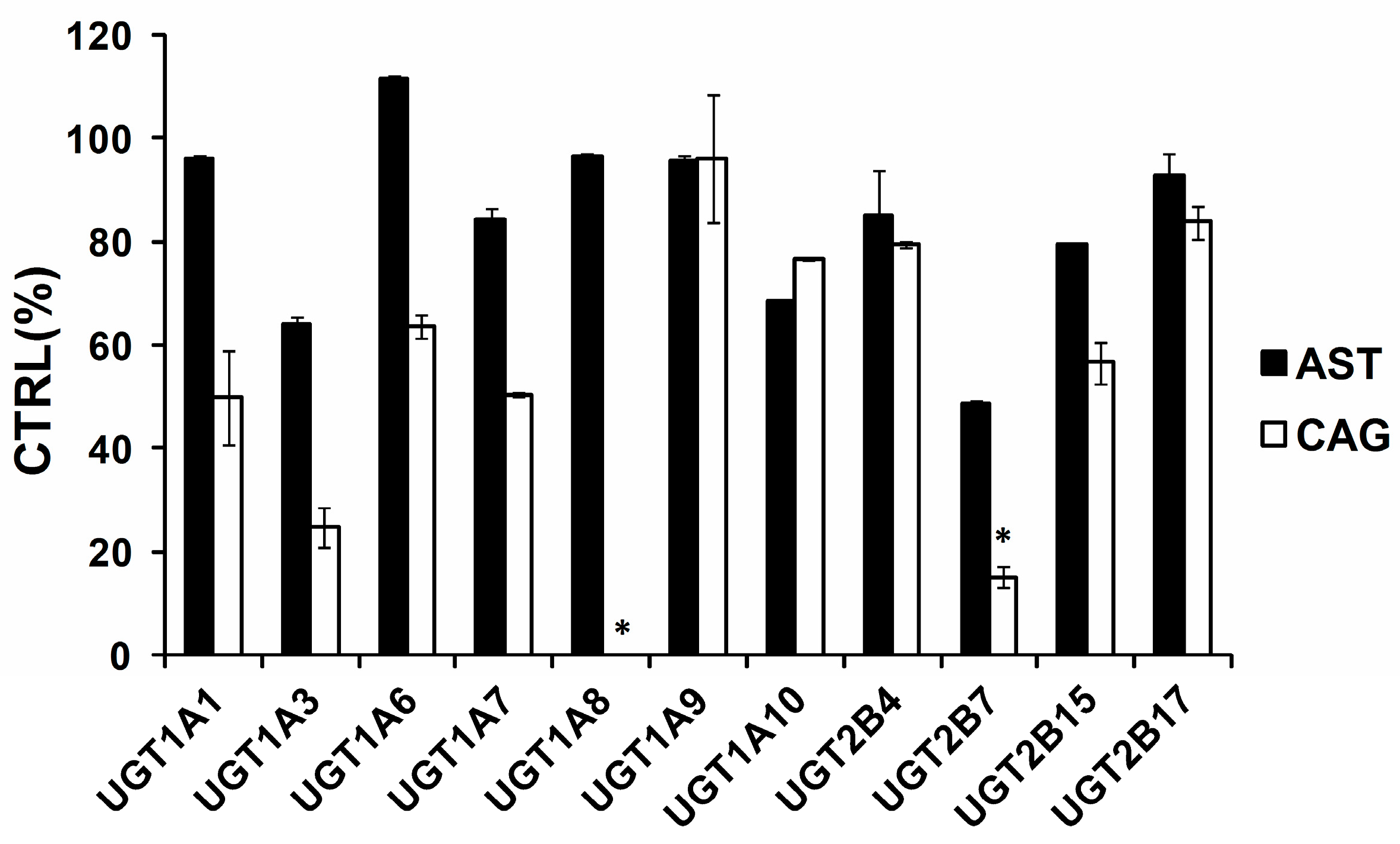
2.1. Comparison of Inhibition Effect of AST and CAG
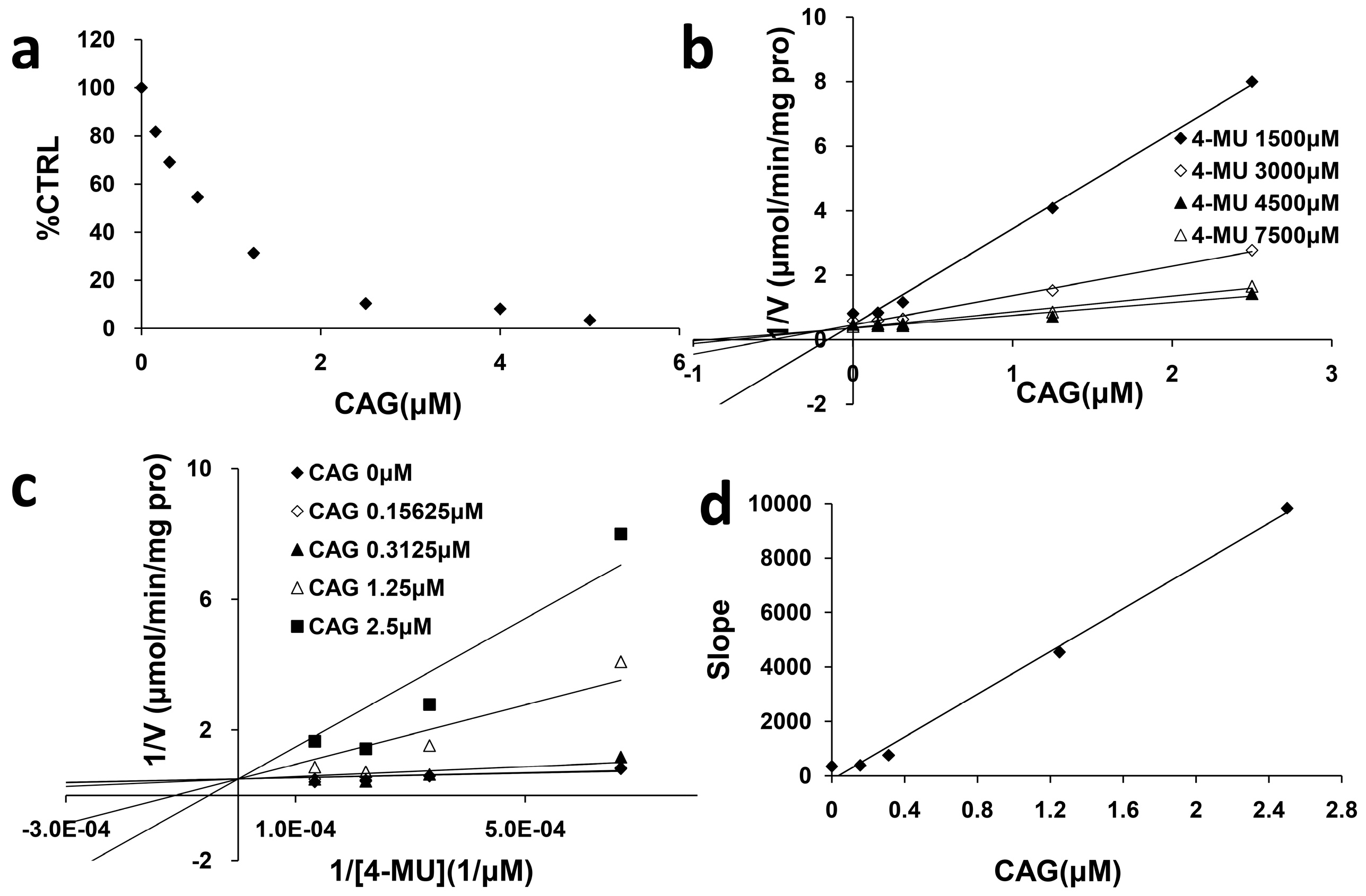
2.2. Inhibition Type and Kinetics of CAG Towards UGT1A8 and UGT2B7
3. Discussion
4. Materials and Methods
4.1. Materials and Reagents
4.2. Inhibition of UGT Activity Assay
4.3. Analytical Methods
4.4. Data Analysis
4.5. Inhibition Kinetics
4.6. In Vitro–In Vivo Extrapolation (IVIVE)
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Zhou, R.N.; Song, Y.L.; Ruan, J.Q.; Wang, Y.T.; Yan, R. Pharmacokinetic evidence on the contribution of intestinal bacterial conversion to beneficial effects of astragaloside IV, a marker compound of astragali radix, in traditional oral use of the herb. Drug. Metab. Pharmacokinet. 2012, 27, 586–597. [Google Scholar] [CrossRef] [PubMed]
- Fu, J.; Wang, Z.H.; Huang, L.F.; Zheng, S.H.; Wang, D.M.; Chen, S.L.; Zhang, H.T.; Yang, S.H. Review of the botanical characteristics, phytochemistry, and pharmacology of astragalus membranaceus (Huangqi). Phytother. Res. 2014, 28, 1275–1283. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.Y.; Ouyang, H.T.; Yang, J.Y.; Huang, X.L.; Yang, T.; Duan, J.P.; Cheng, J.P.; Chen, Y.X.; Yang, Y.J.; Qiong, P. Subchronic toxicity studies of Radix Astragali extract in rats and dogs. J. Ethnopharmacol. 2007, 110, 352–355. [Google Scholar] [CrossRef] [PubMed]
- Lu, S.; Chen, K.J.; Yang, Q.Y.; Sun, H.R. Progress in the research of Radix Astragali in treating chronic heart failure: Effective ingredients, dose-effect relationship and adverse reaction. Chin. J. Integr. Med. 2011, 17, 473–477. [Google Scholar] [CrossRef] [PubMed]
