
The Chiral Pool in the Pictet–Spengler Reaction for the Synthesis of β-Carbolines


Abstract
:
{kind=link}
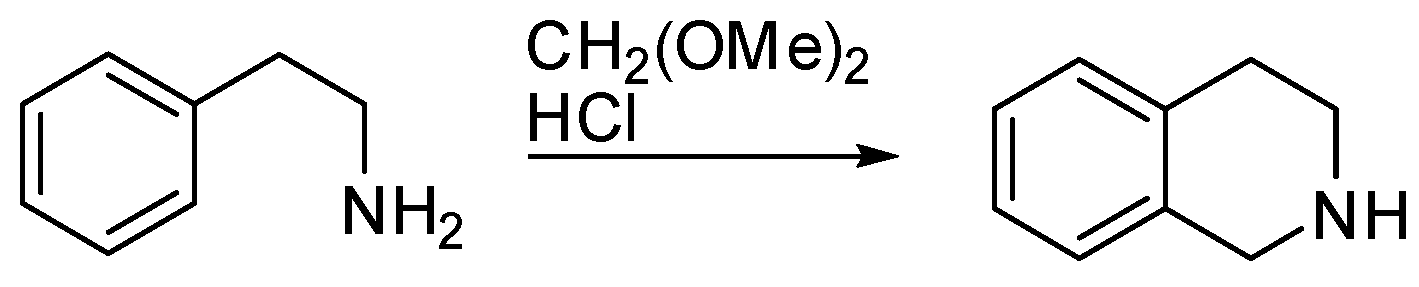{kind=link}
{kind=link}
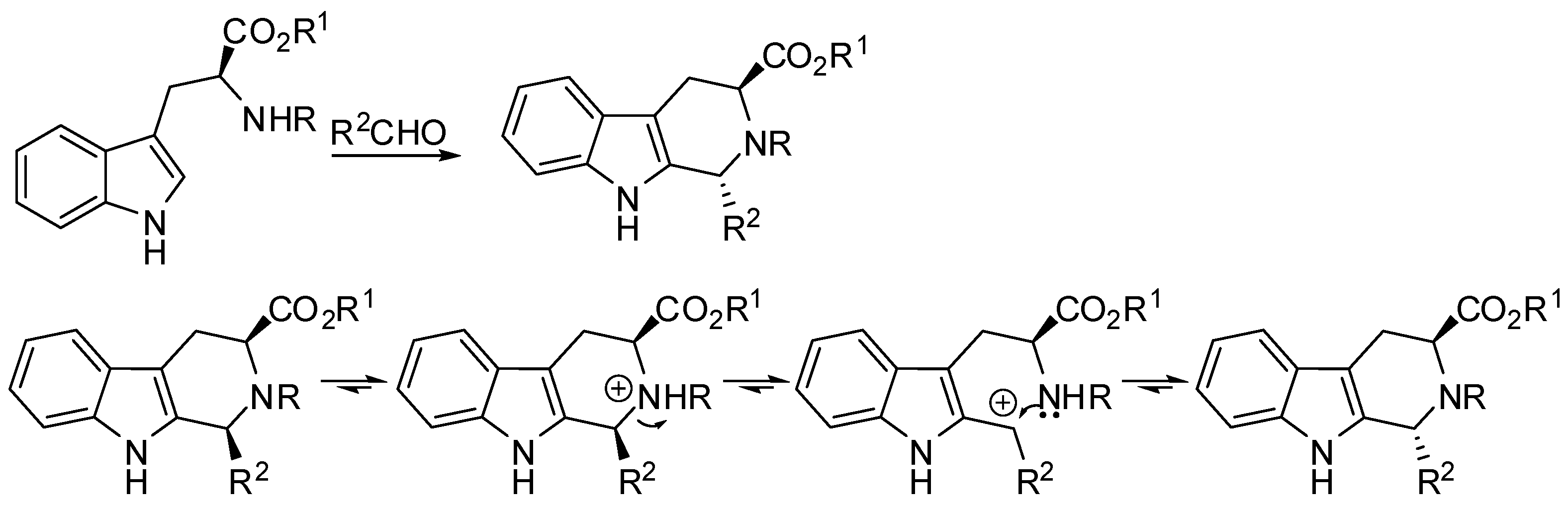{kind=link}
{kind=link}
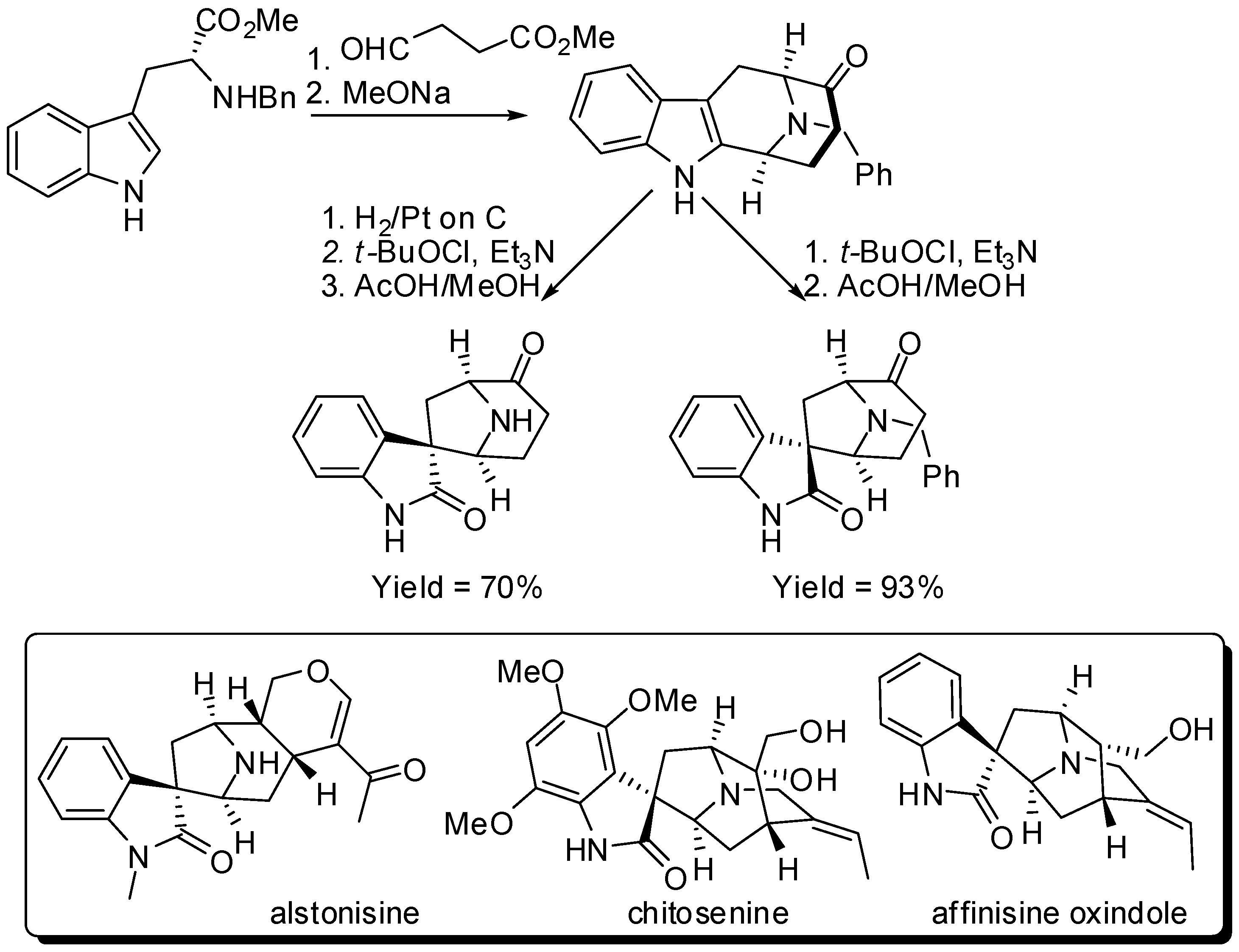
{kind=link}
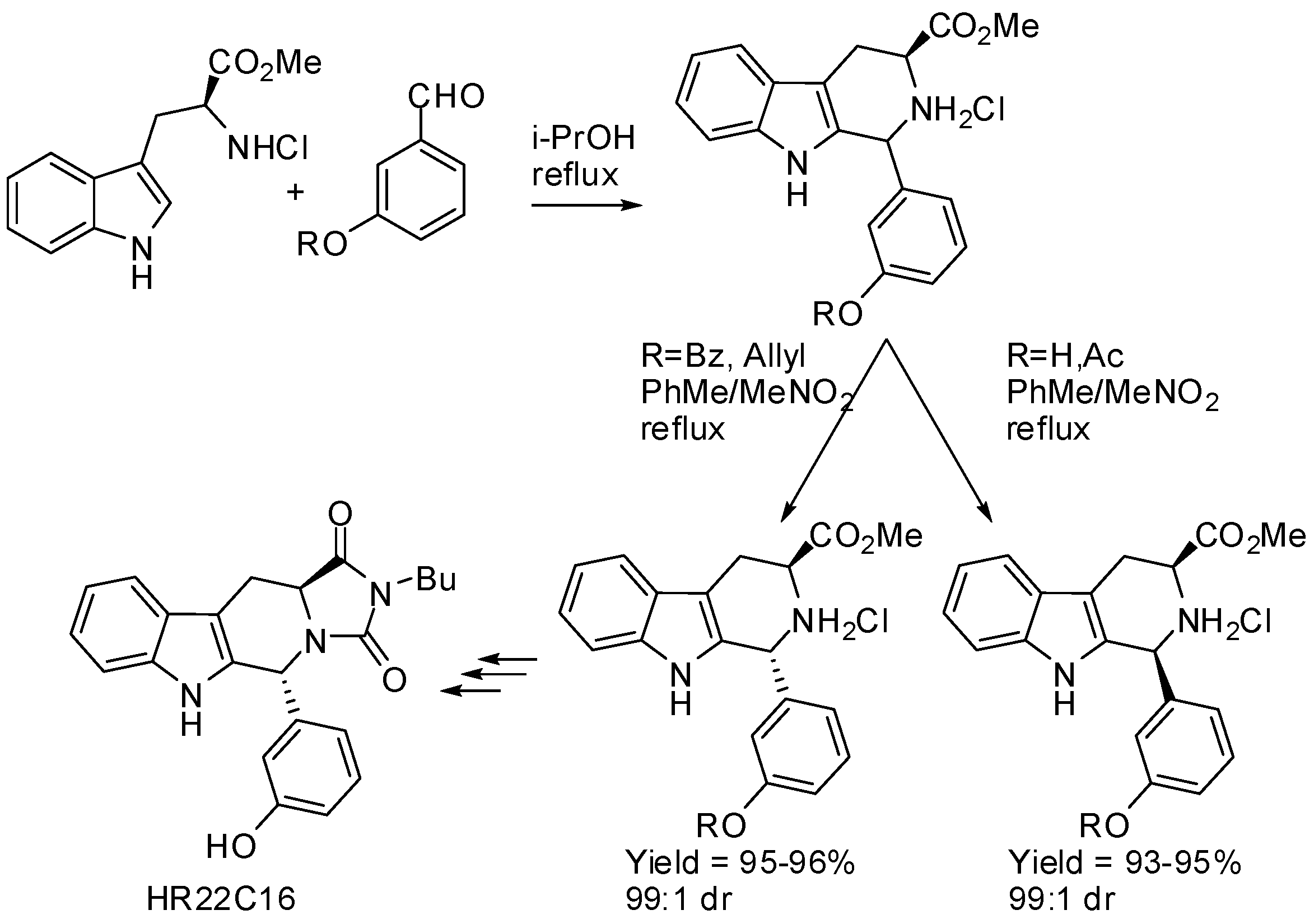{kind=link}
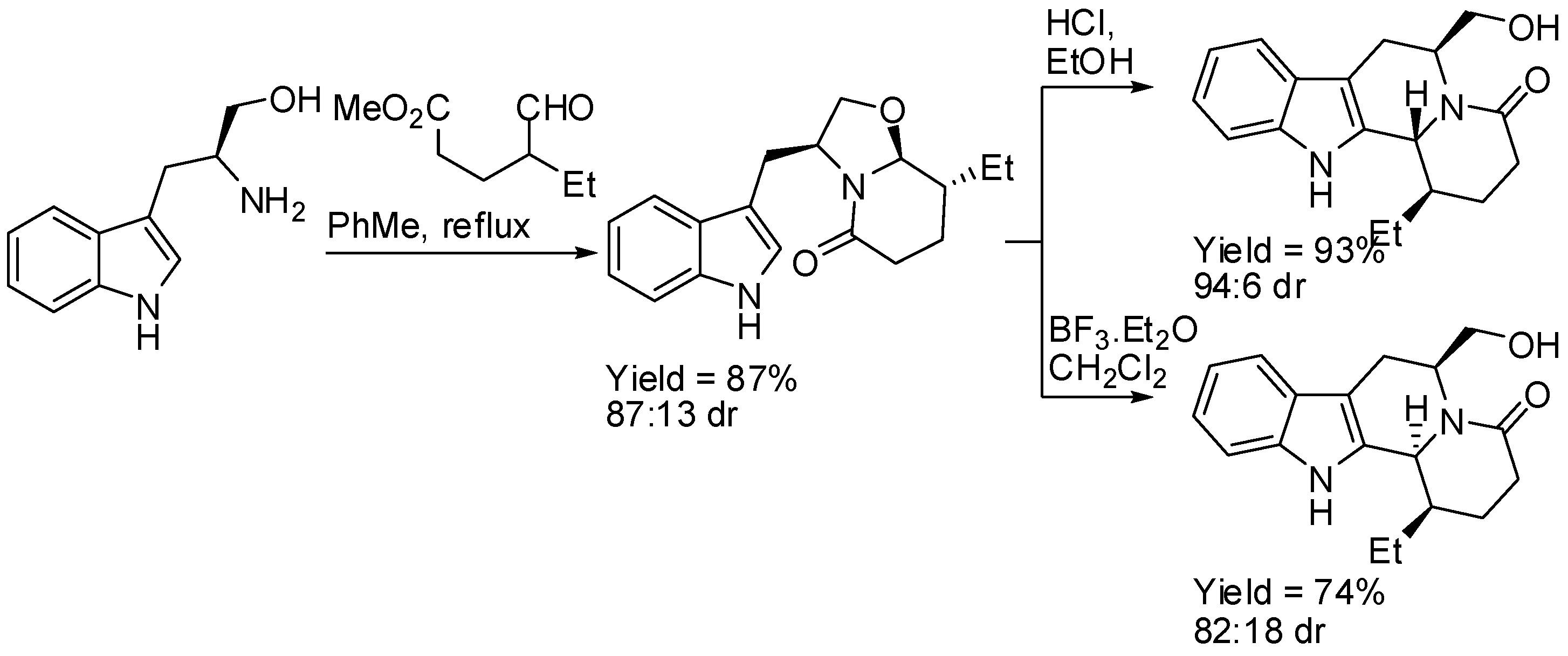{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
