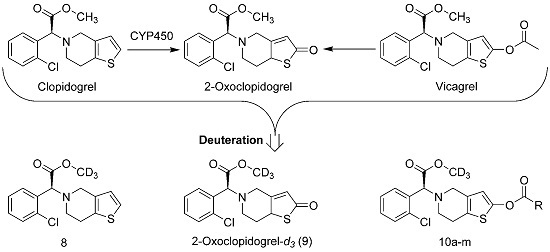
3.2. Synthesis of Deuterated Clopidogrel- and Vicagrel-Related Compounds
Methyl-d3 (R)-2-(2-chlorophenyl)-2-hydroxyacetate (4). To a stirred mixture of (R)-2-(2-chlorophenyl)-2-hydroxyacetic acid (3) (110.0 g, 0.59 mol, 99.5% ee) and methanol-d4 (110.0 g, 3.05 mol, 99.5% atom) at room temperature, a 1,6-dioxane solution of hydrogen chloride (3.0 mL, 3.75 mol/L) was added. The mixture was heated and refluxed for 5 h. After adding sodium bicarbonate (1.0 g), the mixture was distilled under atmospheric pressure and the methanol-d4 (74 g) recovered. The residue was mixed with dichloromethane (300 mL), washed sequentially with 5% sodium bicarbonate solution (300 mL) and brine (300 mL), dried over anhydrous sodium sulfate, and concentrated under vacuum to afford the title compound as a colorless oil (111.8 g, 93.0% yield). 1H-NMR (400 MHz, CDCl3): δ 3.69 (d, 1H, J = 5.2 Hz), 5.60 (d, 1H, J = 5.2 Hz), 7.27–7.32 (m, 2H), 7.39–7.44 (m, 2H). LRMS m/z 226.1 [M + Na]+.
Methyl-d3 (R)-2-(2-Chlorophenyl)-2-(4-nitrophenylsulfonyloxy)-acetate (5). To a stirred mixture of 4 (110 g, 0.540 mol), 4-nitrobenzenesulfonyl chloride (131.6 g, 0.594 mol), 2,6-dimethylpyridine (6.59 g, 0.054 mol) and dichloromethane (800 mL) at 0 °C was slowly added Et3N (60.6 g, 0.600 mol). After stirring for 2 h at the same temperature, the mixture was quenched with 0.5 N hydrochloric acid (400 mL). The organic layer was separated, washed with brine (800 mL), dried over anhydrous sodium sulfate, and concentrated under vacuum. The residue was recrystallized in methanol to afford the title