
Activating and Attenuating the Amicoumacin Antibiotics


Abstract
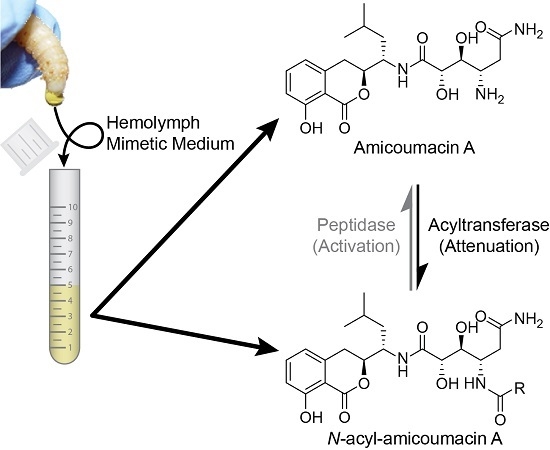
:
1. Introduction
2. Results and Discussion
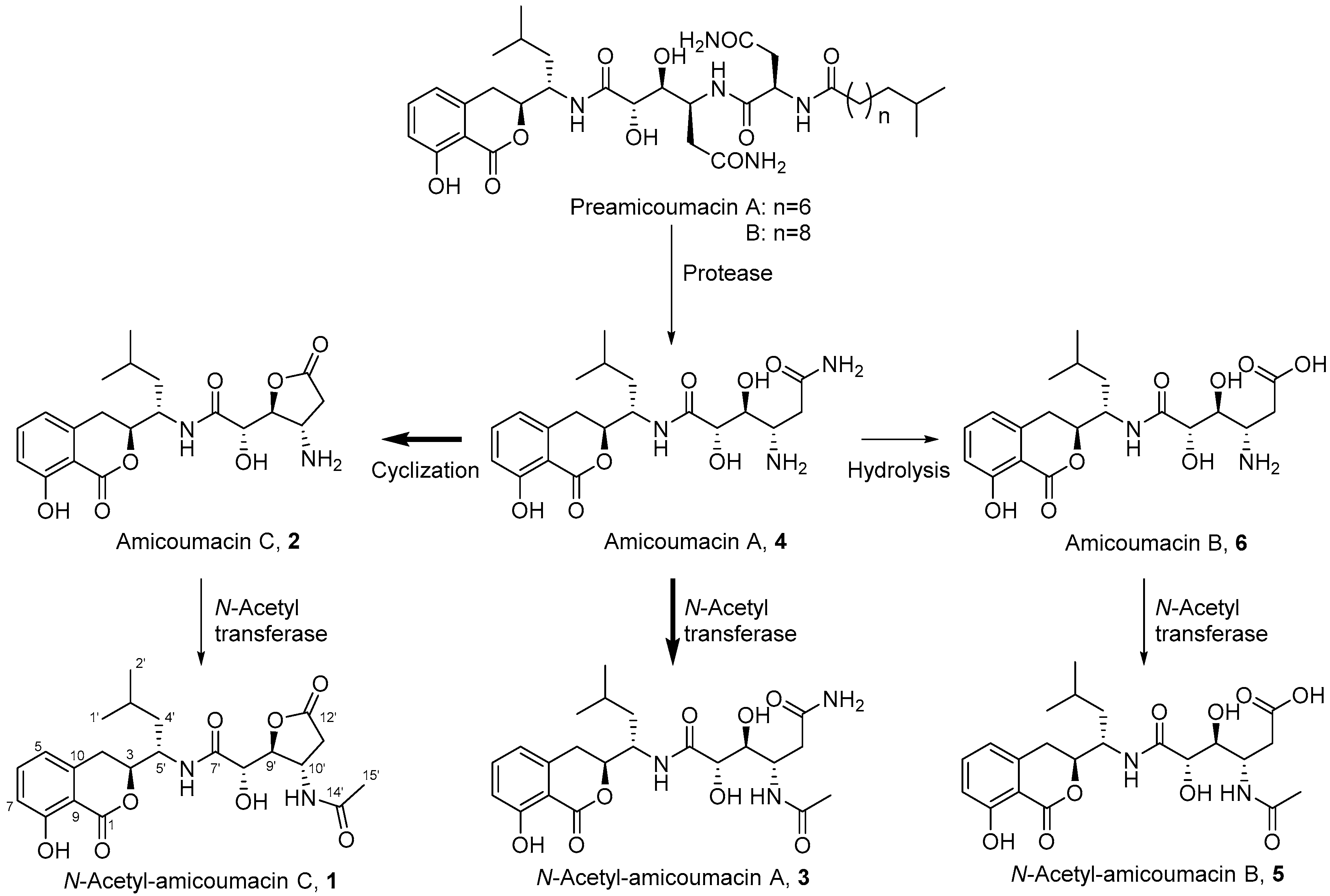
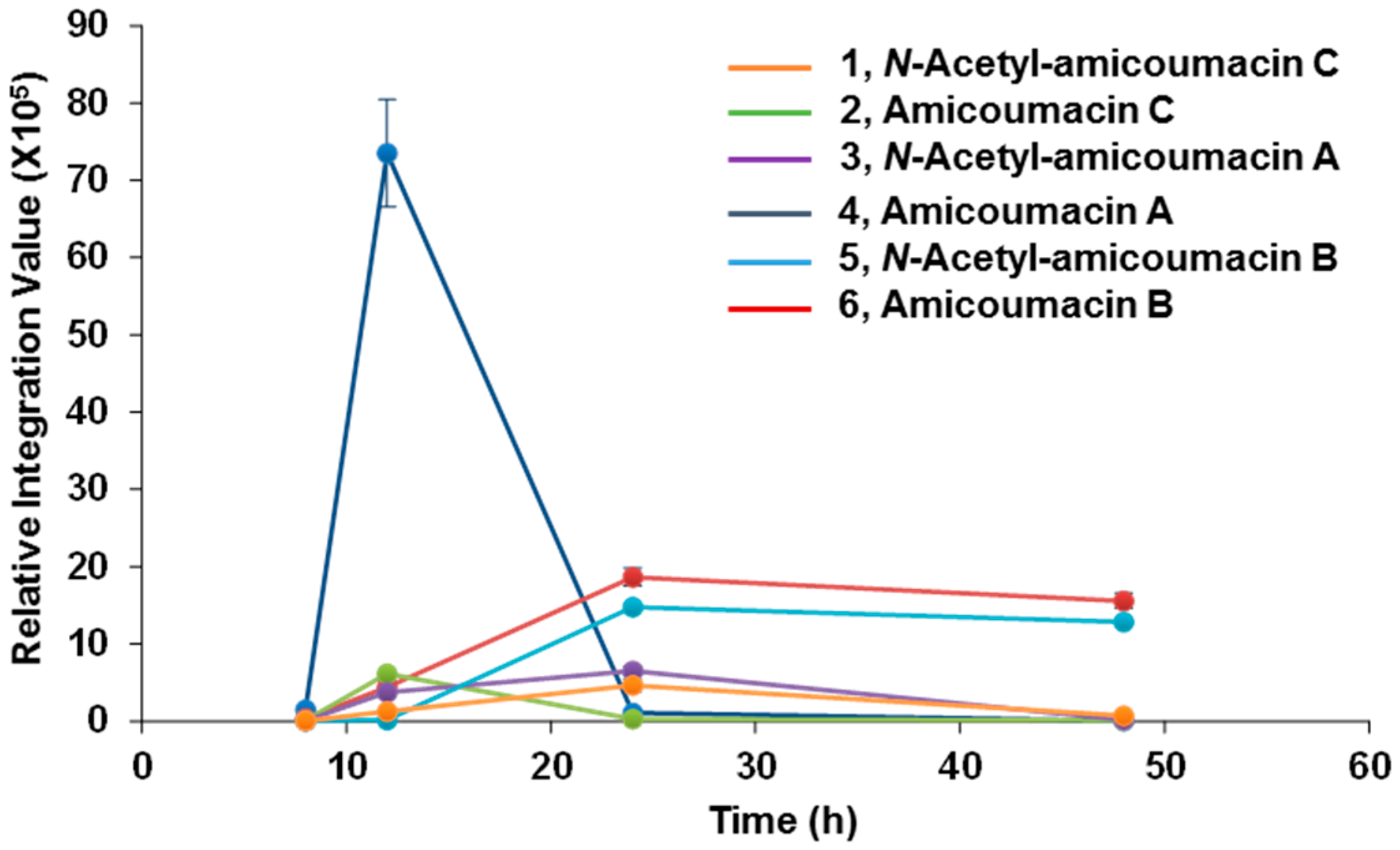
2.1. Structural Identification of Amicoumacin Metabolites from Hemolymph-Mimetic Medium
2.2. Antibacterial Evaluation of Amicoumacins
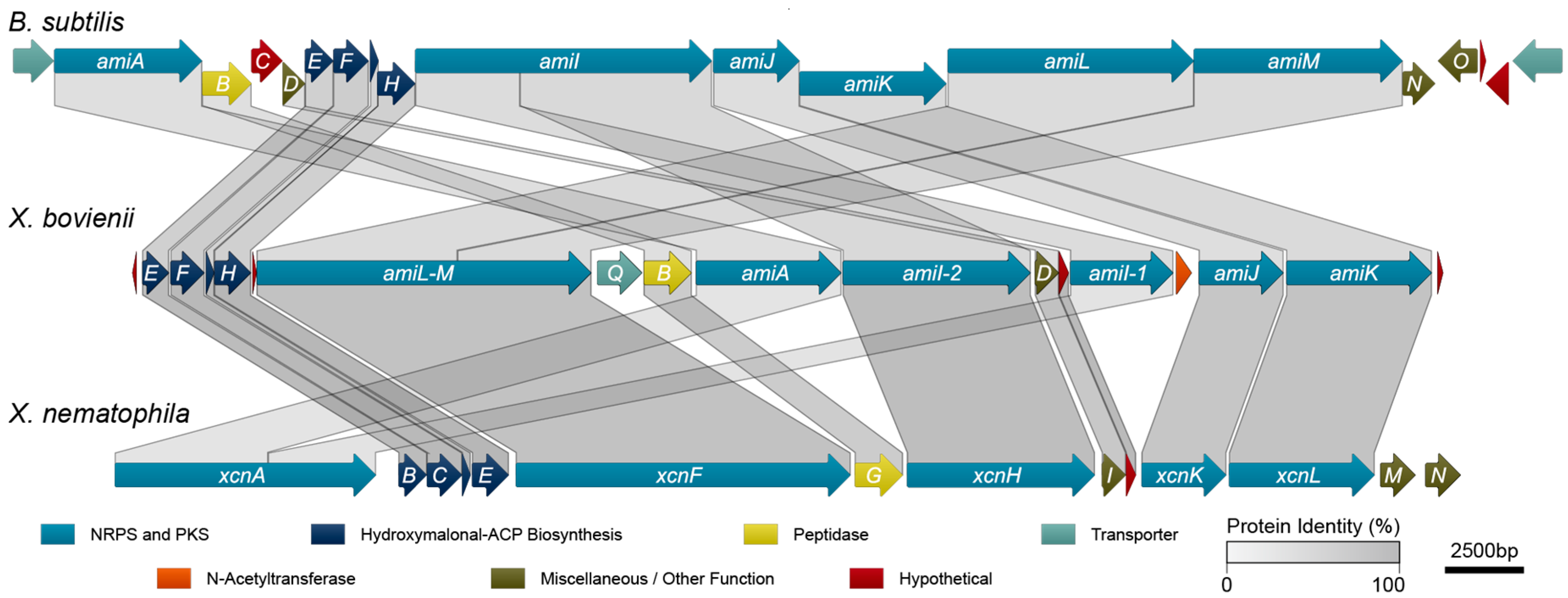
2.3. Amicoumacin Biosynthetic Gene Cluster Analysis
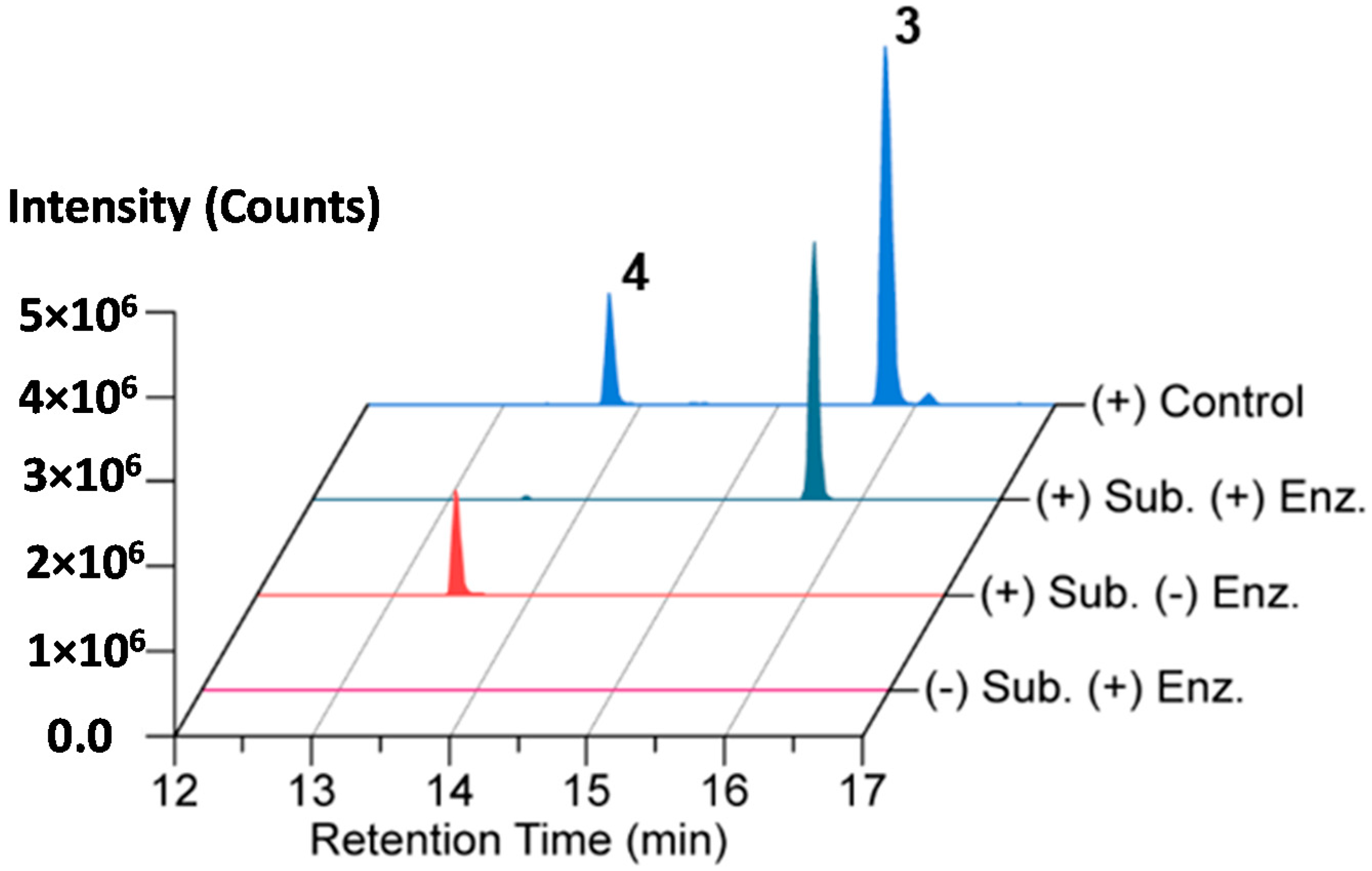
2.4. In Vitro N-Acetylation of Amicoumacin A
2.5. Ribosomal Structural Modeling of N-Acetyl-Amicoumacin A
3. Materials and Methods
3.1. General Procedures
3.2. Bacterial Strain, Growth Condition and Analytical-Scale Cultivation
3.3. Time-Course Analysis of Amicoumacin Production
3.4. Larger-Scale Cultivation and Extraction
3.5. Isolation of Amicoumacin Metabolites 1, 3 and 4
3.6. Minimum Inhibitory Concentration Determination of Amicoumacin A and N-Acetylamicoumacins
3.7. Identification of the Amicoumacin Biosynthetic Pathway in X. bovienii Moldova
3.8. Construction of 6×His-Tagged AmiS
3.9. Preparation of pEAmiS Expression Strains
3.10. Overexpression, Isolation, and Purification of 6 × His-AmiS
3.11. In Vitro Acetylation of Amicoumacin A
3.12. Modeling of N-Acetyl-Amicoumacin A Interactions with the T. thermophilus Ribosome
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Pinchuk, I.V.; Bressollier, P.; Sorokulova, I.B.; Verneuil, B.; Urdaci, M.C. Amicoumacin antibiotic production and genetic diversity of Bacillus subtilis strains isolated from different habitats. Res. Microbiol. 2002, 153, 269–276. [Google Scholar] [CrossRef]