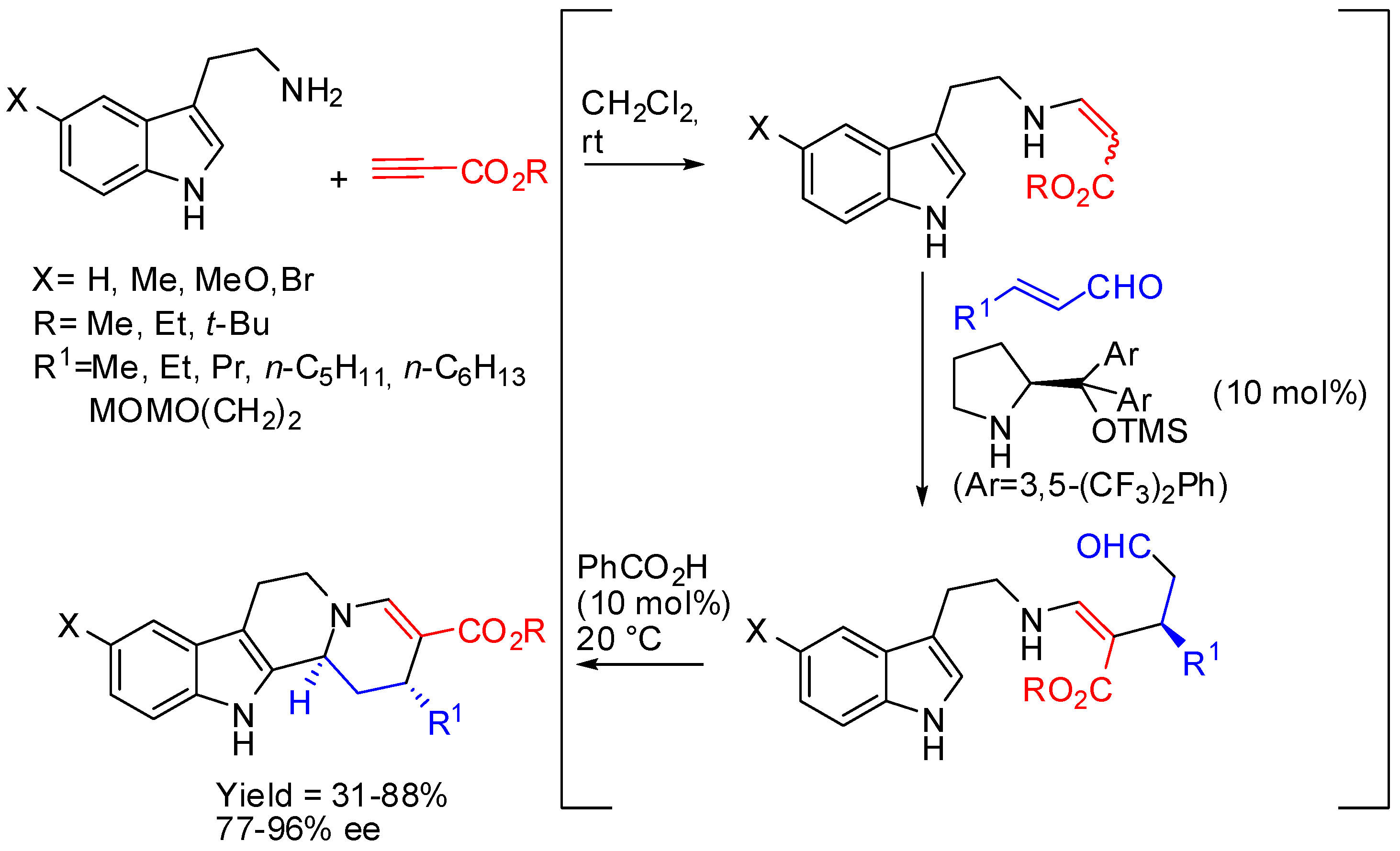{kind=link}
{kind=link}
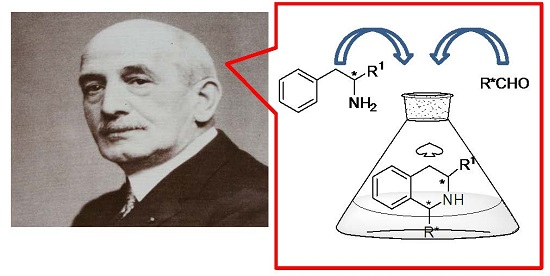
1. Introduction
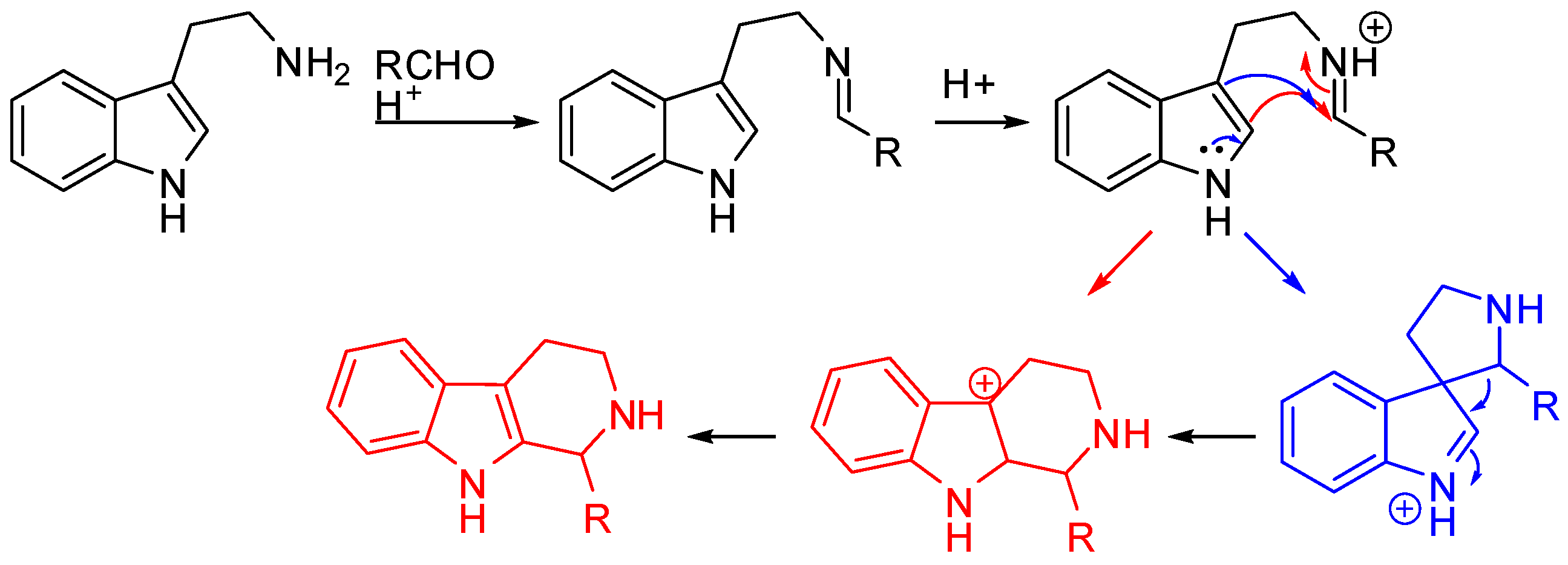
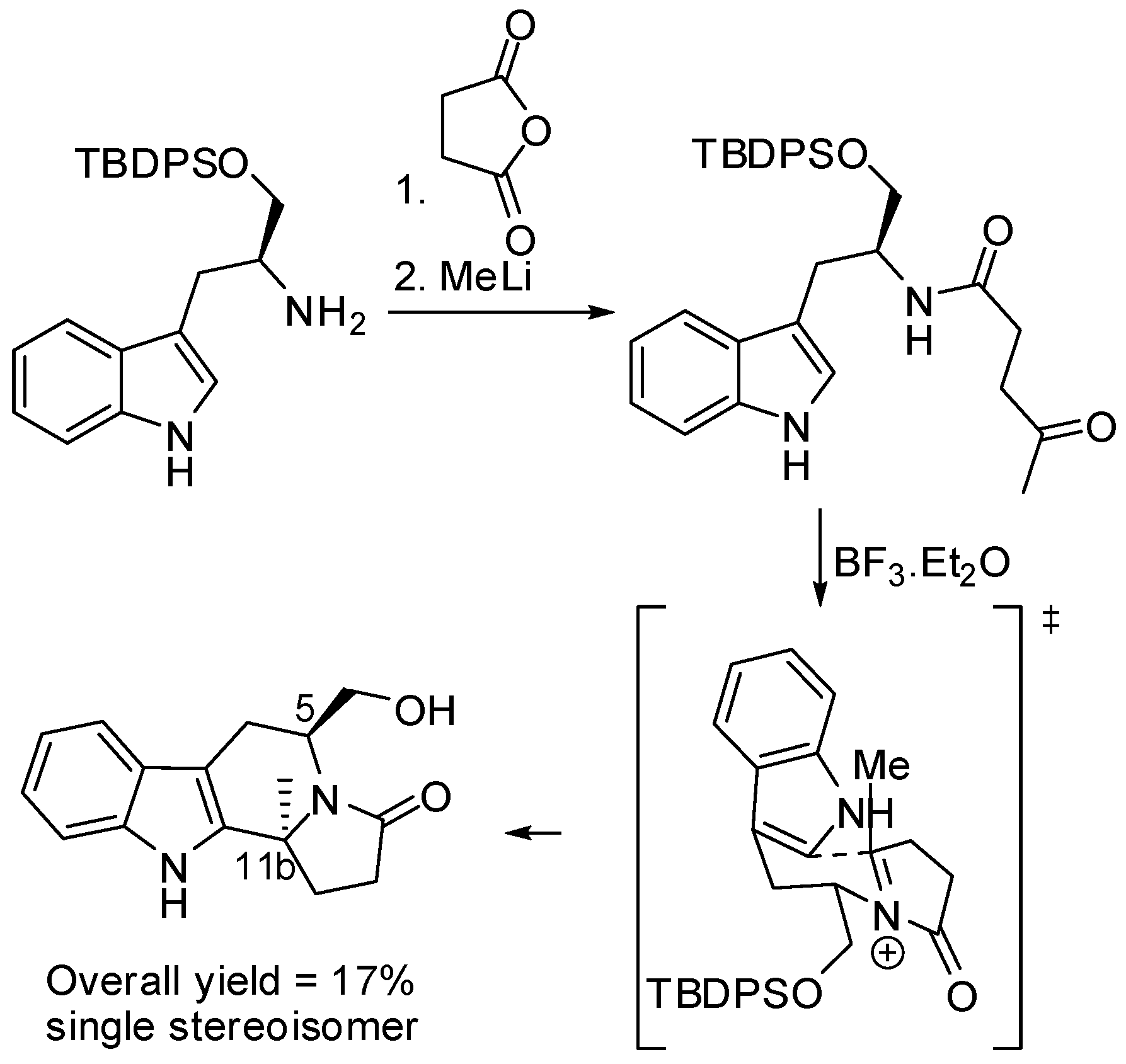
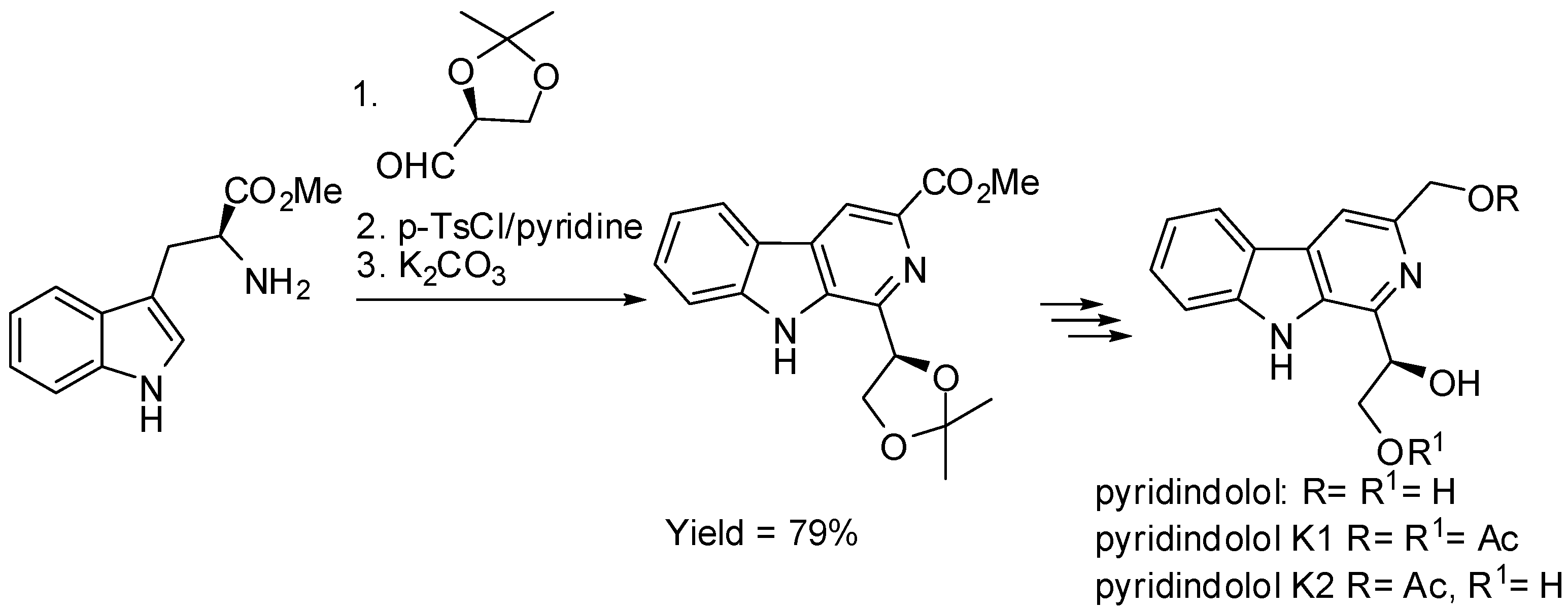
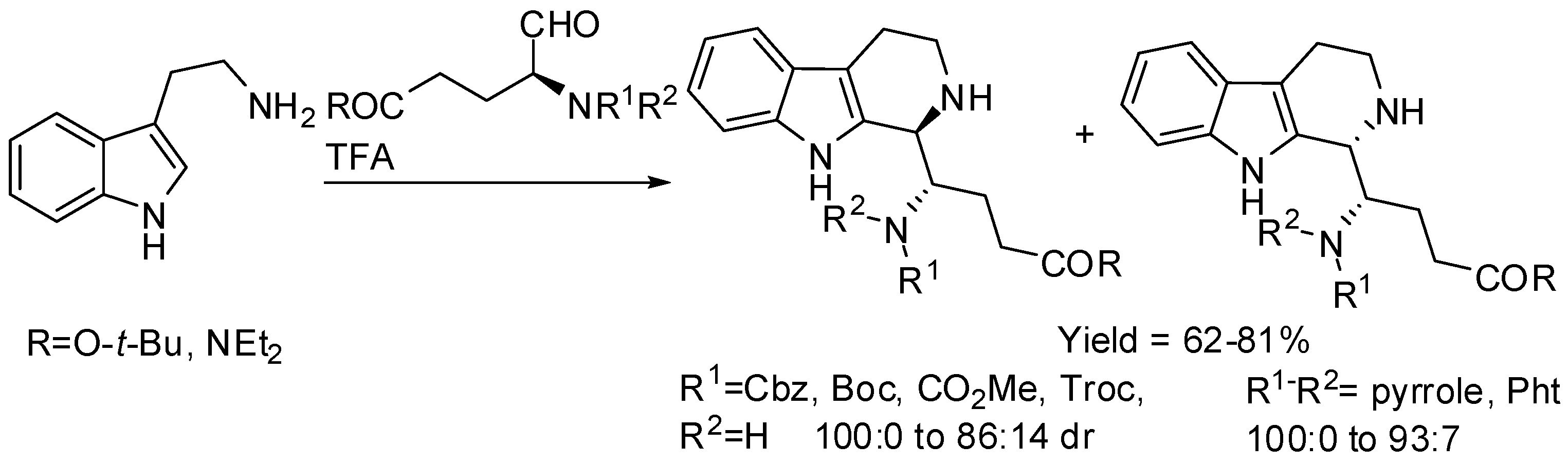
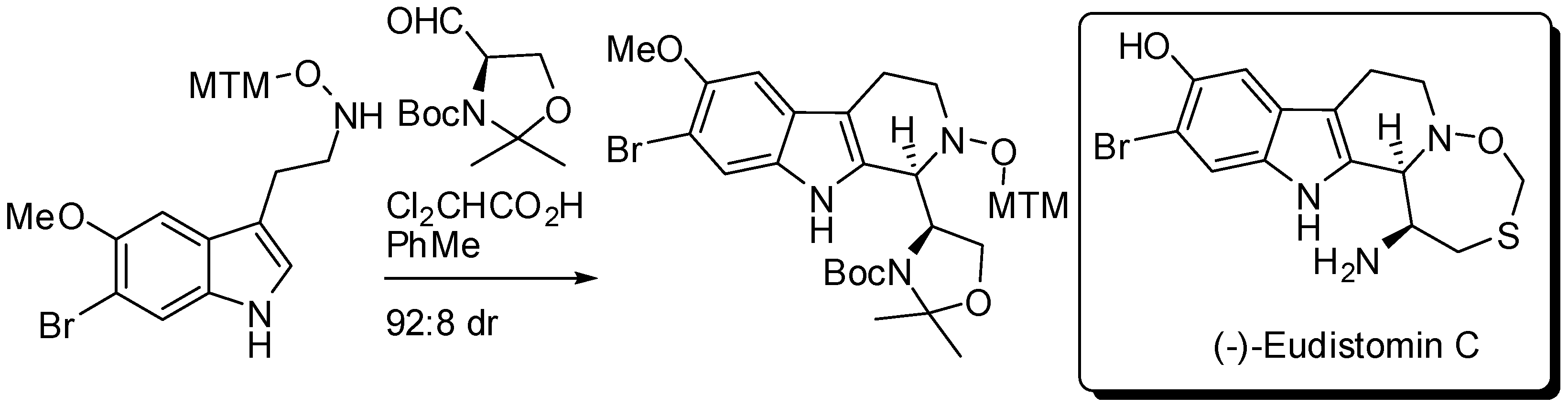
2. Starting from Tryptophan Derivatives
3. Starting from Chiral Carbonyl Compounds
4. Chiral Auxiliaries
5. Preparing Chiral Compounds Beforehand
6. Conclusions
