1. Introduction
Nowadays, improving the traditional methods for organic synthesis is an important issue. Ultrasound is widely used to avoid using expensive reagents, highly acidic solutions, long reaction times and high temperatures as well as to improve reaction yields and incompatibility with other functional groups [
1].
The use of ultrasound irradiation in the organic synthesis offers many benefits, which include milder reaction conditions, decrease in the reaction times, and increase in selectivity, thus enhancing the purity and reaction efficiency [
2,
3,
4]. Ultrasound irradiation has been used for the synthesis of simple heterocyles [
5,
6,
7], various ionic liquids [
8], and poly functionalized heterocyclic compounds [
9] and has been reported to promote the reaction rates and yields. One of the fascinating chemical reactions of pyridin-3-ol is the exhibition of 1,3-dipolar characters.
Based on the spectroscopic data [
10,
11,
12,
13,
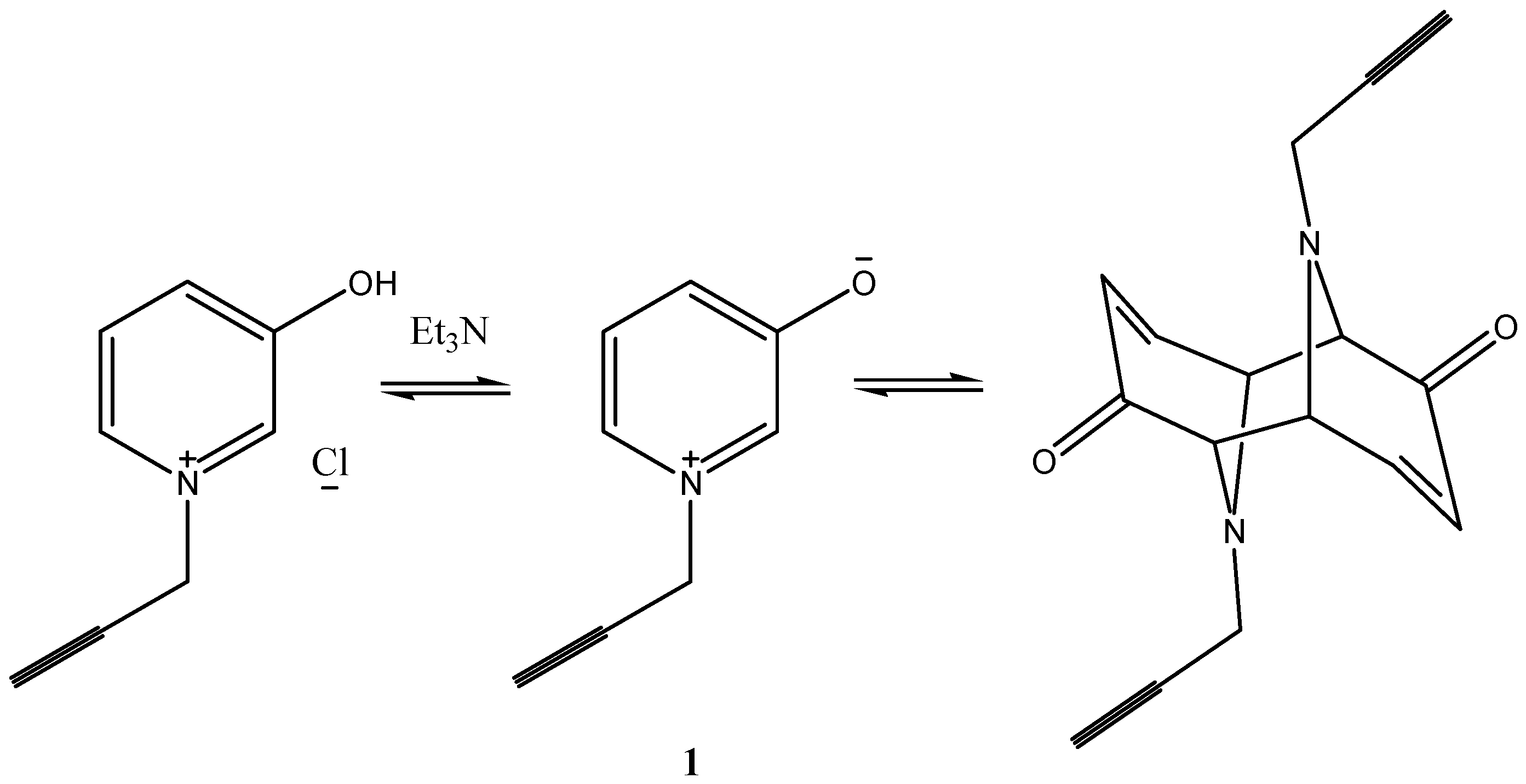
14], pyridin-3-ol
Ia in polar solvents exists to a considerable extent in the unstable mesomeric form, pyridinium-3-olate
Ib, via prototropic rearrangement [
15] (which is being considered as a resonance hybrid between the indicated canonical structures) (
Scheme 1).