- Sun, W.; Zhang, Y.-Q.; Huang, Y.; Zhang, Y.-Q.; Yang, Z.-Y.; Liu, Z.-H. Nocardia jinanensis sp. nov., an amicoumacin B-producing actinomycete. Int. J. Syst. Evol. Microbiol. 2009, 59, 417–420. [Google Scholar] [CrossRef] [PubMed]
- Itoh, J.; Omoto, S.; Shomura, T.; Nishizawa, N.; Miyado, S.; Yuda, Y.; Shibata, U.; Inouye, S. Amicoumacin-A, a new antibiotic with strong antiinflammatory and antiulcer activity. J. Antibiot. 1981, 34, 611–613. [Google Scholar] [CrossRef] [PubMed]
- Itoh, J.; Shomura, T.; Omoto, S.; Miyado, S.; Yuda, Y.; Shibata, U.; Inouye, S. Isolation, physicochemical properties and biological activities of amicoumacins produced by Bacillus pumilus. Agric. Biol. Chem. 1982, 46, 1255–1259. [Google Scholar] [CrossRef]
- Okazaki, H.; Kishi, T.; Beppu, T.; Arima, K. A new antibiotic, baciphelacin. J. Antibiot. 1975, 28, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Xu, Y.; Liu, L.; Han, Z.; Lai, P.Y.; Guo, X.; Zhang, X.; Lin, W.; Qian, P.-Y. Five new amicoumacins isolated from a marine-derived bacterium Bacillus subtilis. Mar. Drugs 2012, 10, 319–328. [Google Scholar] [CrossRef] [PubMed]
- Itoh, J.; Omoto, S.; Nishizawa, N.; Kodama, Y.; Inouye, S. Chemical structures of amicoumacins produced by Bacillus pumilus. Agric. Biol. Chem. 1982, 46, 2659–2665. [Google Scholar] [CrossRef]