- Jin, Y.; Guo, X.J.; Yuan, B.; Yu, W.; Suo, H.; Li, Z.; Xu, H. Disposition of Astragaloside IV via enterohepatic circulation is affected by the activity of the intestinal microbiome. J. Agric. Food. Chem. 2015, 63, 6084–6093. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Zhang, Q.; Chen, G.G.; Wei, P.; Tu, C.Y. Pharmacokinetics of Astragaloside IV in rats by liquid chromatography coupled with tandem mass spectrometry. Eur. J. Drug Metab. Pharmacokinet. 2005, 30, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.C.; Wang, G.J.; Pan, G.Y.; Fawcett, J.P.; A, J.; Sun, J.G. Transport and bioavailability studies of astragaloside IV, an active ingredient in radix astragali. Basic Clin. Pharmacol. Toxicol. 2004, 95, 295–298. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Zhu, L.L.; Chen, G.G.; Du, Y. Pharmacokinetics of astragaloside IV in beagle dogs. Eur. J. Drug. Metab. Pharm. 2007, 32, 75–79. [Google Scholar] [CrossRef]
- Cheng, X.D.; Wei, M.G. Profiling the metabolism of astragaloside IV by ultra performance liquid chromatography coupled with quadrupole/time-of-flight mass spectrometry. Molecules 2014, 19, 18881–18896. [Google Scholar] [CrossRef] [PubMed]
- Shan, W.Y.; Zhang, Y.F.; Zhu, J.Q.; Shao, Q.; Fan, X.H. Inhibitory effects of astragaloside IV on cytochrome P450 enzyme of rat liver microsomes. Chin. J. Chin. Mater. Med. 2012, 37, 85–88. [Google Scholar]
- Zhang, Y.H.; Zhang, Y.J.; Guo, Y.L.; Li, W.J.; Yu, C. Astragaloside IV inhibited the activity of CYP1A2 in liver microsomes and influenced theophylline pharmacokinetics in rats. J. Pharm. Pharmacol. 2013, 65, 149–155. [Google Scholar] [CrossRef] [PubMed]
- Bourcier, K.; Hyland, R.; Kempshall, S.; Jones, R.; Maximilien, J.; Irvine, N.; Jones, B. Investigation into UDP-Glucuronosyltransferase (UGT) enzyme kinetics of imidazole- and triazole-containing antifungal drugs in human Liver microsomes and recombinant UGT enzymes. Drug. Metab. Dispos. 2010, 38, 923–929. [Google Scholar] [CrossRef] [PubMed]
- Chang, J.H.; Plise, E.; Cheong, J.; Ho, Q.; Lin, M. Evaluating the in vitro inhibition of UGT1A1, OATP1B1, OATP1B3, MRP2 and BSEP in predicting drug-induced hyperbilirubinemia. Mol. Pharm. 2013, 10, 3067–3075. [Google Scholar] [CrossRef] [PubMed]
