
Clematichinenoside (AR) Attenuates Hypoxia/Reoxygenation-Induced H9c2 Cardiomyocyte Apoptosis via a Mitochondria-Mediated Signaling Pathway

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
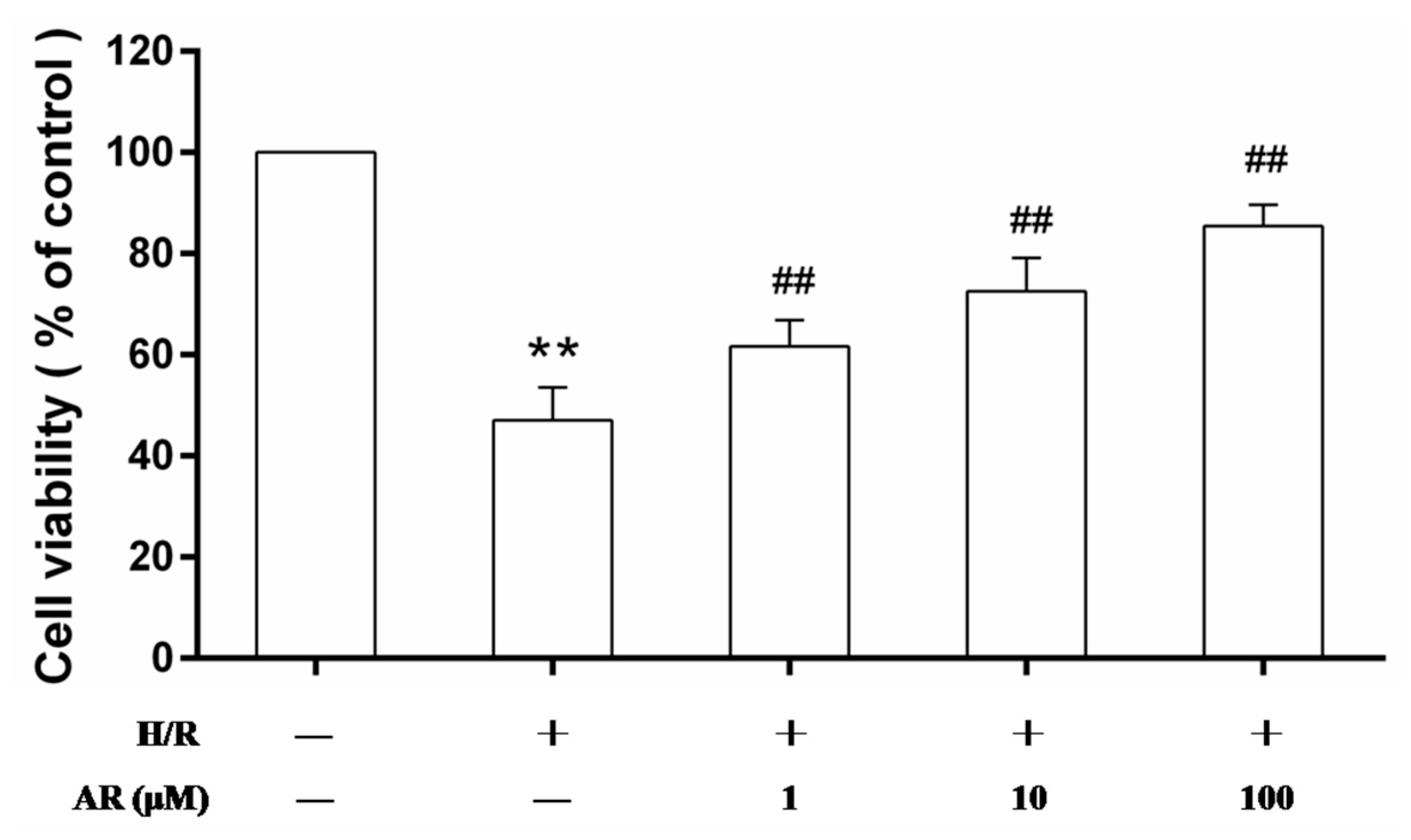
2.1. AR Pretreatment Increases Cell Viability
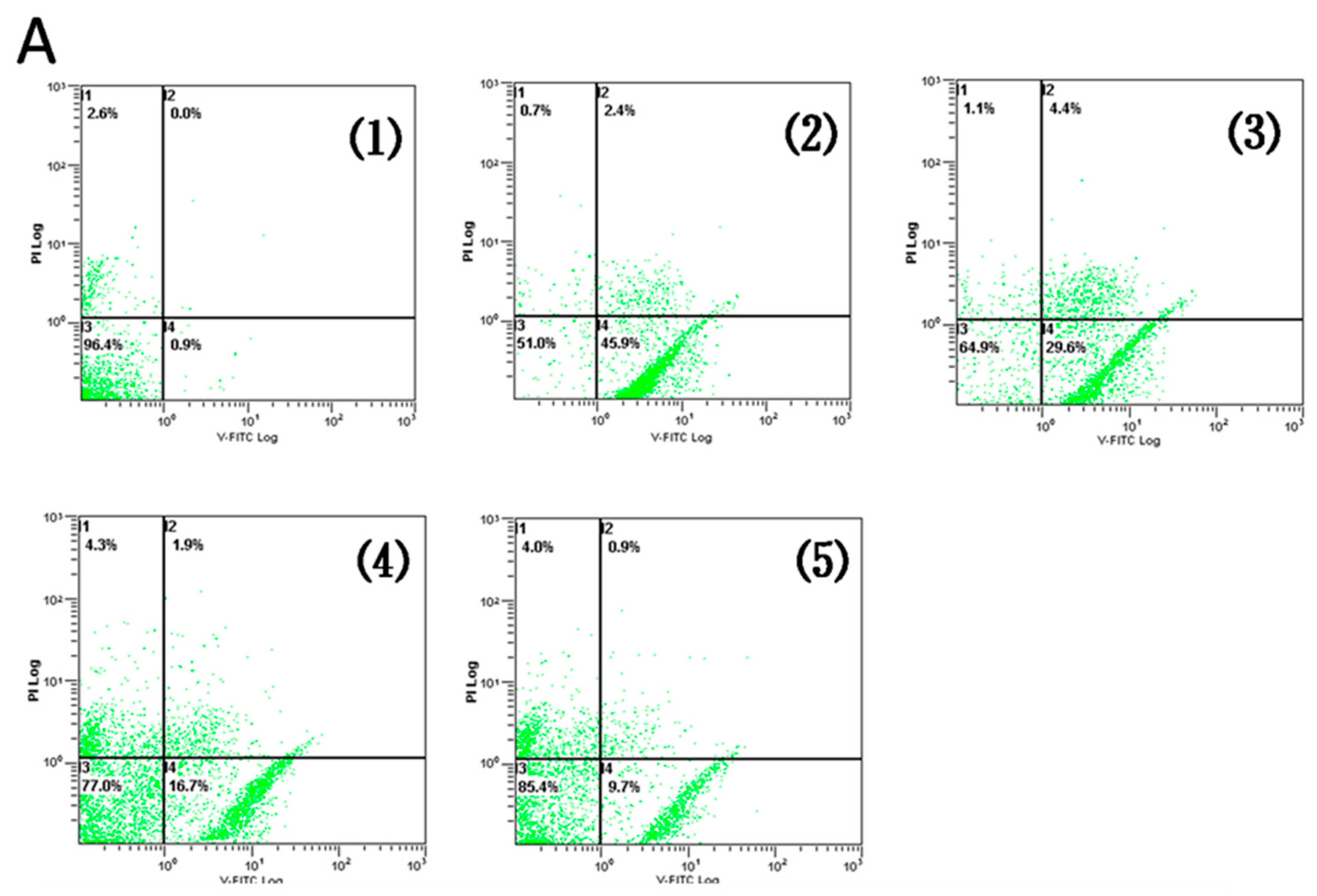
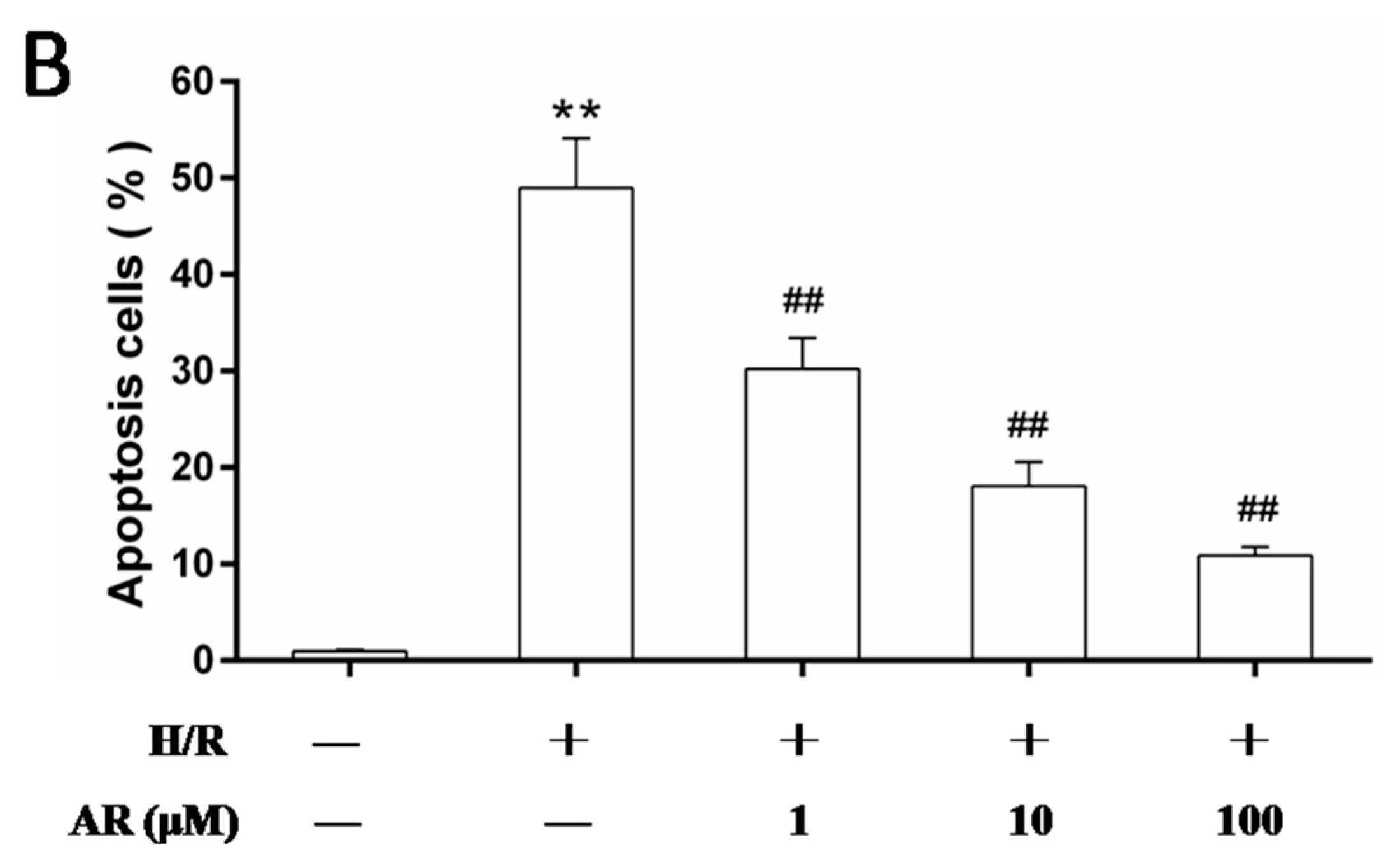
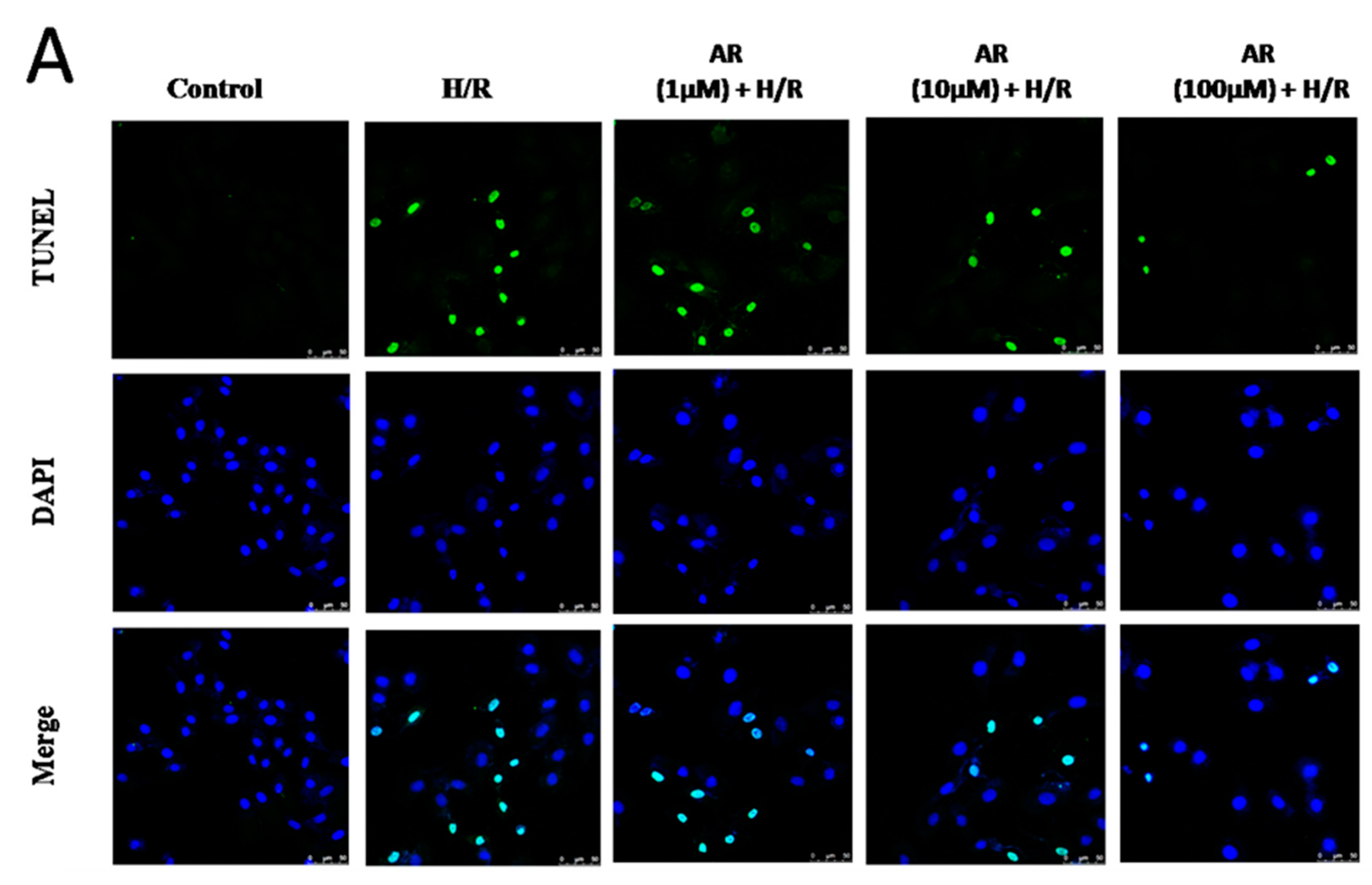
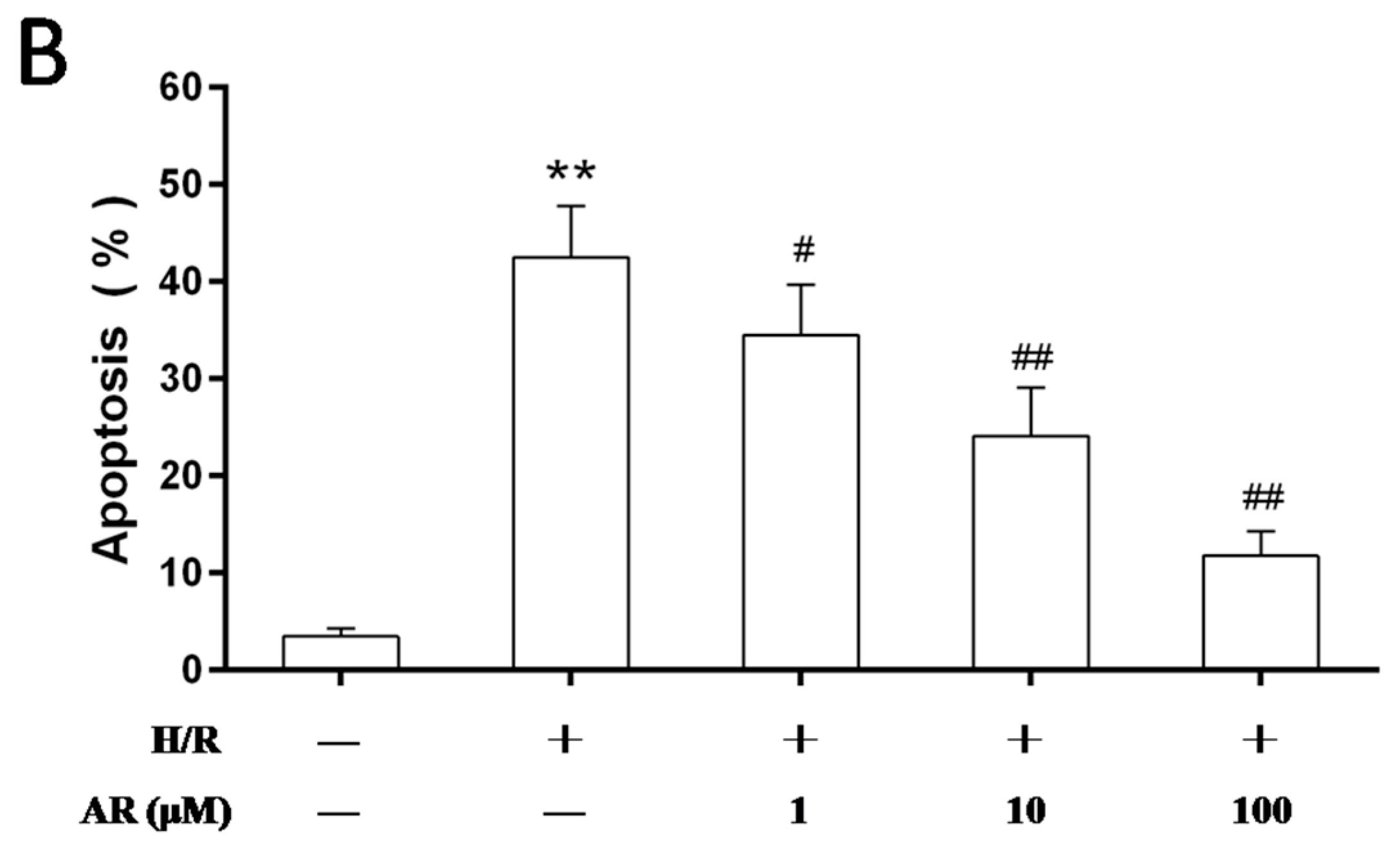
2.2. AR Pretreatment Inhibits H9c2 Cardiomyocytes Apoptosis against H/R Injury
2.3. AR Pretreatment Protects H9c2 Cardiomyocytes against H/R Injury
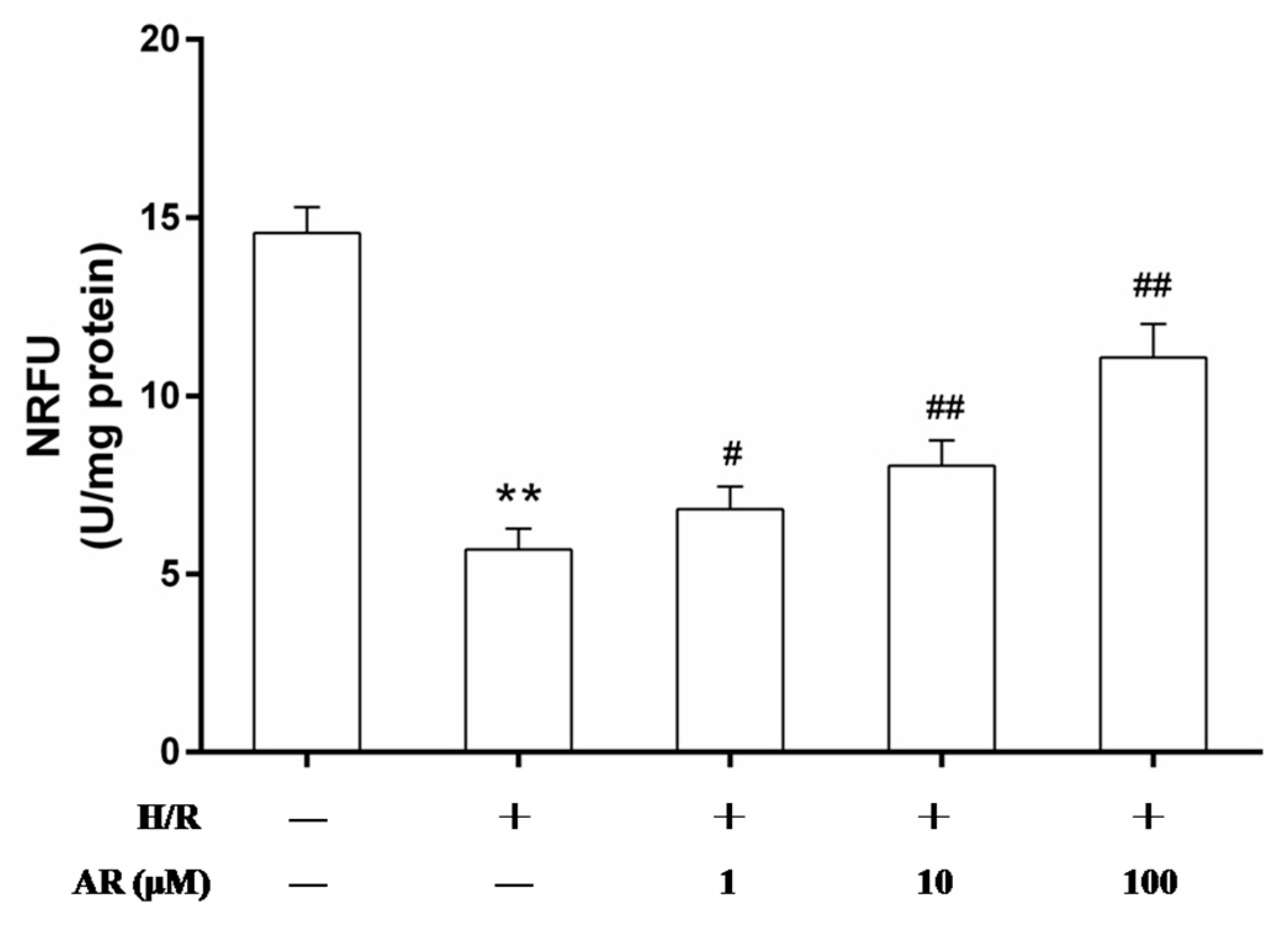
2.4. AR Pretreatment Inhibits mPTP Opening Induced by H/R Injury
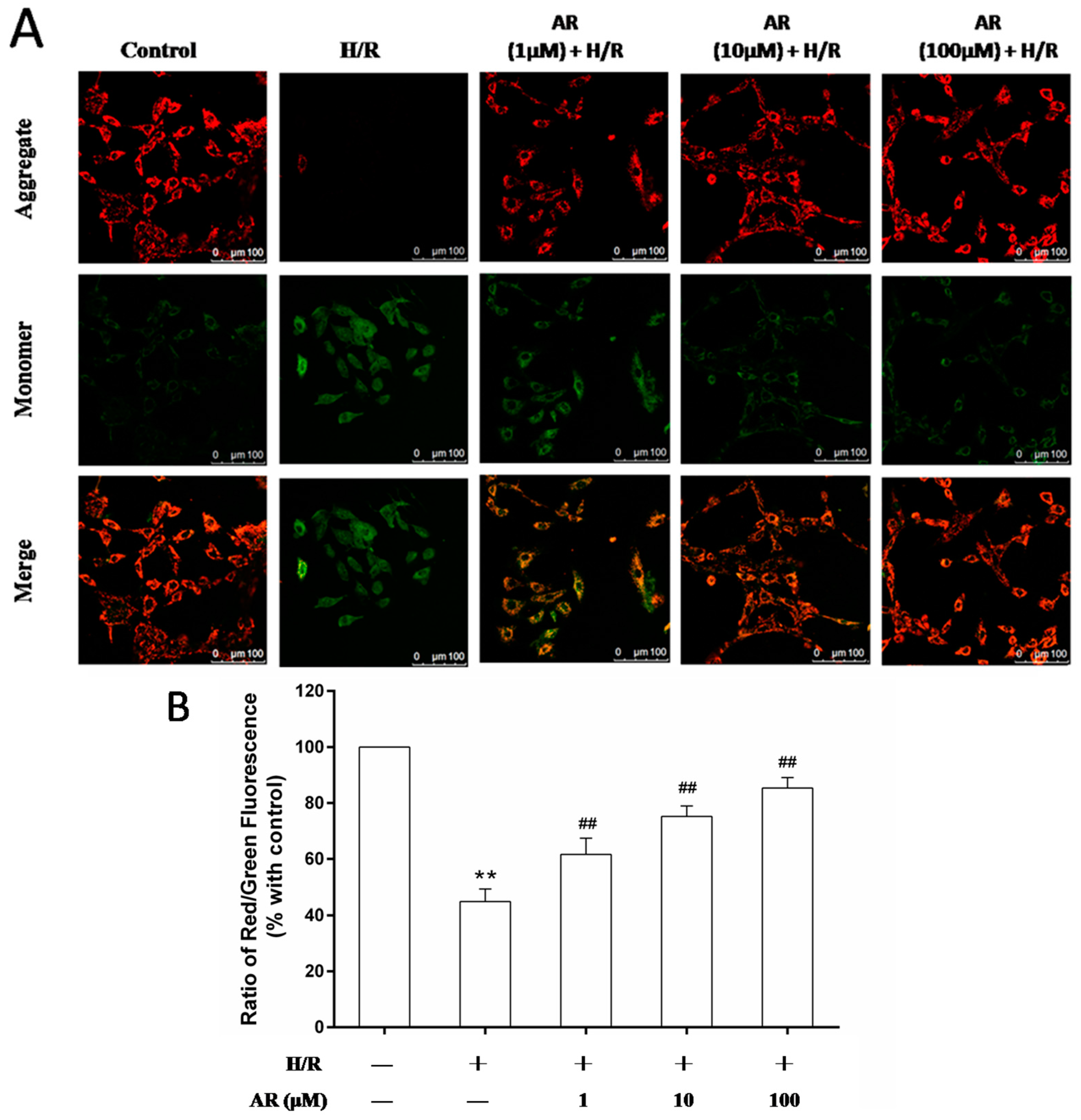
2.5. AR Pretreatment Maintains the Mitochondrial Membrane Potential (ΔΨm) in H/R-Treated Cells
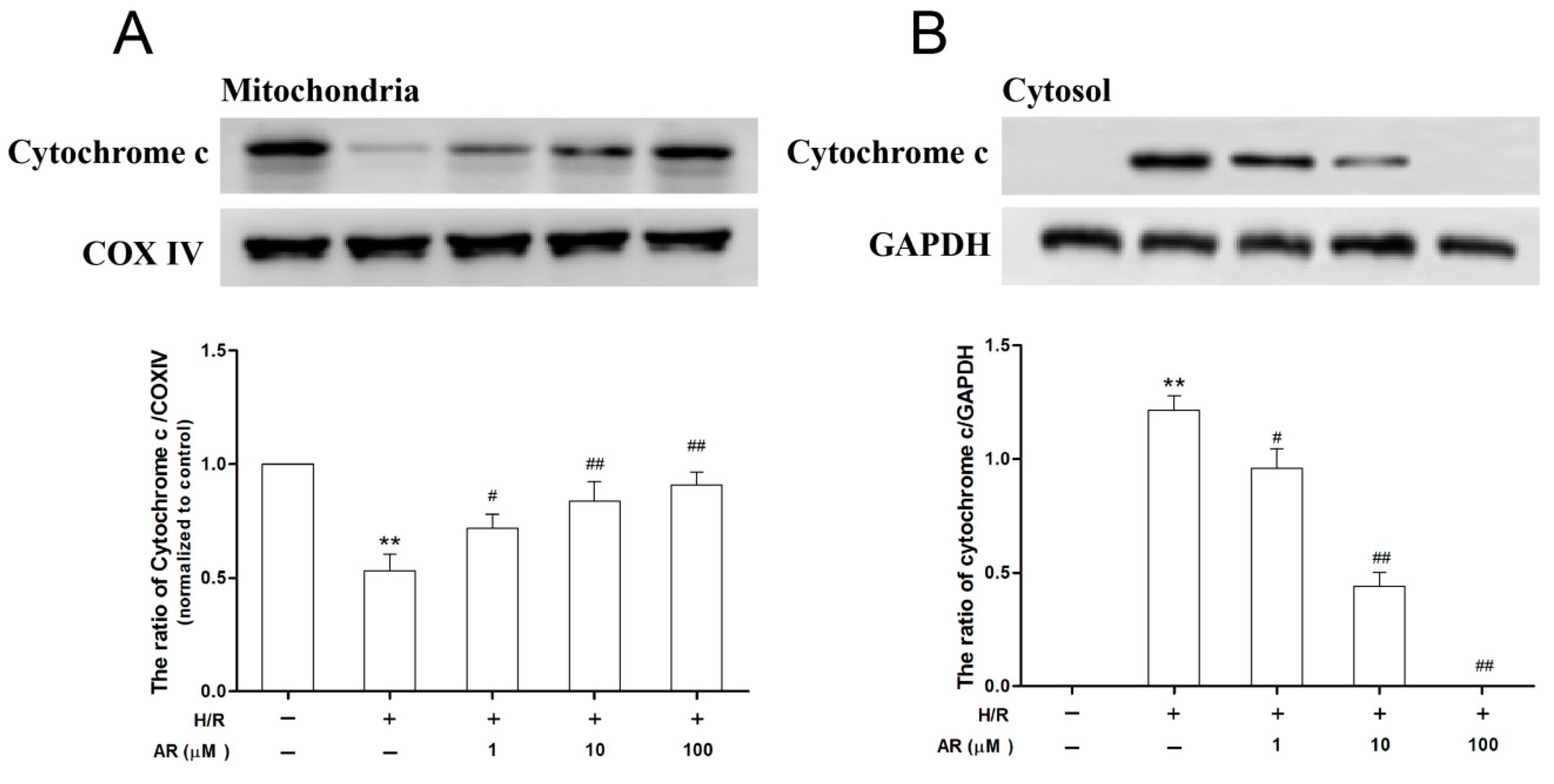
2.6. AR Pretreatment Prevents Cytochrome c Release
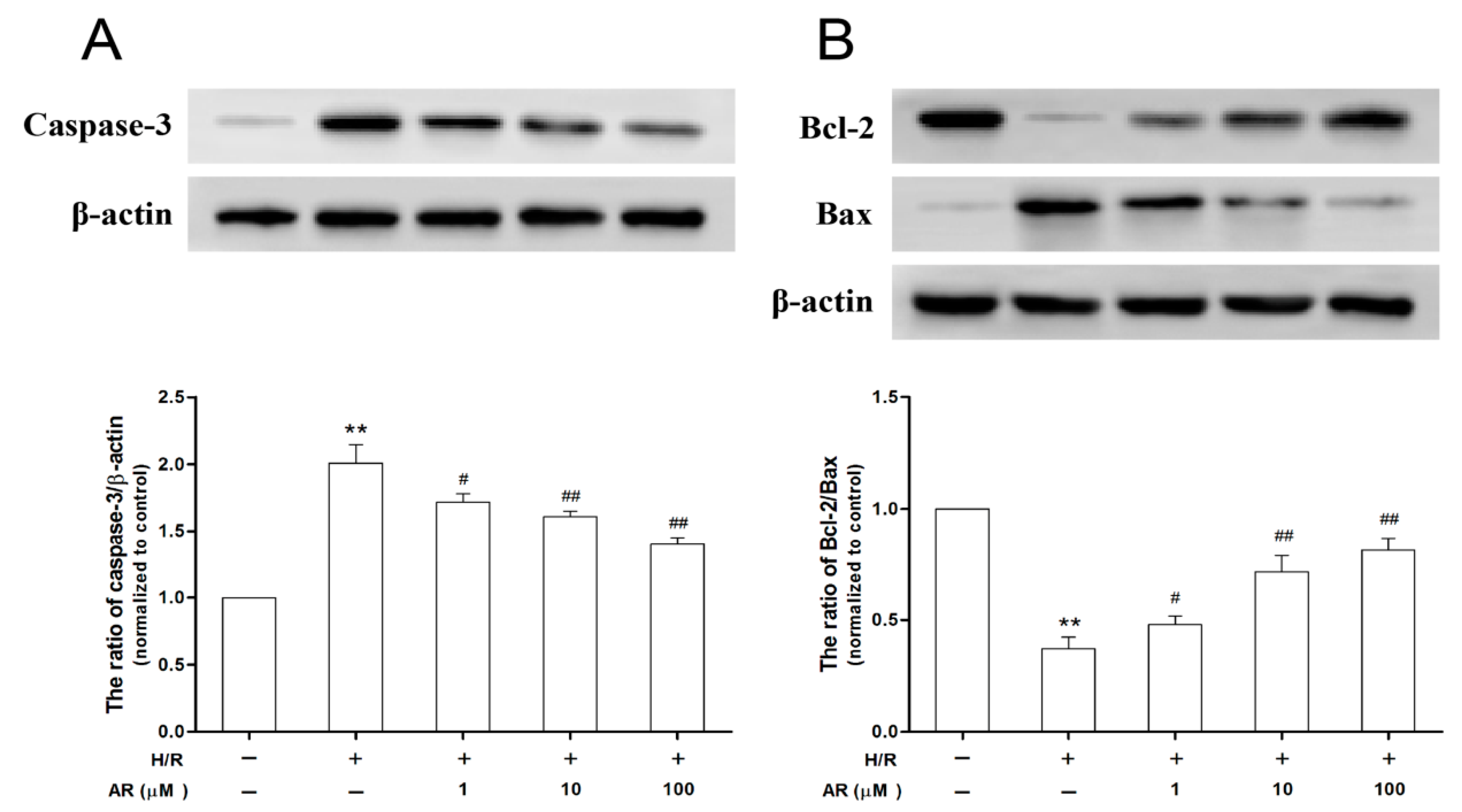
2.7. AR Pretreatment Suppresses Caspase-3 Activity and Increases the Ratio of Bcl-2 to Bax Exposed to H/R Injury
3. Discussion
4. Experimental Section
4.1. Materials
4.2. Cell Culture
4.3. Detect Cell Viability by MTT Assay
4.4. Determination of Apoptosis by Flow Cytometry
4.5. Determination of Apoptosis by TUNEL Assay
4.6. The Opening of Mitochondrial Permeability Transition Pore (mPTP) Detection
4.7. Measurement of Mitochondrial Membrane Potential (ΔΨm)
4.8. Analysis of Cytochrome c Leakage from Mitochondrial