compound as a light yellow solid (172.7 g, 82.2% yield), mp 70.8–72.1 °C, −60.5 ° (c 0.5, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ 6.41 (s, 1H), 7.24–7.28 (m, 1H), 7.30–7.41 (m, 3H), 8.09 (d, 2H, J = 8.8 Hz), 8.33 (d, 2H, J = 8.8 Hz). LRMS m/z 411.1 [M + Na]+.
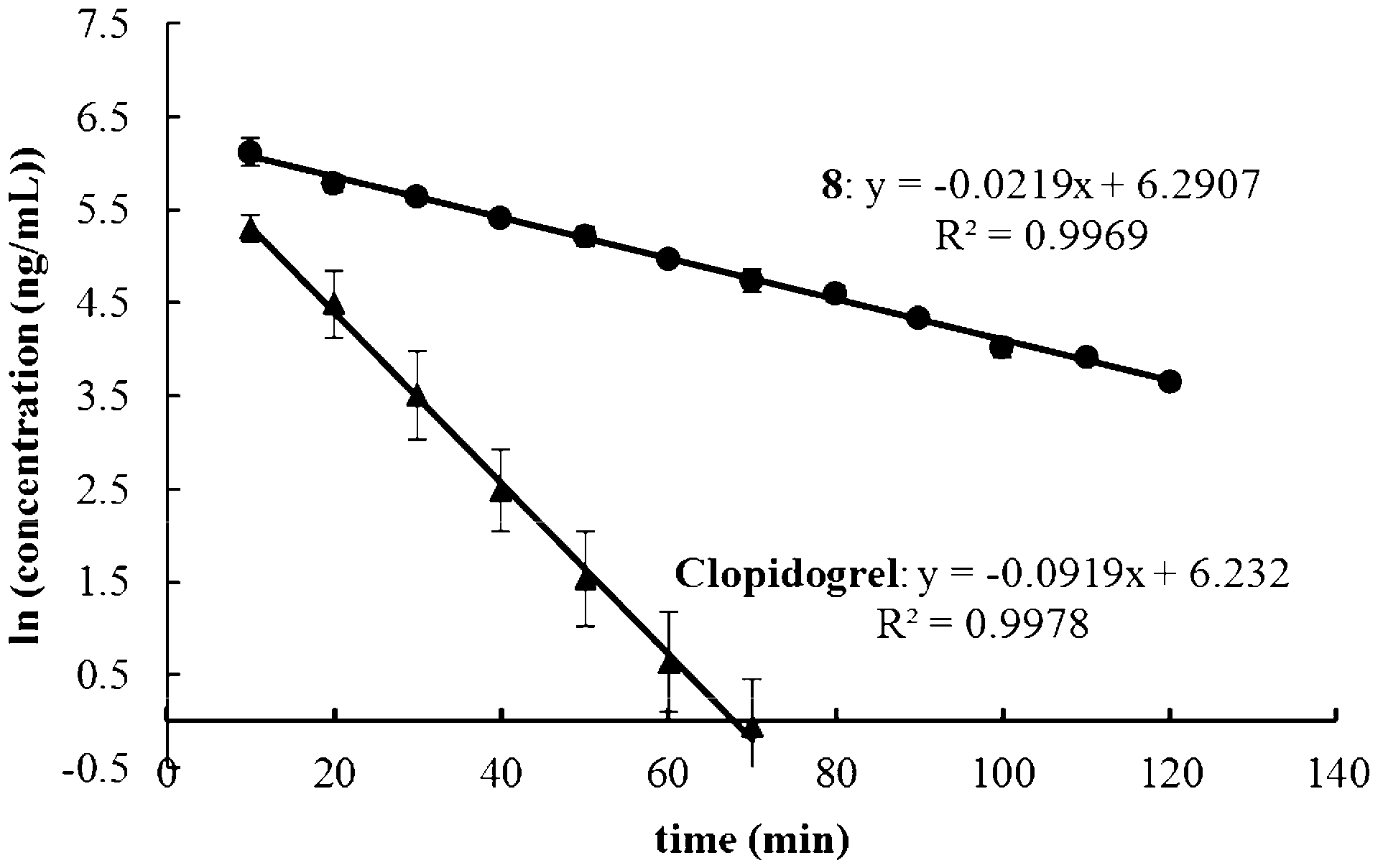
Methyl-d3 (S)-2-(2-chlorophenyl)-2-(6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)acetate bisulfate (8). To a solution of 5 (10.00 g, 25.7 mmol) in acetone (80 mL) were added 4,5,6,7-tetrahydrothieno[3,2-c]pyridine hydrochloride (6) (4.39 g, 25.0 mmol) and potassium carbonate (7.00 g). After refluxing for 6 h, the mixture was cooled, filtered and mixed with sulfuric acid (2.60 g). After stirring for 2 h at 0 °C, the precipitate was filtered and recrystallized in purified water and acetone to afford the title compound as an off-white crystalline powder (8.88 g, 84.6% yield), mp 177.3–178.2 °C, +44.1° (c 0.5, MeOH), 99.6% ee (chiral HPLC analytical conditions: Shinwa ULTRON ES-OVM, 10 μm × 4.6 mm × 250 mm, eluting with 20% acetonitrile +80% 0.01 mol/L potassium dihydrogen phosphate solution, flow rate 1.0 mL/ min, column temperature 30 °C, detection UV 220 nm). 1H-NMR (400 MHz, CD3OD): δ 3.15–3.21 (m, 2H), 3.63–3.70 (m, 1H), 3.79 (s, 1H), 4.13–4.16 (m, 1H), 4.33 (s, 1H), 5.76 (s, 1H), 6.73 (d, 1H, J = 5.2 Hz), 7.30 (d, 1H, J = 5.2 Hz), 7.42–7.46 (m, 1H), 7.49–7.60 (m, 3H). 13C-NMR (100 MHz, CD3OD): δ 167.2, 135.3, 132.7, 131.5, 130.9, 130.1, 128.5, 126.8, 126.7, 125.5, 124.8, 65.9, 48.1, 47.9, 47.7, 21.7. HRMS calculated for C16H14D3ClNO2S [M + H]+ m/z 325.0851, found 325.0915.