- Peer, C.J.; Sissung, T.M.; Kim, A.; Jain, L.; Woo, S.; Gardner, E.R.; Kirkland, C.T.; Troutman, S.M.; English, B.C.; Richardson, E.D.; et al. Sorafenib is an inhibitor of UGT1A1 but is metabolized by UGT1A9: Implications of genetic variants on pharmacokinetics and hyperbilirubinemia. Clin. Cancer Res. 2012, 18, 2099–2107. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Q.; Cao, Y.F.; Ran, R.X.; Li, R.S.; Wu, X.; Dong, P.P.; Zhang, Y.Y.; Hu, C.M.; Wang, W.M. Strong specific inhibition of UDP-glucuronosyltransferase 2B7 by atractylenolide I and III. Phytother. Res. 2016, 30, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Wu, J.; Liu, D.; Liu, M.; Zhang, H.; Huang, S.; Xiong, Y.; Xia, C. In vitro inhibition of UGT1A3, UGT1A4 by ursolic and oleanolic acid and drug–drug interaction risk prediction. Xenobiotica 2016, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.; Zheng, Y.F.; Min, J.S.; Park, J.B.; Bae, S.H.; Yoon, K.D.; Chin, Y.W.; Oh, E.; Bae, S.K. In vitro, Stereoselective Inhibition of Ginsenosides toward UDP-glucuronosyltransferase (UGT) Isoforms. Toxicol. Lett. 2016, 259, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Chen, D.W.; W, X.; Zhao, Z.; Fu, Z.W.; Huang, C.T.; Ye, L.X.; Du, Z.; Yu, Y.; Fang, Z.Z.; Sun, H.Z. The Inhibition of UDP-Glucuronosyltransferase (UGT) Isoforms by Praeruptorin A and B. Phytother. Res. 2016, 30, 1872–1878. [Google Scholar] [CrossRef] [PubMed]
- Guo, B.; Fan, X.R.; Fang, Z.Z.; Cao, Y.F.; Hu, C.M.; Yang, J.L.; Zhang, Y.Y.; He, R.R.; Zhu, X.; Yu, Z.W.; et al. Deglycosylation of liquiritin strongly enhances its inhibitory potential towards UDP-Glucuronosyltransferase (UGT) isoforms. Phytother. Res. 2013, 27, 1232–1236. [Google Scholar] [CrossRef] [PubMed]