Conflicts of Interest
Abbreviations
| Ac | Acetyl |
| Boc | tert-Butoxycarbonyl |
| Bz | Benzoyl |
| Cbz | Benzyloxycarbonyl |
| CSA | Camphorsulfonic acid |
| DABCO | 1,8-Diazabicyclooctane |
| MTM | Methylthiomethyl |
| Pht | Phthalimide |
| TBDMS | tert-Butyldimethylsilyl |
| TBDPS | tert-Butyldiphenylsilyl |
| TES | Triethylsilyl |
| TFA | Trifluoroacetic acid |
| Tol | 4-Methylphenyl (Tolyl) |
| Troc | 2,2,2-Trichloroethoxycarbonyl |
| Ts | Methylphenylsulfonyl (Tosyl) |
References
- Pictet, A.; Spengler, T. Über die bildung von isochinolin-derivaten durch einwirkung von methylal auf phenyl-äthylamin, phenyl-alanin und tyrosin. Ber. Dtsch. Chem. Ges. 1911, 44, 2030–2036. [Google Scholar] [CrossRef]
- Tatsui, G. Über die synthese von carbolinderivaten. J. Pharm. Soc. Jpn. 1928, 48, 453–459. [Google Scholar]
- The Alkaloids: Chemistry and Physiology; Academic Press: London, UK, 1981; Volume 20.