4.9. Western Blot Analysis for Caspase-3, Bcl-2 and Bax
4.10. Statistical Analysis
5. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Seif, A.A. Nigella sativa attenuates myocardial ischemic reperfusion injury in rats. J. Physiol. Biochem. 2013, 69, 937–944. [Google Scholar] [CrossRef] [PubMed]
- Tehrani, D.M.; Seto, A.H. Third universal definition of myocardial infarction: update, caveats, differential diagnoses. Clevel. Clin. J. Med. 2013, 80, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Hausenloy, D.J.; Yellon, D.M. The evolving story of “conditioning” to protect against acute myocardial ischaemia-reperfusion injury. Heart 2007, 93, 649–651. [Google Scholar] [CrossRef] [PubMed]
- Gottlieb, R.A.; Burleson, K.O.; Kloner, R.A.; Babior, B.M.; Engler, R.L. Reperfusion injury induces apoptosis in rabbit cardiomyocytes. J. Clin. Investig. 1994, 94, 1621–1628. [Google Scholar] [CrossRef] [PubMed]
- Movassagh, M.; Foo, R.S. Simplified apoptotic cascades. Heart Fail. Rev. 2008, 13, 111–119. [Google Scholar] [CrossRef] [PubMed]
- Konstantinidis, K.; Whelan, R.S.; Kitsis, R.N. Mechanisms of cell death in heart disease. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 1552–1562. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Ling, Z.; Wang, F.; Chen, W.; Li, H.; Jin, J.; Zhang, H.; Pang, M.; Junjie, Y.U.; Liu, J. Clostridium butyricum pretreatment attenuates cerebral ischemia/reperfusion injury in mice via anti-oxidation and anti-apoptosis. Neurosci. Lett. 2015. [Google Scholar] [CrossRef] [PubMed]