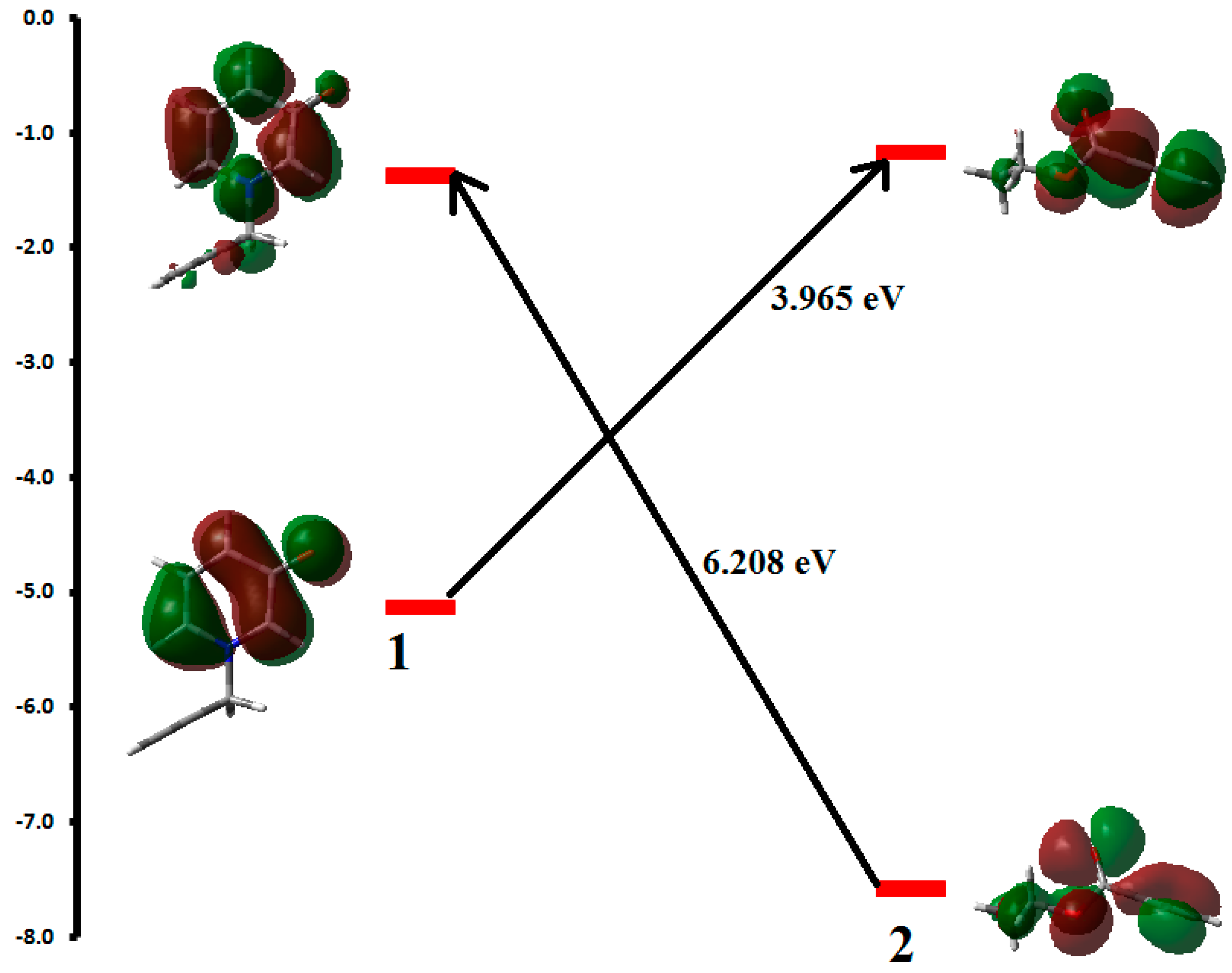
The aim of this work focused on the use of ultrasound to promote 1,3-dipolar cycloaddition reaction of the, 1-(propergyl)-pyridinium-3-olate 1 with acetylene derivatives.
3. Materials and Methods
1-(propergyl)-pyridinium-3-olate (1): A mixture of pyridin-3-ol (1.9 g, 0.02 mol) and propargyl chloride (1.45 g, 0.02 mol) in absolute alcohol (30 mL) was heated under reflux in water bath while stirring for 3 h. The reaction mixture was allowed to