- Stockigt, J.; Husson, H.P.; Kan-Fan, C.; Zenk, M.H. Cathenamine, a central intermediate in the cell free biosynthesis of ajmalicine and related indole alkaloids. J. Chem. Soc. Chem. Commun. 1977, 164–166. [Google Scholar] [CrossRef]
- Stöckigt, J.; Antonchick, A.P.; Wu, F.; Waldmann, H. The Pictet–Spengler reaction in nature and in organic chemistry. Angew. Chem. Int. Ed. 2011, 50, 8538–8564. [Google Scholar] [CrossRef] [PubMed]
- Bartoli, G.; Bencivenni, G.; Dalpozzo, R. Organocatalytic strategies for the asymmetric functionalization of indoles. Chem. Soc. Rev. 2010, 39, 4449–4465. [Google Scholar] [CrossRef] [PubMed]
- Lorenz, M.; Van Linn Michael, L.; James, M.C. The asymmetric Pictet–Spengler reaction. Curr. Org. Synth. 2010, 7, 189–223. [Google Scholar] [CrossRef]
- Laine, A.E.; Lood, C.; Koskinen, A.M.P. Pharmacological importance of optically active tetrahydro-β-carbolines and synthetic approaches to create the C1 stereocenter. Molecules 2014, 19, 1544–1567. [Google Scholar] [CrossRef] [PubMed]
- Dalpozzo, R. Strategies for the asymmetric functionalization of indoles: An update. Chem. Soc. Rev. 2015, 44, 742–778. [Google Scholar] [CrossRef] [PubMed]
- Ingallina, C.; D’Acquarica, I.; Monache, G.D.; Ghirga, F.; Quaglio, D.; Ghirga, P.; Berardozzi, S.; Markovic, V.; Botta, B. The Pictet–Spengler reaction still on stage. Curr. Pharm. Des. 2016, 22, 1808–1850. [Google Scholar] [CrossRef] [PubMed]
- Jackson, A.H.; Smith, A.E. Electrophilic substitution in indoles—II: The formation of 3,3-spirocyclic indole derivatives from tryptamines and their rearrangement to β-carbolines. Tetrahedron 1968, 24, 403–413. [Google Scholar] [CrossRef]
- Ungemach, F.; Cook, J.M. The spiroindolenine intermediate, a review. Heterocycles 1978, 9, 1089–1119. [Google Scholar]
- Liu, J.; Nakagawa, M.; Ogata, K.; Hino, T. The Pictet–Spengler reaction of Nb-hydroxytryptamines and cysteinals. II. Temperature effects, stereochemistry and mechanism. Chem. Pharm. Bull. 1991, 39, 1672–1676. [Google Scholar] [CrossRef]
- Czerwinski, K.M.; Deng, L.; Cook, J.M. Mechanism driven trans stereospecificity in the Pictet–Spengler reaction. Stereospecific formation of trans-1,2,3-trisubstituted-tetrahydro-β-carbolines by condensation of Nb-diphenylmethyl tryptophan isopropyl esters wtm aldehydes. Tetrahedron Lett. 1992, 33, 4721–4724. [Google Scholar] [CrossRef]
- Soerens, D.; Sandrin, J.; Ungemach, F.; Mokry, P.; Wu, G.S.; Yamanaka, E.; Hutchins, L.; DiPierro, M.; Cook, J.M. Study of the Pictet–Spengler reaction in aprotic media: Synthesis of the β-galactosidase inhibitor, pyridindolol. J. Org. Chem. 1979, 44, 535–545. [Google Scholar] [CrossRef]
- Deng, L.; Czerwinski, K.; Cook, J.M. Stereospecificity in the Pictet–Spengler reaction kinetic vs. thermodynamic control. Tetrahedron Lett. 1991, 32, 175–178. [Google Scholar] [CrossRef]
- Cox, E.D.; Hamaker, L.K.; Li, J.; Yu, P.; Czerwinski, K.M.; Deng, L.; Bennett, D.W.; Cook, J.M.; Watson, W.H.; Krawiec, M. Enantiospecific formation of trans 1,3-disubstituted tetrahydro-β-carbolines by the Pictet-Spengler reaction and conversion of cis diastereomers into their trans counterparts by scission C-1/N-2 bond. J. Org. Chem. 1997, 62, 44–61. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Deschamp, J.R.; Cook, J.M. Regiospecific, enantiospecific total synthesis alkoxy-substituted indole bases, 16-epi-Na-methylgardneral, 11-methoxyaffinisine, and 11-methoxymacroline as well as the indole alkaloids alstophylline and macralstonine. Org. Lett. 2002, 4, 3339–3342. [Google Scholar] [CrossRef] [PubMed]
- Han, D.; Försterling, F.H.; Deschamps, J.R.; Parrish, D.; Liu, X.; Yin, W.; Huang, S.; Cook, J.M. Conformational analysis cis- and trans-adducts Pictet–Spengler reaction. Evidence for the structural basis for the C(1)-N(2) scission process in the cis- to trans-isomerization. J. Nat. Prod. 2007, 70, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Kumpaty, H.J.; Van Linn, M.L.; Kabir, M.S.; Försterling, F.H.; Deschamps, J.R.; Cook, J.M. Study of the cis to trans isomerization of 1-phenyl-2,3-disubstituted tetrahydro-β-carbolines at C(1). evidence for the carbocation-mediated mechanism. J. Org. Chem. 2009, 74, 2771–2779. [Google Scholar] [CrossRef] [PubMed]
- Van Linn, M.L.; Cook, J.M. Mechanistic studies on the cis to trans epimerization of trisubstituted 1,2,3,4-tetrahydro-β-carbolines. J. Org. Chem. 2010, 75, 3587–3599. [Google Scholar] [CrossRef] [PubMed]
- Bailey, P.D.; Hollinshead, S.P.; McLay, N.R.; Morgan, K.; Palmer, S.J.; Prince, S.N.; Reynolds, C.D.; Wood, S.D. Diastereo- and enantio-selectivity in the Pictet–Spengler reaction. J. Chem. Soc., Perkin Trans. 1 1993, 3, 431–439. [Google Scholar] [CrossRef]
- Peng, Y.; Wang, T.; Fuxiang, Y.; Cook, J.M. General approach for the synthesis of macroline/sarpagine related indole alkaloids via the asymmetric Pictet–Spengler reaction: The enantiospecific synthesis Na-H, azabicyclo[3.3.1]nonone template. Tetrahedron Lett. 1997, 38, 6819–6822. [Google Scholar] [CrossRef]