Methyl-d3 (2S)-2-(2-chlorophenyl)-2-(2-oxo-2,6,7,7a-tetrahydrothieno[3,2-c]pyridin-5(4H)-yl)acetate (9). To a solution of 5 (155.5 g, 0.400 mol) in acetonitrile (1000 mL), 5,6,7,7a-tetrahydrothieno[3,2-c]pyridin-2(4H)-one hydrochloride (7) (77.7 g, 0.405 mol) and potassium carbonate (110 g) were added. After stirring at room temperature for 22 h, the mixture was filtered and concentrated under vacuum. The residue, a dark-yellow solid, was recrystallized in ethanol to afford the title compound as a light-yellow powder (113.9 g, 83.5% yield), mp 143.9–145.0 °C. +106.6 ° (c 0.5, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ 1.89 (dq, 1H, J1 = 8.8 Hz, J2 = 4.0 Hz), 2.35–2.39 (m, 1H), 2.62 (dt, 1H, J1 = 10.4 Hz, J2 = 2.0 Hz), 3.04 (d, 1H, J = 12.0 Hz), 3.26 (d, 1H, J = 12.0 Hz), 3.94 (dd, 1H, J1 = 10.8 Hz, J2 = 1.6 Hz), 4.16–4.20 (m, 1H), 4.93 (s, 1H), 6.05 (s, 1H), 7.30–7.34 (m, 2H), 7.43–7.46 (m, 1H), 7.54–7.56 (m, 1H). 13C-NMR (100 MHz, CDCl3): δ 198.6, 170.8, 167.1, 134.8, 132.8, 130.1, 129.8, 129.7, 127.2, 126.9, 77.2, 67.3, 51.7, 51.1, 49.6, 33.81. HRMS calculated for C16H14D3ClNO3S [M + H]+ m/z 341.0806, found 341.0817.
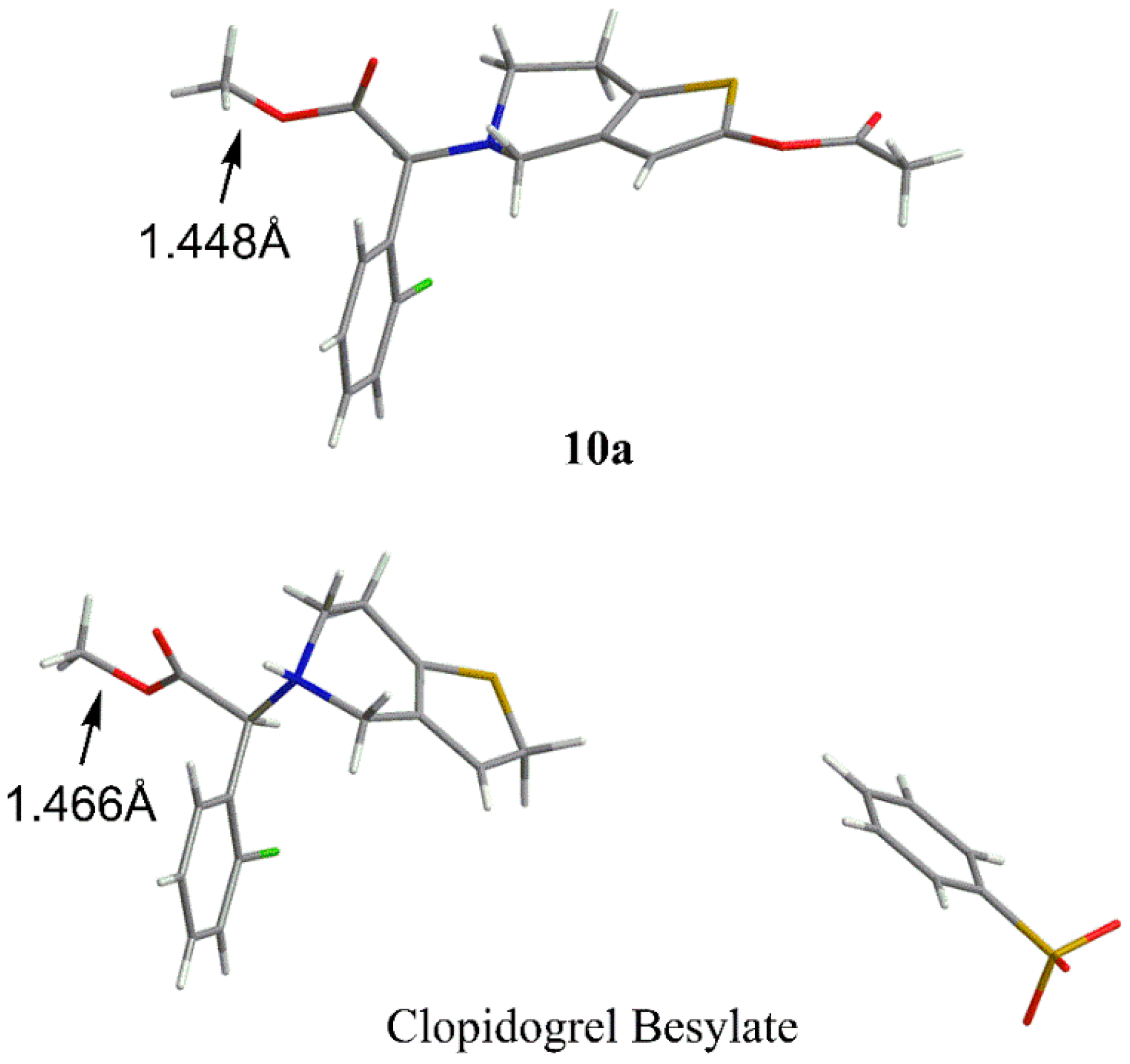
Methyl-d3 (S)-2-(2-acetoxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2chlorophenyl)acetate (10a). To a stirred mixture of 9 (10.00 g, 29.3 mmol) and DIPEN (7.60 g, 58.8 mmol) in dichloromethane (100 mL), acetic anhydride (6.00 g, 58.8 mmol) was slowly added at 0 °C. The mixture was stirred for 3 h at room temperature and quenched with ice water. The organic layer was washed with 5% sodium bicarbonate solution and brine and dried over anhydrous sodium sulfate. The organic layer was concentrated under vacuum and the residue was purified by column chromatography (ethyl acetate and hexane) to afford a light yellow oil which was recrystallized from ethanol to afford the title compound as a white crystalline powder (8.35 g, 74.2% yield), mp 75.9–77.3 °C, 100% ee. Chiral HPLC analytical conditions: Chiralpak AD-H, 4.6 mm × 250 mm, eluting with 90% n-hexane and 10% isopropanol, flow rate 1.0 mL/min, column temperature 30 °C, detection UV 220 nm. +24.6 ° (c 0.5, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ 2.26 (s, 3H), 2.77 (t, 2H, J = 5.2 Hz), 2.89 (t, 2H, J = 5.2 Hz), 3.60 (dd, 2H, J1 = 34.0 Hz, J2 = 14.4 Hz), 4.91 (s, 1H), 6.26 (s, 1H), 7.25–7.32 (m, 2H), 7.40–7.42 (m, 1H), 7.68–7.70 (m, 1H). 13C-NMR (100 MHz, CDCl3): δ 171.3, 167.8, 149.5, 134.7, 133.7, 129.9, 129.8, 129.4, 129.2, 127.2, 125.8, 112.0, 77.2, 67.8, 50.3, 48.1, 25.0, 20.7. HRMS calculated for C18H16D3NO4SCl [M + H]+ m/z 383.0912, found 383.0930.