- Hashimoto, M.; Taguchi, T.; Nishida, S.; Ueno, K.; Koizumi, K.; Aburada, M.; Ichinose, K. Isolation of 8′-phosphate ester derivatives of amicoumacins: Structure-activity relationship of hydroxy amino acid moiety. J. Antibiot. 2007, 60, 752–756. [Google Scholar] [CrossRef] [PubMed]
- Azumi, M.; Ogawa, K.-I.; Fujita, T.; Takeshita, M.; Yoshida, R.; Furumai, T.; Igarashi, Y. Bacilosarcins A and B, novel bioactive isocoumarins with unusual heterocyclic cores from the marine-derived bacterium Bacillus subtilis. Tetrahedron 2008, 64, 6420–6425. [Google Scholar] [CrossRef]
- Suzuki, T.; Nagasawa, T.; Enomoto, M.; Kuwahara, S. Stereoselective total synthesis of amicoumacin C. Tetrahedron 2015, 71, 1992–1997. [Google Scholar] [CrossRef]
- Ward, R.A.; Procter, G. A total synthesis of the natural enantiomer of the gastroprotective natural products AI-77-B and amicoumacin C hydrochloride. Tetrahedron 1995, 51, 12301–12318. [Google Scholar] [CrossRef]
- Lama, A.; Pané-Farré, J.; Chon, T.; Wiersma, A.M.; Sit, C.S.; Vederas, J.C.; Hecker, M.; Nakano, M.M. Response of methicillin-resistant Staphylococcus aureus to amicoumacin A. PLoS ONE 2012, 7, e34037. [Google Scholar] [CrossRef] [PubMed]
- Pinchuk, I.V.; Bressollier, P.; Verneuil, B.; Fenet, B.; Sorokulova, I.B.; Mégraud, F.; Urdaci, M.C. In vitro anti-helicobacter pylori activity of the probiotic strain Bacillus subtilis 3 is due to secretion of antibiotics. Antimicrob. Agents Chemother. 2001, 45, 3156–3161. [Google Scholar] [CrossRef] [PubMed]