- Wu, M.-P.; Zhang, Y.-S.; Zhou, Q.-M.; Xiong, J.; Dong, Y.-R.; Yan, C. Higenamine protects ischemia/reperfusion induced cardiac injury and myocyte apoptosis through activation of β2-AR/PI3K/AKT signaling pathway. Pharmacol. Res. 2016, 104, 115–123. [Google Scholar] [CrossRef] [PubMed]
- Seidlmayer, L.K.; Blatter, L.A.; Pavlov, E.; Dedkova, E.N. Inorganic polyphosphate—An unusual suspect of the mitochondrial permeability transition mystery. Channels Austin Tex. 2012, 6, 463–467. [Google Scholar] [CrossRef] [PubMed]
- Walter, D.H.; Haendeler, J.; Galle, J.; Zeiher, A.M.; Dimmeler, S. Cyclosporin A inhibits apoptosis of human endothelial cells by preventing release of cytochrome C from mitochondria. Circulation 1998, 98, 1153–1157. [Google Scholar] [CrossRef] [PubMed]
- Uchiyama, T.; Otani, H.; Okada, T.; Ninomiya, H.; Kido, M.; Imamura, H.; Nogi, S.; Kobayashi, Y. Nitric oxide induces caspase-dependent apoptosis and necrosis in neonatal rat cardiomyocytes. J. Mol. Cell. Cardiol. 2002, 34, 1049–1061. [Google Scholar] [CrossRef] [PubMed]
- Shamas-Din, A.; Kale, J.; Leber, B.; Andrews, D.W. Mechanisms of action of Bcl-2 family proteins. Cold Spring Harb. Perspect. Biol. 2013, 5, a008714. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.F.; Ma, X.L.; Wang, Y.X.; Li, F.W.; Li, Y.M.; Wan, Z.Q.; Tang, Q.L. Triterpenoid saponins from the roots of Clematis chinensis Osbeck. J. Asian Nat. Prod. Res. 2009, 11, 389–396. [Google Scholar] [CrossRef] [PubMed]