cool until room temperature; the solvent was removed under reduced pressure. The yellow residue obtained was recrystallized from ethanol to give 1 as pale yellow crystals yield 88%, mp 98–100 °C. IR: ν 3401–3250 (broad, OH), 2364 (acetylenic bond), 1571, 1241 cm−1 (ring vibrations). 1H-NMR (CDCl3): δ = 2.05 (s, 1H, acetylenic-H), 3.25 (s, 1H, OH), 3.52 (s, 1H, H-2), 3.75 (d, 1H, H-5), 5.2 (s, 1H, CH2), 5.92 (dd, 1H, H-3), 6.48 (d, 1H, H-4). Anal. Calcd for C8H8NOCl (169.6): MS (EI): m/z = 133 [M+ − HCl], base peak at m/z = 95 [M+ − Cl-CH2CCH].
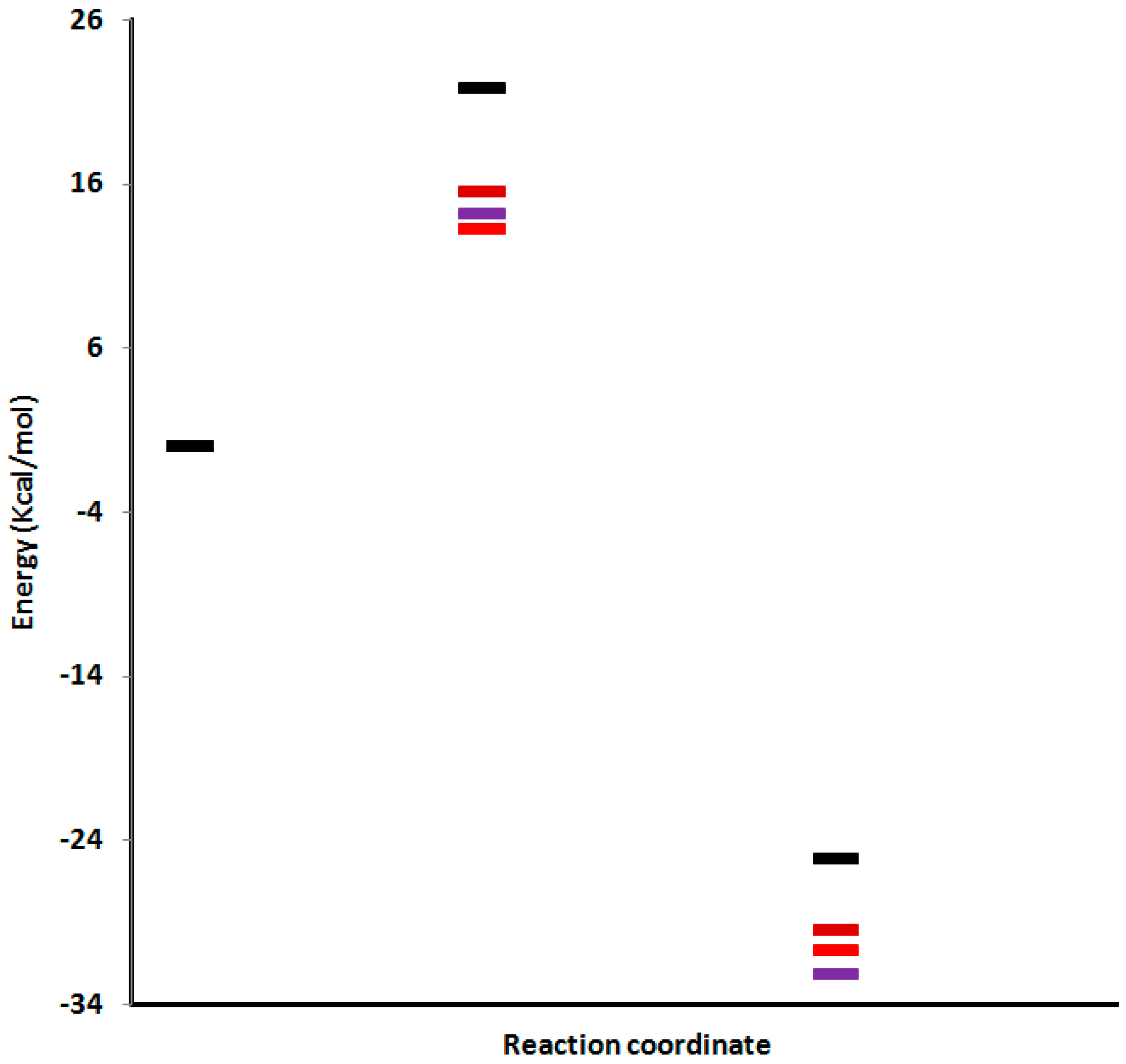
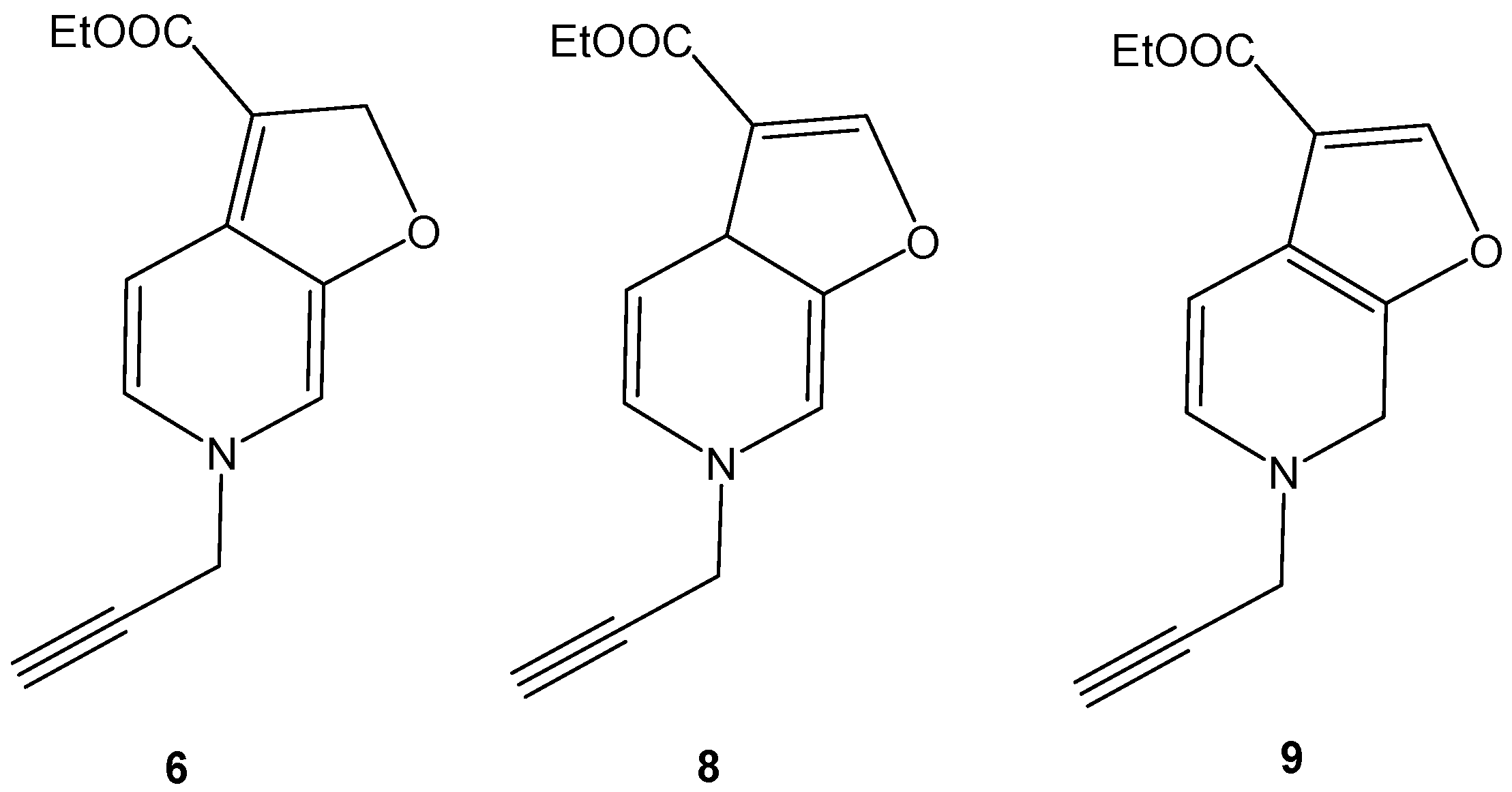
Reaction of 1-(propergyl)-pyridinium-3-olate (1) with ethyl propiolate (2): To a mixture of 3-hydroxy prop-2-yne-1-yl-pyridinium chloride (1.69 g, 0.01 mol) and ethyl propiolate (2 mL, 0.026 mol) in the presence of hydroquinone (0.01 g), triethylamine (1 mL) was added dropwise. The reaction mixture was heated in ultrasonic bath, for 6 h at 40 °C. At the end of the reaction, the brown residue obtained was triturated with distilled water and then extracted with chloroform that was separated, dried over anhydrous sodium sulfate, filtered off, and evaporated under reduced pressure to give a brown sticky residue, which gave three spots on TLC. The residue obtained was eluted through a column of alumina with a mixture of ethyl acetate-light petroleum (1:2). Three products 4–6 were obtained in a combined yield of 80% (1.84 g).