- Polikanov, Y.S.; Osterman, I.A.; Szal, T.; Tashlitsky, V.N.; Serebryakova, M.V.; Kusochek, P.; Bulkley, D.; Malanicheva, I.A.; Efimenko, T.A.; Efremenkova, O.V.; et al. Amicoumacin a inhibits translation by stabilizing mRNA interaction with the ribosome. Mol. Cell 2014, 56, 531–540. [Google Scholar] [CrossRef] [PubMed]
- McInerney, B.V.; Taylor, W.C.; Lacey, M.J.; Akhurst, R.J.; Gregson, R.P. Biologically active metabolites from Xenorhabdus spp., part 2. benzopyran-1-one derivatives with gastroprotective activity. J. Nat. Prod. 1991, 54, 785–795. [Google Scholar] [CrossRef] [PubMed]
- Herbert, E.E.; Goodrich-Blair, H. Friend and foe: The two faces of Xenorhabdus nematophila. Nat. Rev. Microbiol. 2007, 5, 634–646. [Google Scholar] [CrossRef] [PubMed]
- Murfin, K.E.; Dillman, A.R.; Foster, J.M.; Bulgheresi, S.; Slatko, B.E.; Sternberg, P.W.; Goodrich-Blair, H. Nematode-bacterium symbioses—Cooperation and conflict revealed in the “Omics” age. Biol. Bull. 2012, 223, 85–102. [Google Scholar] [PubMed]
- Forst, S.; Nealson, K. Molecular biology of the symbiotic-pathogenic bacteria Xenorhabdus spp. and Photorhabdus spp. Microbiol. Rev. 1996, 60, 21–43. [Google Scholar] [PubMed]
- Reimer, D.; Pos, K.M.; Thines, M.; Grün, P.; Bode, H.B. A natural prodrug activation mechanism in nonribosomal peptide synthesis. Nat. Chem. Biol. 2011, 7, 888–890. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Li, Z.; Yamanaka, K.; Xu, Y.; Zhang, W.; Vlamakis, H.; Kolter, R.; Moore, B.S.; Qian, P.-Y. Directed natural product biosynthesis gene cluster capture and expression in the model bacterium Bacillus subtilis. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.M.; Portmann, C.; Kontnik, R.; Walsh, C.T.; Clardy, J. NRPS substrate promiscuity diversifies the xenematides. Org. Lett. 2011, 13, 5144–5147. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Chen, G.; Webster, J.M.; Czyzewska, E. Antimicrobial metabolites from a bacterial symbiont. J. Nat. Prod. 1995, 58, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Gualtieri, M.; Villain-Guillot, P.; Givaudan, A.; Pages, S. Novel Peptide Derivatives as Antibiotics. Google Patent CN103827138 A, 28 May 2014. [Google Scholar]
- Houard, J.; Aumelas, A.; Noel, T.; Pages, S.; Givaudan, A.; Fitton-Ouhabi, V.; Villain-Guillot, P.; Gualtieri, M. Cabanillasin, a new antifungal metabolite, produced by entomopathogenic Xenorhabdus cabanillasii JM26. J. Antibiot. 2013, 66, 617–620. [Google Scholar] [CrossRef] [PubMed]
- Crawford, J.M.; Kontnik, R.; Clardy, J. Regulating alternative lifestyles in entomopathogenic bacteria. Curr. Biol. 2010, 20, 69–74. [Google Scholar] [CrossRef] [PubMed]
- Scherlach, K.; Hertweck, C. Triggering cryptic natural product biosynthesis in microorganisms. Org. Biomol. Chem. 2009, 7, 1753–1760. [Google Scholar] [CrossRef] [PubMed]
- Nollmann, F.I.; Dauth, C.; Mulley, G.; Kegler, C.; Kaiser, M.; Waterfield, N.R.; Bode, H.B. Insect-specific production of new GameXPeptides in Photorhabdus luminescens TTO1, widespread natural products in entomopathogenic bacteria. ChemBioChem 2015, 16, 205–208. [Google Scholar] [CrossRef] [PubMed]