- Han, W.; Xiong, Y.; Li, Y.; Fang, W.; Ma, Y.; Liu, L.; Li, F.; Zhu, X. Anti-arthritic effects of clematichinenoside (AR-6) on PI3K/Akt signaling pathway and TNF-alpha associated with collagen-induced arthritis. Pharm. Biol. 2013, 51, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Peng, C.; Perera, P.K.; Li, Y.M.; Fang, W.R.; Liu, L.F.; Li, F.W. Anti-inflammatory effects of Clematis chinensis Osbeck extract(AR-6) may be associated with NF-kappaB, TNF-alpha, and COX-2 in collagen-induced arthritis in rat. Rheumatol. Int. 2012, 32, 3119–3125. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Fang, W.; Han, D.; Sha, L.; Wei, J.; Liu, L.; Li, Y. Clematichinenoside attenuates myocardial infarction in ischemia/reperfusion injury both in vivo and in vitro. Planta Medica 2013, 79, 1289–1297. [Google Scholar] [CrossRef] [PubMed]
- Javadov, S.; Karmazyn, M.; Escobales, N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J. Pharmacol. Exp. Ther. 2009, 330, 670–678. [Google Scholar] [CrossRef] [PubMed]
- Sarkey, J.P.; Chu, M.; McShane, M.; Bovo, E.; Ait Mou, Y.; Zima, A.V.; de Tombe, P.P.; Kartje, G.L.; Martin, J.L. Nogo-A knockdown inhibits hypoxia/reoxygenation-induced activation of mitochondrial-dependent apoptosis in cardiomyocytes. J. Mol. Cell. Cardiol. 2011, 50, 1044–1055. [Google Scholar] [CrossRef] [PubMed]