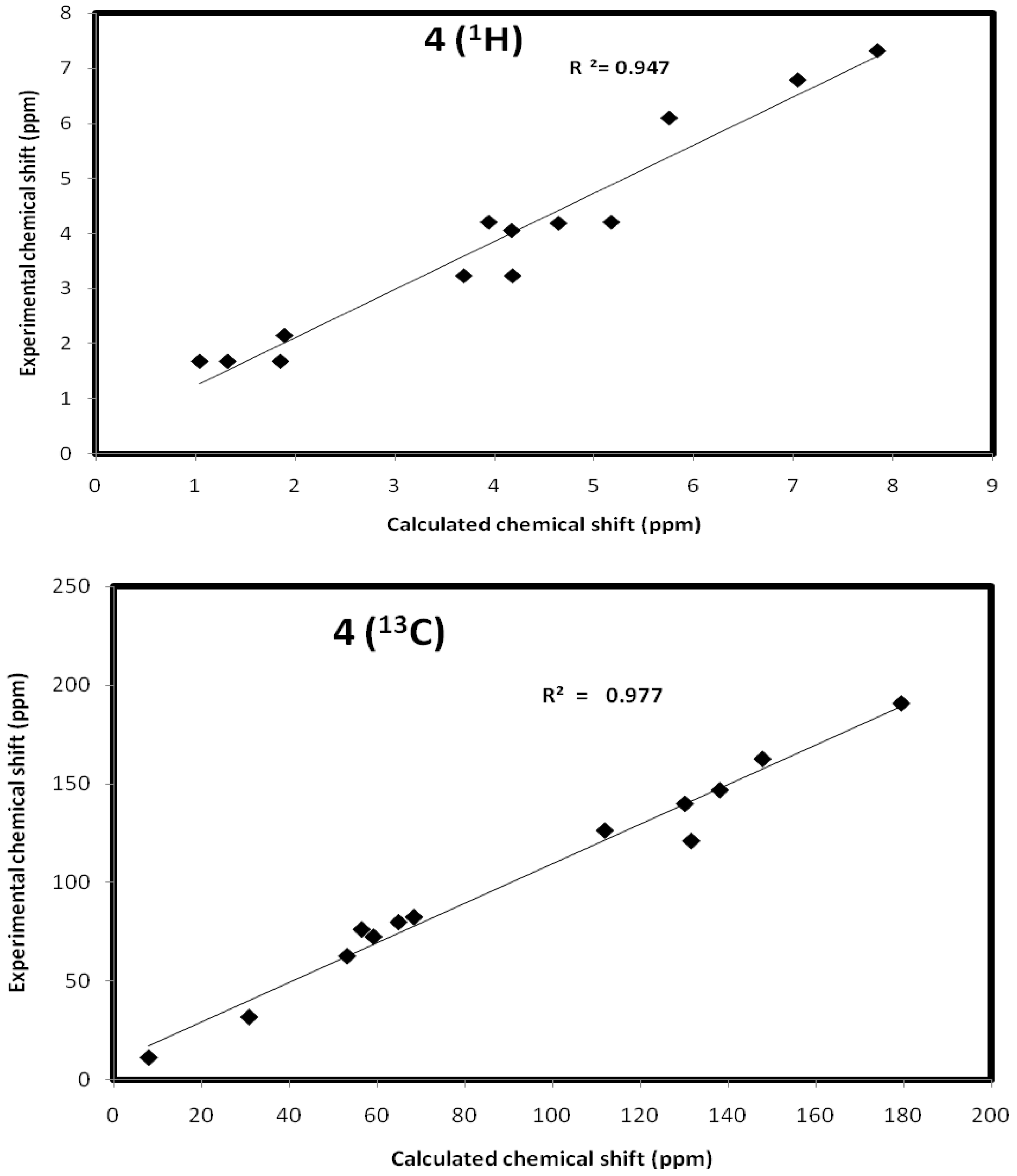
Ethyl 4-oxo-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-2,6-diene-6-carboxylate (4): Yellow crystals (0.61 g, 32%), mp 176–177 °C, IR ν = 2929 (CH), 1698 (conjugated CO), 1735 cm−1 (CO ester), C13H13NO3 (231.25), [M+] at m/z = 231, base peak at m/z = 80.