- Reimer, D.; Luxenburger, E.; Brachmann, A.O.; Bode, H.B. A new type of pyrrolidine biosynthesis is involved in the late steps of xenocoumacin production in Xenorhabdus nematophila. ChemBioChem 2009, 10, 1997–2001. [Google Scholar] [CrossRef] [PubMed]
- Shimojima, Y.; Hayashi, H.; Ooka, T.; Shibukawa, M. Production, isolation and pharmacological studies of AI-77s. Agric. Biol. Chem. 1982, 46, 1823–1829. [Google Scholar] [CrossRef]
- Weber, T.; Blin, K.; Duddela, S.; Krug, D.; Kim, H.U.; Bruccoleri, R.; Lee, S.Y.; Fischbach, M.A.; Muller, R.; Wohlleben, W.; et al. antiSMASH 3.0-a comprehensive resource for the genome mining of biosynthetic gene clusters. Nucleic Acids Res. 2015, 43, W237–W243. [Google Scholar] [CrossRef] [PubMed]
- Morales-Soto, N.; Synder, H.; Forst, S. Interspecies competition in a bacteria-nematode mutualism. In Defensive Mutualism in Microbial Symbiosis; CRC Press: Boca Raton, FL, USA, 2009; pp. 117–128. [Google Scholar]
- Bai, J.; Liu, D.; Yu, S.; Proksch, P.; Lin, W. Amicoumacins from the marine-derived bacterium Bacillus sp. with the inhibition of NO production. Tetrahedron Lett. 2014, 55, 6286–6291. [Google Scholar] [CrossRef]
- Park, D.; Ciezki, K.; van der Hoeven, R.; Singh, S.; Reimer, D.; Bode, H.B.; Forst, S. Genetic analysis of xenocoumacin antibiotic production in the mutualistic bacterium Xenorhabdus nematophila. Mol. Microbiol. 2009, 73, 938–949. [Google Scholar] [CrossRef] [PubMed]
- NRPSpredictor2. Available online: http://nrps.informatik.uni-tuebingen.de/Controller?cmd=SubmitJob (accessed on 15 January 2016).