- Ong, S.B.; Hall, A.R.; Hausenloy, D.J. Mitochondrial dynamics in cardiovascular health and disease. Antioxid. Redox Signal. 2013, 19, 400–414. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Fang, W.; Zhang, R.; Wei, J.; Kodithuwakku, N.D.; Sha, L.; Ma, W.; Liu, L.; Li, F.; Li, Y. Clematichinenoside protects blood brain barrier against ischemic stroke superimposed on systemic inflammatory challenges through up-regulating A20. Brain Behav. Immun. 2016, 51, 56–69. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Morselli, E.; Kepp, O.; Kroemer, G. Targeting post-mitochondrial effectors of apoptosis for neuroprotection. Biochim. Biophys. Acta 2009, 1787, 402–413. [Google Scholar] [CrossRef] [PubMed]
- Di Lisa, F.; Canton, M.; Menabo, R.; Kaludercic, N.; Bernardi, P. Mitochondria and cardioprotection. Heart Fail. Rev. 2007, 12, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Yue, R.; Hu, H.; Yiu, K.H.; Luo, T.; Zhou, Z.; Xu, L.; Zhang, S.; Li, K.; Yu, Z. Lycopene protects against hypoxia/reoxygenation-induced apoptosis by preventing mitochondrial dysfunction in primary neonatal mouse cardiomyocytes. PLoS ONE 2012, 7, e50778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yao, K.; Ye, P.; Zhang, L.; Tan, J.; Tang, X.; Zhang, Y. Epigallocatechin gallate protects against oxidative stress-induced mitochondria-dependent apoptosis in human lens epithelial cells. Mol. Vis. 2008, 14, 217–223. [Google Scholar] [PubMed]