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl propionate hydrochloride (10b). To a solution of 9 (2.00 g, 5.87 mmol) and DIPEN (1.78 g, 17.6 mmol) in dichloromethane (20 mL), propionyl chloride (1.63 g, 17.6 mmol) was slowly added at 0 °C. After stirring at room temperature for 1 h, the mixture was poured into ice and 5% bicarbonate solution. The organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated under vacuum. The residue was purified by column chromatography (ethyl acetate and hexane) to give a light yellow oil which was dissolved in ethyl ether to which 1,6-dioxane solution of hydrogen chloride (1 mL, 3.75 M) was added. The precipitate was filtered to offer the title compound as a light yellow powder (1.64 g, 64.4% yield), mp 87.9–90.3 °C, 98.7% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 1.27 (t, 3H, J = 7.6 Hz), 2.61 (q, 2H, J = 7.6 Hz), 2.88–3.08 (m, 2H), 3.28 (bs, 1H), 3.67–3.83 (m, 2H), 4.24 (bs, 1H), 5.55 (s, 1H), 6.39 (s, 1H), 7.43–7.52 (m, 3H), 8.31 (s, 1H), 14.1 (bs, 1H). 13C-NMR (100 MHz, CDCl3): δ 170.8, 166.3, 151.5, 135.0, 132.0, 131.0, 130.6, 130.5, 128.8, 128.7, 123.0, 110.8, 77.3, 67.1, 58.4, 53.5, 27.3, 18.4, 8.8. HRMS calculated for C19H18D3NO4SCl [M + H]+ m/z 397.1069, found 397.1237.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl butyrate (10c). To a solution of 9 (2.00 g, 5.87 mmol) and DIPEN (1.78 g, 17.6 mmol) in dichloromethane (20 mL), butyryl chloride (1.87, 17.6 mmol) was slowly added at 0 °C. After stirring at room temperature for 1 h, the mixture was poured into ice and 5% bicarbonate solution. The organic layer was washed with brine, dried over anhydrous sodium sulfate, and concentrated under vacuum. The residue was purified by column chromatography (ethyl acetate and hexane) to give a light yellow oil which was recrystallized from ethanol to offer the title compound as an off-white crystalline powder (0.88 g, 36.5% yield), mp 64.2–65.4 °C, 99.8% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 1.02 (t, 3H, J = 7.6 Hz), 1.77 (sext, 2H, J = 7.6 Hz), 2.53 (t, 2H, J = 7.6 Hz), 2.81 (s, 2H), 2.91 (s, 2H), 3.58 (s, 1H), 3.66 (s, 1H), 4.94 (s, 1H), 6.28 (s, 1H), 7.30–7.35 (m, 2H), 7.42–7.45 (m, 1H), 7.72 (s, 1H). 13C-NMR (100 MHz, CDCl3): δ 171.2, 170.4, 149.7, 134.7, 133.6, 130.0, 129.8, 129.5, 127.2, 125.6, 111.7, 100.0, 77.2, 67.8, 50.3, 48.2, 35.8, 25.0, 18.2, 13.6. HRMS calculated for C20H20D3NO4SCl [M + H]+ m/z 411.1225, found 411.1292.