- Cao, X.; Li, B.; Li, S.; Wang, J.; Liu, F.; Li, W.P. Deglycosylation of glucoaurantio-obtusin affects its inhibition capability towards drug metabolizing enzymes (DMEs). Lat. Am. J. Pharm. 2013, 32, 1249–1251. [Google Scholar]
- Liu, Y.; Zhang, J.W.; Li, W.; Ma, H.; Sun, J.; Deng, M.C.; Yang, L. Ginsenoside metabolites, rather than naturally occurring ginsenosides, lead to inhibition of human cytochrome P450 enzymes. Toxicol. Sci. 2006, 91, 356–364. [Google Scholar] [CrossRef] [PubMed]
- Cao, Y.F.; He, R.R.; Cao, J.; Chen, J.X.; Huang, T.; Liu, Y. Drug-Drug Interactions Potential of Icariin and Its Intestinal Metabolites via Inhibition of Intestinal UDP-Glucuronosyltransferases. Evid. Based Complement. Alternat. Med. 2012, 2012, 395912. [Google Scholar] [CrossRef] [PubMed]
- Wang, X. Structure, mechanism and engineering of plant natural product glycosyltransferases. FEBS Lett. 2009, 583, 3303–3309. [Google Scholar] [CrossRef] [PubMed]
- Back, D.J.; Rogers, S.M. Review: First-pass metabolism by the gastrointestinal mucosa. Aliment. Pharm. Ther. 1987, 1, 339–357. [Google Scholar] [CrossRef]
- Cheng, Z.; Radominska-Pandya, A.; Tephly, T.R. Studies on the substrate specificity of human intestinal UDP-glucuronosyltransferases 1A8 and 1A10. Drug. Metab. Dispos. 1999, 27, 1165–1170. [Google Scholar] [PubMed]
- Oda, S.; Fukami, T.; Yokoi, T.; Nakajima, M. A comprehensive review of UDP-glucuronosyltransferase and esterases for drug development. Drug. Metab. Pharmacokinet. 2015, 30, 30–51. [Google Scholar] [CrossRef] [PubMed]
- Rowland, A.; Miners, J.O.; Mackenzie, P.I. The UDP-glucuronosyltransferases: Their role in drug metabolism and detoxification. Int. J. Biochem. Cell. B 2013, 45, 1121–1132. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.Y.; Qiu, Z.X.; Zhang, Q.Y.; Chen, J.Y.; Chen, X.J. Inhibitory Effects of Calf Thymus DNA on Metabolism Activity of CYP450 Enzyme in Human Liver Microsomes. Drug. Metab. Pharmacokinet. 2014, 29, 475–481. [Google Scholar] [CrossRef] [PubMed]