- Zhang, T.; Saghatelian, A. Emerging roles of lipids in BCL-2 family-regulated apoptosis. Biochim. Biophys. Acta BBA Mol. Cell Biol. Lipids 2013, 1831, 1542–1554. [Google Scholar] [CrossRef] [PubMed]
- Kumar, D.; Jugdutt, B.I. Apoptosis and oxidants in the heart. J. Lab. Clin. Med. 2003, 142, 288–297. [Google Scholar] [CrossRef]
- Lee, Y.; Gustafsson, A.B. Role of apoptosis in cardiovascular disease. Apoptosis 2009, 14, 536–548. [Google Scholar] [CrossRef] [PubMed]
- Borutaite, V.; Brown, G.C. Mitochondria in apoptosis of ischemic heart. FEBS Lett. 2003, 541, 1–5. [Google Scholar] [CrossRef]
- Cook, S.A.; Sugden, P.H.; Clerk, A. Regulation of bcl-2 family proteins during development and in response to oxidative stress in cardiac myocytes: Association with changes in mitochondrial membrane potential. Circ. Res. 1999, 85, 940–949. [Google Scholar] [CrossRef] [PubMed]
- Nishihara, M.; Miura, T.; Miki, T.; Tanno, M.; Yano, T.; Naitoh, K.; Ohori, K.; Hotta, H.; Terashima, Y.; Shimamoto, K. Modulation of the mitochondrial permeability transition pore complex in GSK-3beta-mediated myocardial protection. J. Mol. Cell. Cardiol. 2007, 43, 564–570. [Google Scholar] [CrossRef] [PubMed]
- Fang, W.; Deng, Y.; Li, Y.; Shang, E.; Fang, F.; Lv, P.; Bai, L.; Qi, Y.; Yan, F.; Mao, L. Blood brain barrier permeability and therapeutic time window of Ginkgolide B in ischemia-reperfusion injury. Eur. J. Pharm. Sci. 2010, 39, 8–14. [Google Scholar] [CrossRef] [PubMed]