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl isobutyrate hydrochloride (10d). A similar procedure to that described for the preparation of 10b was followed, except that an equivalent amount of isobutyryl chloride was used in place of propionyl chloride. The title compound was obtained as a white powder (1.94 g, 73.9% yield), mp 146.7–149.9 °C, 99.2% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 1.30 (d, 6H, J = 6.8 Hz), 2.80 (hept, 2H, J = 6.8 Hz), 3.04–4.76 (m, 6H), 5.56 (s, 1H), 6.39 (s, 1H), 7.42–7.51 (m, 3H), 8.32 (d, 1H, J = 6.0 Hz), 14.08 (bs, 1H). 13C-NMR (100 MHz, CDCl3): δ 173.3, 166.2, 151.8, 134.9, 132.1, 131.1, 130.5, 128.8, 126.7, 122.8, 121.7, 110.6, 77.3, 62.6, 53.5, 53.4, 33.9, 19.9, 18.7. HRMS calculated for C20H20D3NO4SCl [M + H]+ m/z 411.1225, found 411.1299.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl cyclopro-panecarboxylate (10e). A similar procedure to that described for the preparation of 10c as followed, except that an equivalent amount of cyclopropanecarbonyl chloride was used in place of butyryl chloride. The title compound was obtained as a white powder (0.70 g, 29.2% yield), mp 58.4–60.8 °C, 99.7% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 1.02–1.07 (m, 2H), 1.16–1.20 (m, 2H), 1.78–1.84 (m, 1H), 2.79 (t, 2H, J = 5.6 Hz), 2.89 (t, 2H, J = 5.2 Hz), 3.61 (dd, 2H, J1 = 36.8 Hz, J2 = 14.4 Hz), 4.92 (s, 1H), 6.28 (s, 1H), 7.26–7.34 (m, 2H), 7.42–7.44 (m, 1H), 7.70 (dd, 1H, J1 = 5.6 Hz, J2 = 1.6 Hz). 13C-NMR (100 MHz, CDCl3): δ 171.9, 171.2, 149.8, 134.7, 133.6, 129.9, 129.8, 129.5, 129.0, 127.2, 125.6, 111.7, 77.3, 67.8, 50.4, 48.2, 24.9, 12.7, 9.7. HRMS calculated for C20H18D3NO4SCl [M + H]+ m/z 409.1069, found 409.1157.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl pivalate (10f). A similar procedure to that described for the preparation of 10c was followed, except that an equivalent amount of pivaloyl chloride was used in place of butyryl chloride. The title compound was obtained as a light yellow powder (1.57 g, 62.9% yield), mp 105.4–107.1 °C, 99.4% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 1.33 (s, 9H), 2.80 (t, 2H, J = 5.2 Hz), 2.90 (t, 2H, J = 4.4 Hz), 3.61 (dd, 2H, J1 = 36.4 Hz, J2 = 14.0 Hz), 4.93 (s, 1H), 6.28 (s, 1H), 7.27–7.34 (m, 2H), 7.42–7.44 (m, 1H), 7.71 (d, 1H, J = 6.0 Hz). 13C-NMR (100MHz, CDCl3): δ 175.2, 171.2, 150.1, 134.7, 133.7, 129.9, 129.8, 129.5, 129.0, 127.2, 125.6, 111.4, 77.3, 67.8, 50.4, 48.2, 39.1, 27.0, 24.9. HRMS calculated for C21H22D3NO4SCl [M + H]+ m/z 425.1382, found 411.1472.