- EMBOSS Water. Available online: http://www.ebi.ac.uk/Tools/psa/emboss_water/ (accessed on 6 January 2016).
- Sample Availability: Not available.






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Protein | Size a | Predicted Function | Homolog b | X. nematophila (%) | B. subtilis (%) | ||
|---|---|---|---|---|---|---|---|
| Identity | Similarity | Identity | Similarity | ||||
| AmiA | 1488 | NRPS | XcnA c | 23.3 | 38.8 | 30.3 | 49.2 |
| AmiB | 497 | Peptidase d | XcnG | 55.1 | 71.8 | 30.1 | 46.9 |
| AmiD | 243 | Thioesterase | XcnI | 60.5 | 74.5 | 31.3 | 52.0 |
| AmiE | 282 | Dehydrogenase e | XcnB | 74.8 | 86.2 | 49.3 | 67.8 |
| AmiF | 352 | Acyl carrier protein f | XcnC | 79.8 | 90.6 | 53.0 | 74.0 |
| AmiG | 85 | Acyl carrier protein | XcnD | 76.5 | 92.9 | 44.7 | 73.7 |
| AmiH | 382 | Dehydrogenase g | XcnE | 83.1 | 93.4 | 49.0 | 70.8 |
| AmiI-1 | 1051 | NRPS | XcnA c | 24.5 | 43.5 | 33.5 | 54.1 |
| AmiI-2 | 1924 | PKS | XcnH | 72.6 | 85.2 | 37.6 | 55.1 |
| AmiJ | 858 | NRPS | XcnK | 61.9 | 76.5 | 30.9 | 49.6 |
| AmiK | 1487 | PKS | XcnL | 66.3 | 81.2 | 36.0 | 54.7 |
| AmiL-M | 3419 | PKS | XcnF | 68.1 | 81.8 | 34.8 i | 50.3 i |
| AmiP | 46 | Hypothetical | -- | -- | -- | -- | -- |
| AmiQ | 461 | Drug transporter h | -- | -- | -- | -- | -- |
| AmiR | 107 | Hypothetical | XcnJ | 78.5 | 88.8 | -- | -- |
| AmiS | 152 | N-acetyltransferase | -- | -- | -- | -- | -- |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Park, H.B.; Perez, C.E.; Perry, E.K.; Crawford, J.M. Activating and Attenuating the Amicoumacin Antibiotics. Molecules 2016, 21, 824. https://doi.org/10.3390/molecules21070824
Park HB, Perez CE, Perry EK, Crawford JM. Activating and Attenuating the Amicoumacin Antibiotics. Molecules. 2016; 21(7):824. https://doi.org/10.3390/molecules21070824
Chicago/Turabian StylePark, Hyun Bong, Corey E. Perez, Elena Kim Perry, and Jason M. Crawford. 2016. "Activating and Attenuating the Amicoumacin Antibiotics" Molecules 21, no. 7: 824. https://doi.org/10.3390/molecules21070824
APA StylePark, H. B., Perez, C. E., Perry, E. K., & Crawford, J. M. (2016). Activating and Attenuating the Amicoumacin Antibiotics. Molecules, 21(7), 824. https://doi.org/10.3390/molecules21070824