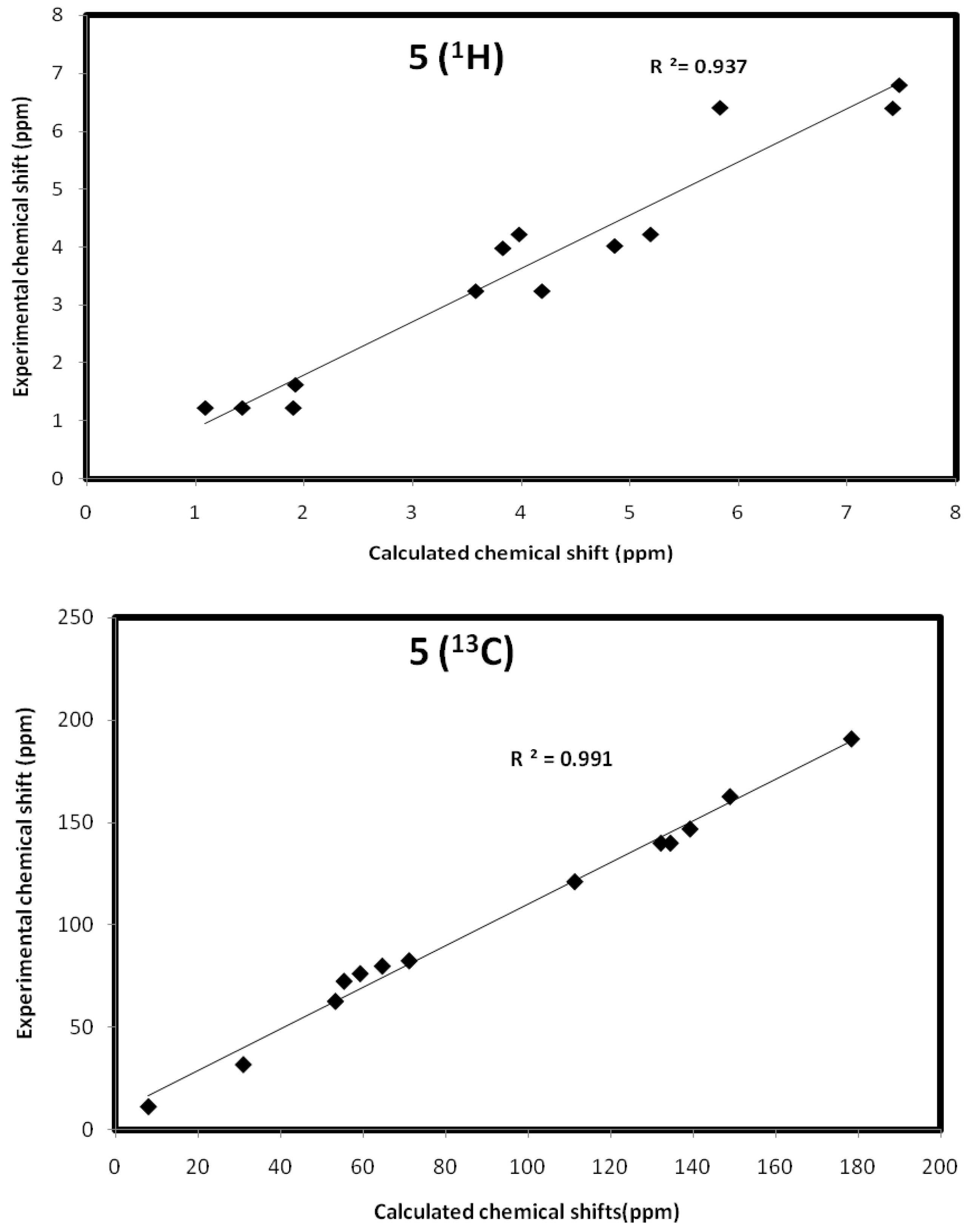
Ethyl 2-oxo-8-(prop-2-ynyl)-8-aza-bicyclo[3.2.1]octa-3,6-diene-6-carboxylate (5): Orange crystals (0.52 g, 27.5%), mp 180–182 °C, IR ν = 2931 (CH), 1686 (conjugated CO), 1726 cm−1 (CO ester), C13H13NO3 (231.25), [M+] at m/z = 231, base peak at m/z = 80.