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl hexanoate (10g). A similar procedure to that described for the preparation of 10c was followed, except that an equivalent amount of hexanoyl chloride was used in place of butyryl chloride. The title compound was obtained as a white powder (0.89 g, 34.5% yield), mp 49.4–52.7 °C, 99.8% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 0.93 (t, 3H, J = 7.2 Hz), 1.32–1.41 (m, 4H), 1.73 (quint, 2H, J = 7.2 Hz), 2.53 (t, 2H, J = 7.2 Hz), 2.79 (t, 2H, J = 4.8 Hz), 2.90 (t, 2H, J = 5.2 Hz), 3.61 (dd, 2H, J1 = 36.0 Hz, J2 = 14.4 Hz), 4.92 (s, 1H), 6.28 (s, 1H), 7.26–7.34 (m, 2H), 7.42–7.44 (m, 1H), 7.70 (dd, 1H, J1 = 4.8 Hz, J2 = 2.0 Hz). 13C-NMR (100 MHz, CDCl3): δ 171.3, 170.7, 149.7, 134.7, 133.7, 129.9, 129.8, 129.5, 129.2, 127.2, 125.7, 111.7, 77.3, 67.9, 50.4, 48.2, 33.9, 31.2, 25.0, 24.4, 22.3, 13.9. HRMS calculated for C22H24D3NO4SCl [M + H]+ m/z 439.1538, found 439.1651.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl 2,2-dimethylbutanoate (10h). A similar procedure to that described for the preparation of 10c was followed, except that an equivalent amount of 2,2-dimethylbutanoyl chloride was used in place of butyryl chloride. The title compound was obtained as a white powder (1.85 g, 71.8% yield), mp 99.4–100.3 °C, 99.8% ee. Chiral HPLC analytical conditions were the same as those for 10a. 1H-NMR (400 MHz, CDCl3): δ 0.91 (t, 3H, J = 7.6 Hz), 1.28 (s, 6H), 1.79 (q, 2H, J = 7.6 Hz), 2.79 (t, 2H, J = 5.2 Hz), 2.90 (t, 2H, J = 4.8 Hz), 3.61 (dd, 2H, J1 = 34.8 Hz, J2 = 14.4 Hz), 4.92 (s, 1H), 6.28 (s, 1H), 7.26–7.33 (m, 2H), 7.41–7.44 (m, 1H), 7.70 (dd, 1H, J1 = 5.2 Hz, J2 = 2.0 Hz). 13C-NMR (100 MHz, CDCl3): δ 174.8, 171.3, 150.0, 134.7, 133.8, 129.9, 129.8, 129.5, 129.1, 127.2, 125.6, 111.5, 77.3, 67.8, 50.4, 48.2, 43.1, 33.4, 25.0, 24.5, 9.3. HRMS calculated for C22H24D3NO4SCl [M + H]+ m/z 439.1538, found 439.1628.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl laurate (10i). A similar procedure to that described for the preparation of 10c was followed, except that an equivalent amount of lauroyl chloride was used in place of butyryl chloride. The title compound was obtained as a white crystalline powder (0.72 g, 23.4% yield), mp 35.8–37.6 °C, 99.7% ee. Chiral HPLC analytical conditions were the same as those for 10a. 1H-NMR (400 MHz, CDCl3): δ 0.90 (t, 3H, J = 7.2 Hz), 1.25–1.39 (m, 16H), 1.73 (quint, 2H, J = 7.6 Hz), 2.53 (t, 2H, J = 7.6 Hz), 2.79 (s, 2H), 2.90 (s, 2H), 3.61 (dd, 2H, J1 = 35.6 Hz, J2 = 14.4 Hz), 4.92 (s, 1H), 6.28 (s, 1H), 7.28–7.34 (m, 2H), 7.42–7.44 (m, 1H), 7.71 (d, 1H, J = 6.0 Hz). 13C-NMR (100 MHz, CDCl3): δ 170.7, 155.1, 149.7, 134.7, 133.7, 129.9, 129.8, 129.5, 127.2, 125.7, 125.6, 111.7, 77.2, 67.9, 50.4, 48.2, 34.0, 31.9, 29.7, 29.6, 29.4, 29.3, 29.2, 29.0, 25.0, 24.7, 22.7, 14.1. HRMS calculated for C28H36D3NO4SCl [M + H]+ m/z 523.2477, found 523.2624.