- Petronilli, V.; Miotto, G.; Canton, M.; Brini, M.; Colonna, R.; Bernardi, P.; Di Lisa, F. Transient and long-lasting openings of the mitochondrial permeability transition pore can be monitored directly in intact cells by changes in mitochondrial calcein fluorescence. Biophys. J. 1999, 76, 725–734. [Google Scholar] [CrossRef]
- Smiley, S.T.; Reers, M.; Mottola-Hartshorn, C.; Lin, M.; Chen, A.; Smith, T.W.; Steele, G.D., Jr.; Chen, L.B. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. PNAS 1991, 88, 3671–3675. [Google Scholar] [CrossRef] [PubMed]
- Li, H.Z.; Guo, J.; Gao, J.; Han, L.P.; Jiang, C.M.; Li, H.X.; Bai, S.Z.; Zhang, W.H.; Li, G.W.; Wang, L.N.; et al. Role of dopamine D2 receptors in ischemia/reperfusion induced apoptosis of cultured neonatal rat cardiomyocytes. J. Biomed. Sci. 2011, 18. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.N.; Wang, C.; Lin, Y.; Xi, Y.H.; Zhang, W.H.; Zhao, Y.J.; Li, H.Z.; Tian, Y.; Lv, Y.J.; Yang, B.F.; et al. Involvement of calcium-sensing receptor in cardiac hypertrophy-induced by angiotensinII through calcineurin pathway in cultured neonatal rat cardiomyocytes. Biochem. Biophys. Res. Commun. 2008, 369, 584–589. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zheng, X.; Xiang, H.L.; Fu, X.H.; Cao, J.G. 7-Difluoromethyl-5,4′-dimethoxygenistein inhibits oxidative stress induced adhesion between endothelial cells and monocytes via NF-kappaB. Eur. J. Pharmacol. 2009, 605, 31–35. [Google Scholar] [CrossRef] [PubMed]