- Li, J.; Wang, T.; Yu, P.; Peterson, A.; Weber, R.; Soerens, D.; Grubisha, D.; Bennett, D.; Cook, J.M. General approach for the synthesis of ajmaline/sarpagine indole alkaloids: Enantiospecific total synthesis of (+)-ajmaline, alkaloid G, and norsuaveoline via the asymmetric Pictet–Spengler reaction. J. Am. Chem. Soc. 1999, 121, 6998–7010. [Google Scholar] [CrossRef]
- Yu, S.; Berner, O.M.; Cook, J.M. General approach for the synthesis of indole alkaloids via the asymmetric Pictet–Spengler reaction: First enantiospecific total synthesis of (−)-corynantheidine as well as the enantiospecific total synthesis of (−)-corynantheidol, (−)-geissoschizol, and (+)-geissoschizine. J. Am. Chem. Soc. 2000, 122, 7827–7828. [Google Scholar]
- Wang, T.; Cook, J.M. General approach for the synthesis of sarpagine/ajmaline indole alkaloids. stereospecific total synthesis of the sarpagine alkaloid (+)-vellosimine. Org. Lett. 2000, 2, 2057–2059. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Wang, T.; Wearing, X.Z.; Ma, J.; Cook, J.M. Enantiospecific total synthesis of (−)-(E)16-epiaffinisine, (+)-(E)16-epinormacusine B, and (+)-dehydro-16-epiaffinisine as well as the stereocontrolled total synthesis of alkaloid G. J. Org. Chem. 2003, 68, 5852–5859. [Google Scholar] [CrossRef] [PubMed]
- Yin, W.; Kabir, M.S.; Wang, Z.; Rallapalli, S.K.; Ma, J.; Cook, J.M. Enantiospecific total synthesis of the important biogenetic intermediates along the ajmaline pathway, (+)-polyneuridine and (+)-polyneuridine aldehyde, as well as 16-epivellosimine and macusine A. J. Org. Chem. 2010, 75, 3339–3349. [Google Scholar] [CrossRef] [PubMed]
- Nören-Müller, A.; Wilk, W.; Saxena, K.; Schwalbe, H.; Kaiser, M.; Waldmann, H. Discovery of a new class of inhibitors of mycobacterium tuberculosis protein tyrosine phosphatase B by biology-oriented synthesis. Angew. Chem. Int. Ed. 2008, 47, 5973–5977. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, G.O.; Wang, Z.-J.; Namjoshi, O.A.; Deschamps, J.R.; Cook, J.M. First stereospecific total synthesis of (−)-affinisine oxindole as well as facile entry into the C(7)-diastereomeric chitosenine stereochemistry. Tetrahedron Lett. 2015, 56, 3052–3056. [Google Scholar] [CrossRef]
- Bailey, P.D.; McLay, N.R. Use of the kinetically controlled Pictet–Spengler reaction in the asymmetric synthesis of indole alkaloids: Formal syntheses of (−)-ajmaline, (−)-koumine, (−)-taberpsychine, (−)-koumidine and (−)-suavoline. J. Chem. Soc. Perkin Trans. 1 1993, 441–449. [Google Scholar] [CrossRef]
- Bailey, P.D.; Moore, M.H.; Morgan, K.M.; Smith, D.I.; Vernon, J.M. Enhancing the yield and diastereoselectivity of the Pictet–Spengler reaction: A highly efficient route to cis-1,3-disubstituted tetrahydro−β−carbolines. Tetrahedron Lett. 1994, 35, 3587–3588. [Google Scholar] [CrossRef]
- Alberch, L.; Bailey, P.D.; Clingan, P.D.; Mills, T.J.; Price, R.A.; Pritchard, R.G. The cis-specific Pictet–Spengler reaction. Eur. J. Org. Chem. 2004, 2004, 1887–1890. [Google Scholar] [CrossRef]
- Bailey, P.D.; Beard, M.A.; Phillips, T.R. Unexpected cis selectivity in the Pictet–Spengler reaction. Tetrahedron Lett. 2009, 50, 3645–3647. [Google Scholar] [CrossRef]
- Bailey, P.D.; Clingan, P.D.; Mills, T.J.; Price, R.A.; Pritchard, R.G. Total synthesis of (−)-raumacline. Chem. Commun. 2003, 35, 2800–2801. [Google Scholar] [CrossRef]
- Martin, S.F.; Chen, K.X.; Eary, C.T. An enantioselective total synthesis of (+)-geissoschizine. Org. Lett. 1999, 1, 79–82. [Google Scholar] [CrossRef] [PubMed]
- Deiters, A.; Chen, K.; Eary, C.T.; Martin, S.F. Biomimetic entry to the sarpagan family of indole alkaloids: total synthesis of (+)-geissoschizine and (+)-N-methylvellosimine. J. Am. Chem. Soc. 2003, 125, 4541–4550. [Google Scholar] [CrossRef] [PubMed]
- Vavsari, V.F.; Dianati, V.; Ramezanpour, S.; Balalaie, S. Stereoselective synthesis of functionalized tetrahydro-β-carbolines via Pictet–Spengler reaction. Synlett 2015, 26, 1955–1960. [Google Scholar] [CrossRef]
- Shi, X.-X.; Liu, S.-L.; Xu, W.; Xu, Y.-L. Highly stereoselective Pictet–Spengler reaction of d-tryptophan methyl ester with piperonal: Convenient syntheses of Cialis (Tadalafil), 12a-epi-Cialis, and their deuterated analogues. Tetrahedron Asymmetry 2008, 19, 435–442. [Google Scholar] [CrossRef]
- Xiao, S.; Lu, X.; Shi, X.-X.; Sun, Y.; Liang, L.-L.; Yu, X.-H.; Dong, J. Syntheses of chiral 1,3-disubstituted tetrahydro−β−carbolines via CIAT process: Highly stereoselective Pictet–Spengler reaction of d-tryptophan ester hydrochlorides with various aldehydes. Tetrahedron Asymmetry 2009, 20, 430–439. [Google Scholar] [CrossRef]
- Xiao, S.; Shi, X.-X. The first highly stereoselective approach to the mitotic kinesin Eg5 inhibitor HR22C16 and its analogues. Tetrahedron Asymmetry 2010, 21, 226–231. [Google Scholar] [CrossRef]
- Ma, Y.; Wu, H.; Zhang, J.; Li, Y. Enantioselective synthesis and antimicrobial activities of tetrahydro-β-carboline diketopiperazines. Chirality 2013, 25, 656–662. [Google Scholar] [CrossRef] [PubMed]