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl cinnamate (10j). A similar procedure to that described for the preparation of 10c was followed, except that an equivalent amount of cinnamoyl chloride was used in place of butyryl chloride. The title compound was obtained as a yellow powder (1.12 g, 40.5% yield), mp 121.7–123.1 °C, 99.8% ee. Chiral HPLC analytical conditions were the same as those for 10a. 1H-NMR (400 MHz, CDCl3): δ 2.85 (s, 2H), 2.98 (s, 2H), 3.66 (s, 1H), 3.73–3.76 (m, 1H), 5.00 (s, 1H), 6.39 (s, 1H), 6.59 (d, 1H, J = 16.0 Hz), 7.31–7.36 (m, 2H), 7.44 (s, 1H), 7.46 (s, 3H), 7.59 (t, 2H, J = 4.0 Hz), 7.79 (s, 1H), 7.88 (d, 1H, J = 16.0 Hz). 13C-NMR (100 MHz, CDCl3): δ 171.3, 163.7, 149.7, 147.4, 134.7, 134.0, 133.7, 131.0, 130.0, 129.8, 129.5, 129.1, 129.0, 128.4, 127.2, 125.9, 116.0, 111.8, 77.3, 67.8, 50.4, 48.2, 25.0. HRMS calculated for C25H20D3NO4SCl [M + H]+ m/z 471.1225, found 471.1328.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl benzoate hydrochloride (10k). A similar procedure similar to that described for the preparation of 10b was followed, except that an equivalent amount of benzoyl chloride was used in place of propionyl chloride. The title compound was obtained as an off-white powder (1.04 g, 39.8% yield), mp 158.7–161.9 °C, 97.6% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 3.12–4.85 (m, 6H), 5.63 (s, 1H), 6.57 (s, 1H), 7.44–7.56 (m, 5H), 7.68 (t, 1H, J = 7.6 Hz), 8.17 (d, 2H, J = 7.6 Hz), 8.37 (d, 1H, J = 7.6 Hz), 14.18 (bs, 1H). 13C-NMR (100 MHz, CDCl3): δ 166.1, 163.0, 151.8, 135.0, 134.4, 132.3, 131.3, 131.2, 130.6, 130.3, 128.9, 128.8, 127.8, 126.5, 123.1, 111.1, 77.3, 68.3, 59.4, 53.5, 19.8. HRMS calculated for C23H18D3NO4SCl [M + H]+ m/z 445.1069, found 445.1172.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl 2-(trifluoromethyl)benzoate (10l). A similar procedure to that described for the preparation of 10c was followed, except that an equivalent amount of 2-(trifluoromethyl)benzoyl chloride was used in place of butyryl chloride. The title compound was obtained as an off-white powder (1.37 g, 45.5% yield), mp 74.3–76.2 °C, 98.6% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 2.84 (s, 2H), 2.94 (s, 2H), 3.66 (dd, 2H, J1 = 30.8 Hz, J2 = 13.6 Hz), 4.96 (s, 1H), 6.47 (s, 1H), 7.30–7.35 (m, 2H), 7.43–7.45 (m, 1H), 7.68–7.73 (m, 3H), 7.82–7.86 (m, 1H), 7.96–7.99 (m, 1H). 13C-NMR (100 MHz, CDCl3): δ 170.8, 163.2, 149.4, 134.7, 132.2, 131.9, 131.0, 130.0, 129.9, 129.7, 129.5, 129.2, 129.0, 127.3, 127.1, 127.0, 124.5, 121.8, 112.4, 77.2, 67.5, 50.2, 48.2, 24.7. HRMS calculated for C24H17D3ClF3NO4S [M + H]+ m/z 513.942, found 513.1071.