- Sample Availability: Samples of the compounds clematichinenoside (AR) are available from the authors.










© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ding, H.; Han, R.; Chen, X.; Fang, W.; Liu, M.; Wang, X.; Wei, Q.; Kodithuwakku, N.D.; Li, Y. Clematichinenoside (AR) Attenuates Hypoxia/Reoxygenation-Induced H9c2 Cardiomyocyte Apoptosis via a Mitochondria-Mediated Signaling Pathway. Molecules 2016, 21, 683. https://doi.org/10.3390/molecules21060683
Ding H, Han R, Chen X, Fang W, Liu M, Wang X, Wei Q, Kodithuwakku ND, Li Y. Clematichinenoside (AR) Attenuates Hypoxia/Reoxygenation-Induced H9c2 Cardiomyocyte Apoptosis via a Mitochondria-Mediated Signaling Pathway. Molecules. 2016; 21(6):683. https://doi.org/10.3390/molecules21060683
Chicago/Turabian StyleDing, Haiyan, Rong Han, Xueshan Chen, Weirong Fang, Meng Liu, Xuemei Wang, Qin Wei, Nandani Darshika Kodithuwakku, and Yunman Li. 2016. "Clematichinenoside (AR) Attenuates Hypoxia/Reoxygenation-Induced H9c2 Cardiomyocyte Apoptosis via a Mitochondria-Mediated Signaling Pathway" Molecules 21, no. 6: 683. https://doi.org/10.3390/molecules21060683
APA StyleDing, H., Han, R., Chen, X., Fang, W., Liu, M., Wang, X., Wei, Q., Kodithuwakku, N. D., & Li, Y. (2016). Clematichinenoside (AR) Attenuates Hypoxia/Reoxygenation-Induced H9c2 Cardiomyocyte Apoptosis via a Mitochondria-Mediated Signaling Pathway. Molecules, 21(6), 683. https://doi.org/10.3390/molecules21060683