- Amat, M.; Santos, M.M.M.; Bassas, O.; Llor, N.; Escolano, C.; Gòmez-Esqué, A.; Molins, E.; Allin, S.M.; McKee, V.; Bosch, J. Straightforward methodology for the enantioselective synthesis of benzo[a]- and indolo[2,3-a]quinolizidines. J. Org. Chem. 2007, 72, 5193–5201. [Google Scholar] [CrossRef] [PubMed]
- Ardeo, A.; Garcia, E.; Arrasate, S.; Lete, E.; Sotomayor, N. A practical approach to the fused β-carboline system. Asymmetric synthesis of indolo[2,3-a]indolizidinones via a diastereoselective intramolecular a-amidoalkylation reaction. Tetrahedron Lett. 2003, 44, 8445–8448. [Google Scholar] [CrossRef]
- Horiguchi, Y.; Nakamura, M.; Saitoh, T.; Sano, T. A synthesis of chiral 1,1,3-trisubstituted 1,2,3,4-tetrahydro-β-carbolines by the Pictet–Spengler reaction of tryptophan and ketones: Conversion of (1R,3S)-diastereomers into their (1S,3S)-counterparts by scission C(1)-N(2) bond. Chem. Pharm. Bull. 2003, 51, 1368–1373. [Google Scholar] [CrossRef] [PubMed]
- Singh, K.; Deb, P.K.; Venugopalan, P. Modified Pictet–Spengler reaction. A highly diastereoselective approach to 1,2,3-trisubstituted-1,2,3,4-tetrahydro-β-carbolines using perhydro-1,3-heterocycles. Tetrahedron 2001, 57, 7939–7949. [Google Scholar] [CrossRef]
- Larghi, E.L.; Amongero, M.; Bracca, A.B.J.; Kaufman, T.S. The intermolecular Pictet–Spengler condensation with chiral carbonyl derivatives in the stereoselective syntheses of optically-active isoquinoline and indole alkaloids. Arkivoc 2005, 2005, 98–153. [Google Scholar]
- Zhang, Q.; Fan, Z.; Dong, J.; Shi, X.-X.; Lu, X. Novel asymmetric total syntheses of (R)-(−)-pyridindolol, (R)-(−)-pyridindolol K1, and (R)-(−)-pyridindolol K2 via a mild one-pot aromatization of N-tosyl-tetrahydro-β-carboline with (S)-2,3-O-isopropylidene-l-glyceraldehyde as the source of chirality. Tetrahedron Asymmetry 2013, 24, 633–637. [Google Scholar] [CrossRef]
- Ducrot, P.; Rabhi, C.; Thal, C. Synthesis of tetrahydro-β-carbolines and studies of the Pictet–Spengler reaction. Tetrahedron 2000, 56, 2683–2692. [Google Scholar] [CrossRef]
- Pulka, K.; Kulis, P.; Tymecka, D.; Frankiewicz, L.; Wilczek, M.; Kozminski, W.; Misicka, A. Diastereoselective Pictet–Spengler condensation of tryptophan with α−amino aldehydes as chiral carbonyl components. Tetrahedron 2008, 64, 1506–1514. [Google Scholar] [CrossRef]
- Yamashita, T.; Kawai, N.; Tokuyama, H.; Fukuyama, T. Stereocontrolled total synthesis of (−)-eudistomin C. J. Am. Chem. Soc. 2005, 127, 15038–15039. [Google Scholar] [CrossRef] [PubMed]
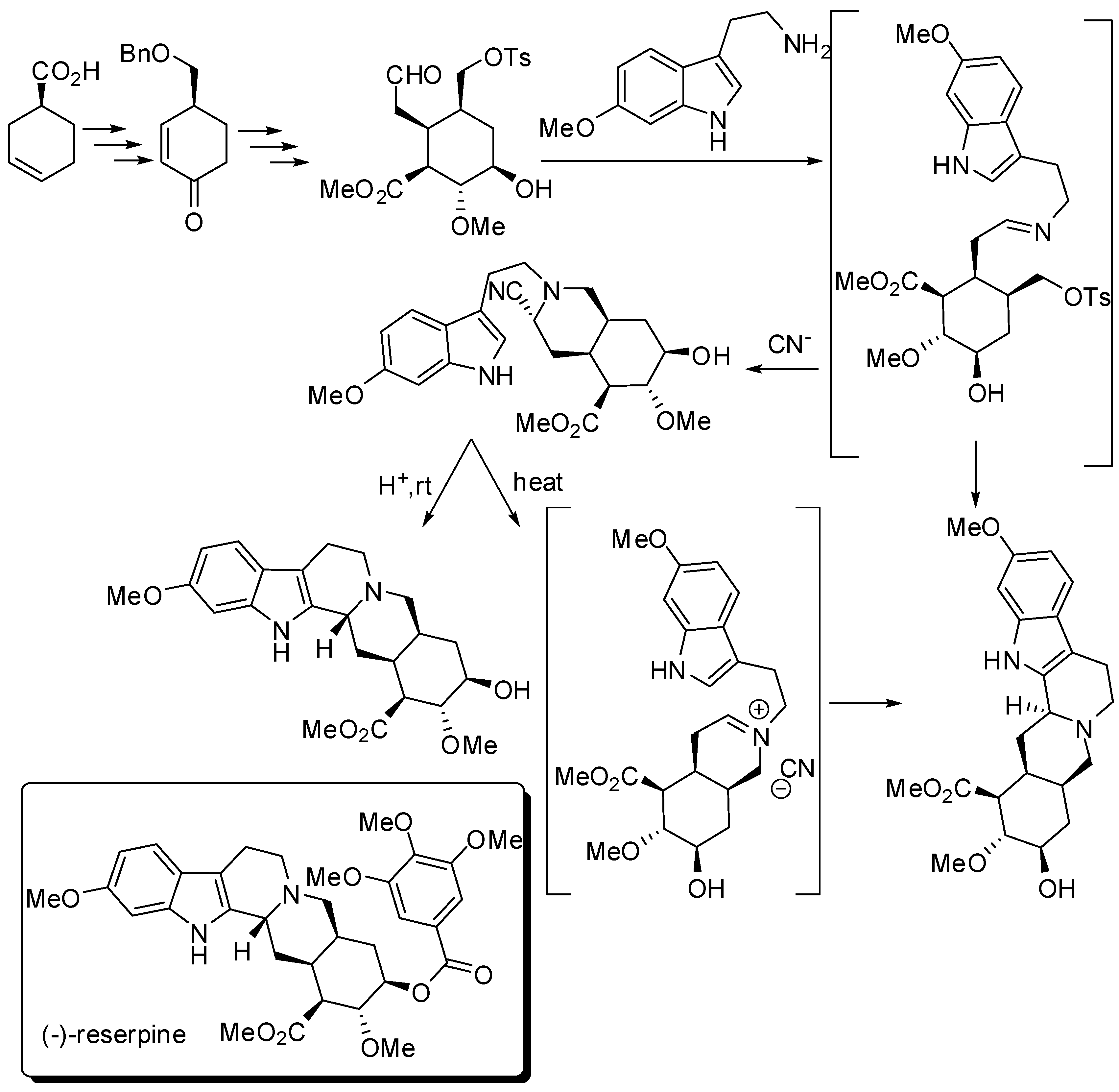
- Stork, G.; Tang, P.C.; Casey, M.; Goodman, B.; Toyota, M. Regiospecific and stereoselective syntheses of (±)-reserpine and (−)-reserpine. J. Am. Chem. Soc. 2005, 127, 16255–16262. [Google Scholar] [CrossRef] [PubMed]
- Lood, C.S.; Nieger, M.; Koskinen, A.M.P. Enantiospecific gram scale synthesis of (S)-eleagnine. Tetrahedron 2015, 71, 5019–5024. [Google Scholar] [CrossRef]
- Lood, C.S.; Koskinen, A.M.P. Synthesis of (S)- and (R)-harmicine from proline: An approach toward tetrahydro-β-carbolines. Eur. J. Org. Chem. 2014, 2014, 2357–2364. [Google Scholar] [CrossRef]
- Reddy, M.S.; Cook, J.M. The synthesis of roeharmine and (−)-1,2,3,4-tetrahydroroeharmine. Tetrahedron Lett. 1994, 35, 5413–5416. [Google Scholar] [CrossRef]
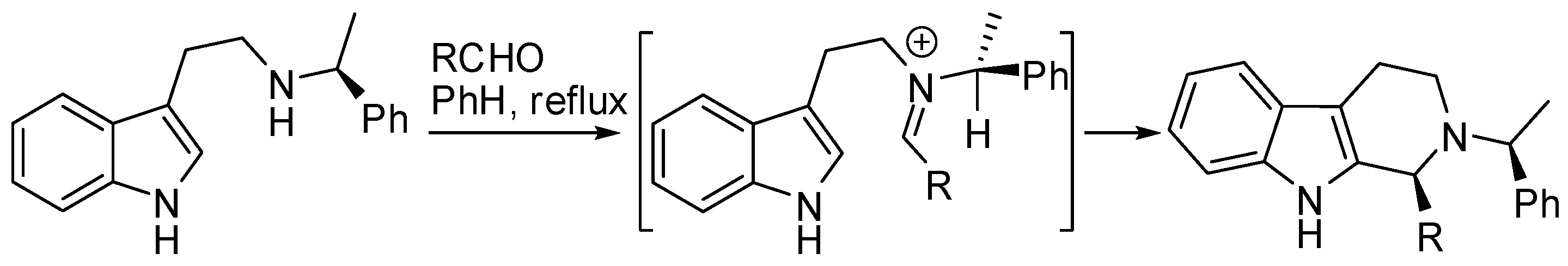
- Soe, T.; Kawate, T.; Fukui, N.; Hino, T.; Nakagawa, M. Asymmetric Pictet–Spengler reaction using α−methylbenzylamine as a chiral auxiliary group. Heterocycles 1996, 42, 347–358. [Google Scholar] [CrossRef]
- Kawate, T.; Yamanaka, M.; Nakagawa, M. Chiral auxiliary approach to the asymmetric Pictet–Spengler reaction of tryptamines. Heterocycles 1999, 50, 1033–1039. [Google Scholar]
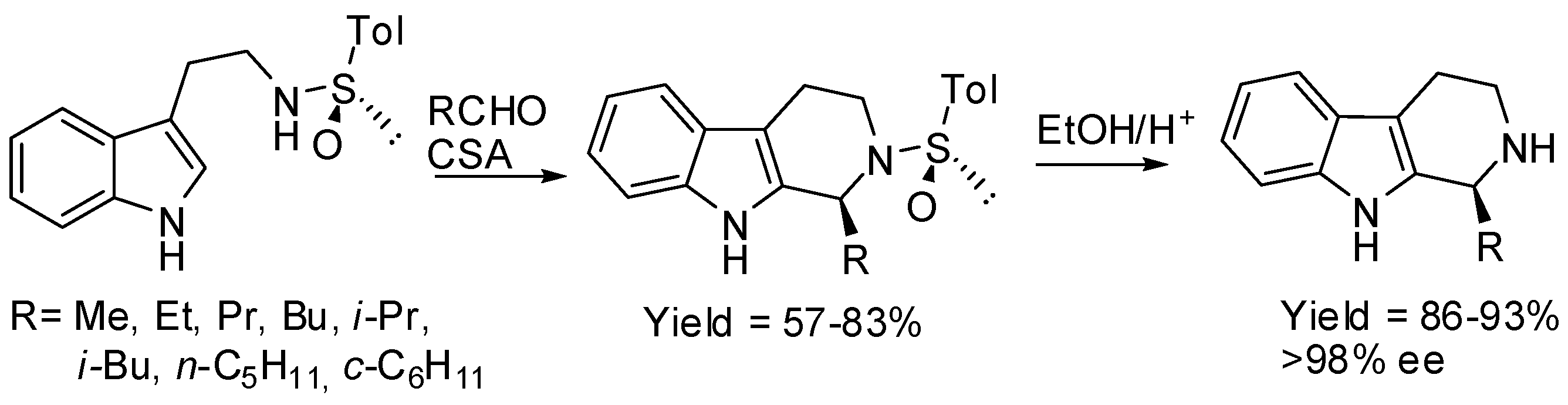
- Gremmen, C.; Willemse, B.; Wanner, M.J.; Koomen, G.-J. Enantiopure tetrahydro−β−carbolines via Pictet–Spengler reactions with N-sulfinyl tryptamines. Org. Lett. 2000, 2, 1955–1958. [Google Scholar] [CrossRef] [PubMed]
- Tsuji, R.; Nakagawa, M.; Nishida, A. An efficient synthetic approach to optically active b-carboline derivatives via Pictet–Spengler reaction promoted by trimethylchlorosilane. Tetrahedron Asymmetry 2003, 14, 177–180. [Google Scholar] [CrossRef]
- Schmidt, G.; Waldmann, H.; Henke, H.; Burkard, M. Asymmetric control in the Pictet–Spengler reaction by means of N-protected amino acids as chiral auxiliary groups. Chem. Eur. J. 1996, 2, 1566–1571. [Google Scholar] [CrossRef]
- Bi, L.; Zhao, M.; Wang, C.; Peng, S.; Winterfeldt, E. Diasteroselective cyclizations with enantiopure malonaldehyde monocycloacetals. J. Org. Chem. 2002, 67, 22–26. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yin, W.; Zhou, H.; Cook, J.M. Total synthesis of the opioid agonistic indole alkaloid mitragynine and the first total syntheses of 9-methoxygeissoschizol and 9-methoxy-Nb-methylgeissoschizol. Org. Lett. 2007, 9, 3491–3494. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Yin, W.; Zhou, H.; Liao, X.; Cook, J.M. General approach to the total synthesis of 9-methoxy-substituted indole alkaloids: Synthesis of mitragynine, as well as 9-methoxygeissoschizol and 9-methoxy-Nb-methylgeissoschizol. J. Org. Chem. 2009, 74, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Edwankar, C.R.; Edwankar, R.V.; Namjoshi, O.A.; Liao, X.; Cook, J.M. Stereospecific approach to the synthesis of ring-A oxygenated sarpagine indole alkaloids. Total synthesis of the dimeric indole alkaloid P-(+)-dispegatrine and six other monomeric indole alkaloids. J. Org. Chem. 2013, 78, 6471–6487. [Google Scholar] [CrossRef] [PubMed]