(S)-5-(1-(2-Chlorophenyl)-2-(methoxy-d3)-2-oxoethyl)-4,5,6,7-tetrahydrothieno[3,2-c]pyridin-2-yl nicotinate (10m). A similar procedure to that described for the preparation of 10c was followed, except that an equivalent amount of nicotinoyl chloride was used in place of butyryl chloride. The title compound was obtained as an off-white powder (0.91 g, 34.7% yield), mp 92.1–94.8 °C, 99.4% ee. Chiral HPLC analytical conditions were the same as for 10a. 1H-NMR (400 MHz, CDCl3): δ 2.84 (t, 2H, J = 5.2 Hz), 2.95 (t, 2H, J = 5.2 Hz), 3.67 (dd, 2H, J1 = 33.2 Hz, J2 = 14.4 Hz), 4.96 (s, 1H), 6.48 (s, 1H), 7.30–7.35 (m, 2H), 7.43–7.50 (m, 2H), 7.72 (dd, 1H, J1 = 5.2 Hz, J2 = 1.6 Hz), 8.42 (dt, 1H, J1 = 8.0 Hz, J2 = 1.6 Hz), 8.85–8.88 (m, 1H), 9.36 (s, 1H). 13C-NMR (100 MHz, CDCl3): δ 171.2, 162.3, 154.3, 151.4, 149.3, 137.6, 134.8, 133.5, 130.0, 129.9, 129.6, 129.2, 127.3, 126.4, 124.7, 123.5, 112.5, 77.3, 67.7, 50.3, 48.1, 25.0. HRMS calculated for C22H16D3ClN2NaO4S [M + Na]+ m/z 468.0841, found 468.0950.
Methyl-d3 (R)-2-(2-acetoxy-6,7-dihydrothieno[3,2-c]pyridin-5(4H)-yl)-2-(2chlorophenyl)acetate (R-10a). A similar procedure to that described for the preparation of 10a was followed, except that an equivalent amount of methyl-d3 (2R)-2-(2-chlorophenyl)-2-(2-oxo-2,6,7,7a-tetrahydrothieno[3,2-c]pyridin-5(4H)-yl) acetate was used in place of 9. The title compound was obtained as an off-white crystalline powder (5.78 g, 52.1% yield), mp 73.8–75.7 °C, 100% ee. Chiral HPLC analytical conditions were the same as for 10a. −24.6 ° (c 0.5, CH2Cl2). 1H-NMR (400 MHz, CDCl3): δ 2.29 (s, 3H), 2.80 (s, 2H), 2.91 (s, 2H), 3.62 (dd, 2H, J1 = 32.4 Hz, J2 = 12.0 Hz), 4.94 (s, 1H), 6.28 (s, 1H), 7.29–7.34 (m, 2H), 7.42–7.44 (m, 1H), 7.72 (s, 1H). 13C-NMR (100 MHz, CDCl3): δ 171.2, 167.8, 149.6, 134.7, 133.6, 129.9, 129.8, 129.5, 129.2, 127.2, 125.8, 112.0, 77.3, 67.8, 50.3, 48.2, 25.0, 20.7. HRMS calculated for C18H16D3NO4SCl [M + H]+ m/z 383.0912, found 383.0927.









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}