- Williams, J.A.; Hyland, R.; Jones, B.C.; Smith, D.A.; Hurst, S.; Goosen, T.C.; Peterkin, V.; Koup, J.R.; Ball, S.E. Drug-drug interactions for UDP-glucuronosyltransferase substrates: A pharmacokinetic explanation for typically observed low exposure (AUCi/AUC) ratios. Drug. Metab. Dispos. 2004, 32, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Not available.




© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ran, R.; Zhang, C.; Li, R.; Chen, B.; Zhang, W.; Zhao, Z.; Fu, Z.; Du, Z.; Du, X.; Yang, X.; et al. Evaluation and Comparison of the Inhibition Effect of Astragaloside IV and Aglycone Cycloastragenol on Various UDP-Glucuronosyltransferase (UGT) Isoforms. Molecules 2016, 21, 1616. https://doi.org/10.3390/molecules21121616
Ran R, Zhang C, Li R, Chen B, Zhang W, Zhao Z, Fu Z, Du Z, Du X, Yang X, et al. Evaluation and Comparison of the Inhibition Effect of Astragaloside IV and Aglycone Cycloastragenol on Various UDP-Glucuronosyltransferase (UGT) Isoforms. Molecules. 2016; 21(12):1616. https://doi.org/10.3390/molecules21121616
Chicago/Turabian StyleRan, Ruixue, Chunze Zhang, Rongshan Li, Bowei Chen, Weihua Zhang, Zhenying Zhao, Zhiwei Fu, Zuo Du, Xiaolang Du, Xiaolong Yang, and et al. 2016. "Evaluation and Comparison of the Inhibition Effect of Astragaloside IV and Aglycone Cycloastragenol on Various UDP-Glucuronosyltransferase (UGT) Isoforms" Molecules 21, no. 12: 1616. https://doi.org/10.3390/molecules21121616
APA StyleRan, R., Zhang, C., Li, R., Chen, B., Zhang, W., Zhao, Z., Fu, Z., Du, Z., Du, X., Yang, X., & Fang, Z. (2016). Evaluation and Comparison of the Inhibition Effect of Astragaloside IV and Aglycone Cycloastragenol on Various UDP-Glucuronosyltransferase (UGT) Isoforms. Molecules, 21(12), 1616. https://doi.org/10.3390/molecules21121616