- Amat, M.; Subrizi, F.; Elias, V.; Llor, N.; Molins, E.; Bosch, J. Cyclocondensation reactions between 2-acyl-3-indoleacetic acid derivatives and phenylglycinol: Enantioselective synthesis of 1-substituted tetrahydro-β-carboline alkaloids. Eur. J. Org. Chem. 2012, 2012, 1835–1842. [Google Scholar] [CrossRef]
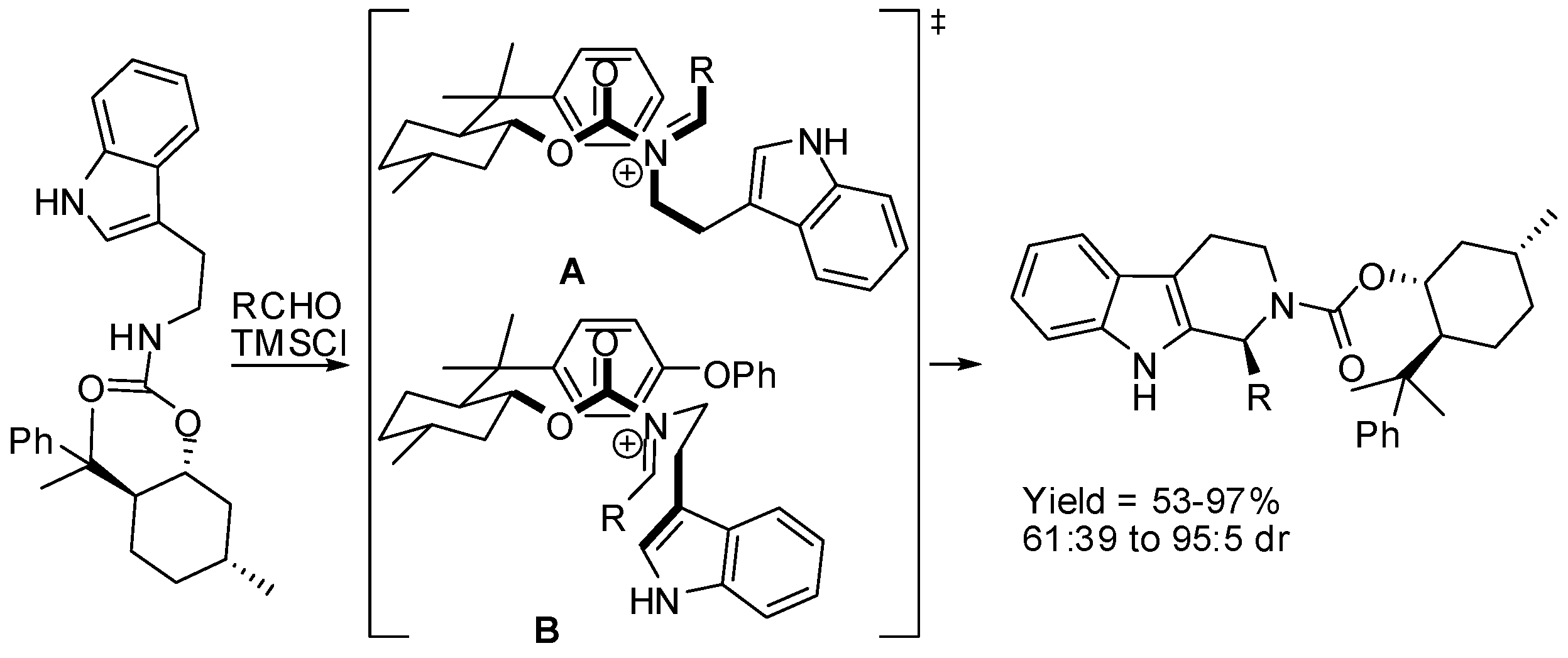
- Franzén, J.; Fisher, A. Asymmetric Alkaloid Synthesis: A one-pot organocatalytic reaction to quinolizidine derivatives. Angew. Chem. Int. Ed. 2009, 48, 787–791. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Franzén, J. Diverse asymmetric quinolizidine synthesis: A stereodivergent one-pot approach. Adv. Synth. Catal. 2010, 352, 499–518. [Google Scholar] [CrossRef]
- Zhang, W.; Bah, J.; Wohlfarth, A.; Franzén, J. A stereodivergent strategy for the preparation of corynantheine and ipecac alkaloids, their epimers, and analogues: Efficient total synthesis of (−)-dihydrocorynantheol, (−)-corynantheol, (−)-protoemetinol, (−)-corynantheal, (−)-protoemetine, and related natural and nonnatural compounds. Chem. Eur. J. 2011, 17, 13814–13824. [Google Scholar] [PubMed]
- Dai, X.; Wu, X.; Fang, H.; Nie, L.; Chen, J.; Deng, H.; Cao, W.; Zhao, G. Enantioselective organocatalyzed cascade reactions to highly functionalized quinolizidines. Tetrahedron 2011, 67, 3034–3040. [Google Scholar] [CrossRef]
- Fang, H.; Wu, X.; Nie, L.; Dai, X.; Chen, J.; Cao, W.; Zhao, G. Diastereoselective syntheses of indoloquinolizidines by a Pictet-Spengler/lactamization cascade. Org. Lett. 2010, 12, 5366–5369. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dai, X.; Nie, L.; Fang, H.; Chen, J.; Ren, Z.; Cao, W.; Zhao, G. Organocatalyzed enantioselective one-pot three-component access to indoloquinolizidines by a Michael addition-Pictet–Spengler sequence. Chem. Commun. 2010, 46, 2733–2735. [Google Scholar] [CrossRef] [PubMed]
- Rueping, M.; Volla, C.M.R.; Bolte, M.; Raabe, G. General and efficient organocatalytic synthesis of indoloquinolizidines, pyridoquinazolines and quinazolinones through a one-pot domino Michael addition-cyclization- Pictet–Spengler or 1,2-amine addition reaction. Adv. Synth. Catal. 2011, 353, 2853–2859. [Google Scholar] [CrossRef]
- Zhu, H.-L.; Ling, J.-B.; Xu, P.-F. α-Oxo-γ-butyrolactam, N-containing pronucleophile in organocatalytic one-pot assembly of butyrolactam-fused indoloquinolizidines. J. Org. Chem. 2012, 77, 7737–7743. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.; Luan, H.-L.; Lin, H.; Sun, X.-W.; Yang, X.-D.; Dong, H.-Q.; Lin, G.-Q. One-pot enantioselective construction of indoloquinolizidine derivatives bearing five contiguous stereocenters using aliphatic aldehydes, nitroethylenes, and tryptamine. Chem. Commun. 2014, 50, 10027–10030. [Google Scholar] [CrossRef] [PubMed]
- Arai, T.; Wasai, M.; Yokoyama, N. Easy access to fully functionalized chiral tetrahydro-β-carboline alkaloids. J. Org. Chem. 2011, 76, 2909–2912. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.-R.; Wu, H.; Xiang, B.; Yu, W.-B.; Han, L.; Jia, Y.-X. Highly enantioselective construction of trifluoromethylated all-carbon quaternary stereocenters via nickel-catalyzed Friedel-Crafts alkylation reaction. J. Am. Chem. Soc. 2013, 135, 2983–2986. [Google Scholar] [CrossRef] [PubMed]
- Wu, X.; Dai, X.; Fang, H.; Nie, L.; Chen, J.; Cao, W.; Zhao, G. One-pot three-component syntheses of indoloquinolizidine derivatives using an organocatalytic Michael addition and subsequent Pictet–Spengler cyclization. Chem. Eur. J. 2011, 17, 10510–10514. [Google Scholar] [CrossRef] [PubMed]
- Fischereder, E.-M.; Pressnitz, D.; Kroutil, W. Stereoselective cascade to C3-methylated strictosidine derivatives employing transaminases and strictosidine synthases. ACS Catal. 2016, 6, 23–30. [Google Scholar] [CrossRef]


























© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dalpozzo, R. The Chiral Pool in the Pictet–Spengler Reaction for the Synthesis of β-Carbolines. Molecules 2016, 21, 699. https://doi.org/10.3390/molecules21060699
Dalpozzo R. The Chiral Pool in the Pictet–Spengler Reaction for the Synthesis of β-Carbolines. Molecules. 2016; 21(6):699. https://doi.org/10.3390/molecules21060699
Chicago/Turabian StyleDalpozzo, Renato. 2016. "The Chiral Pool in the Pictet–Spengler Reaction for the Synthesis of β-Carbolines" Molecules 21, no. 6: 699. https://doi.org/10.3390/molecules21060699
APA StyleDalpozzo, R. (2016). The Chiral Pool in the Pictet–Spengler Reaction for the Synthesis of β-Carbolines. Molecules, 21(6), 699. https://doi.org/10.3390/molecules21060699