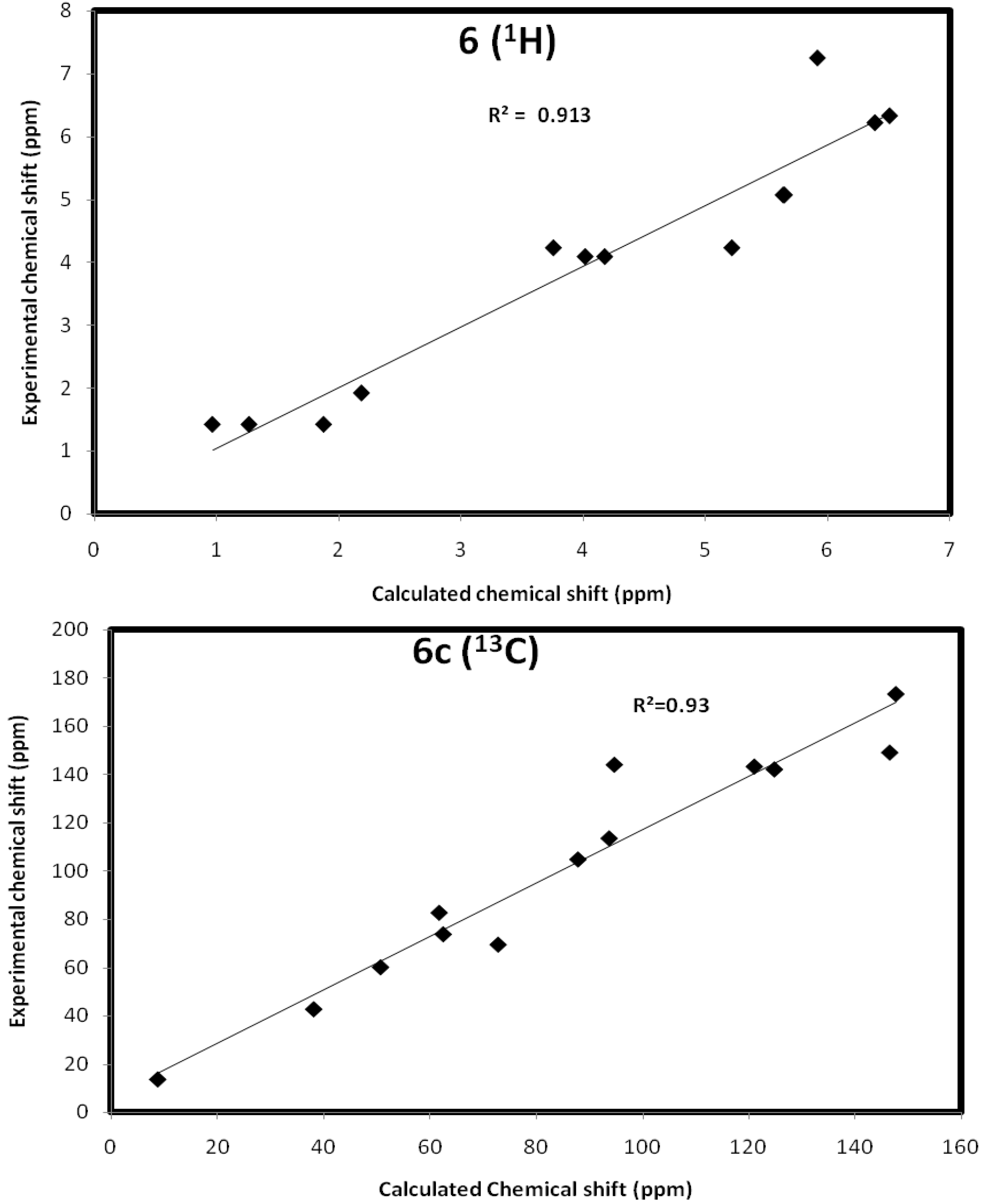
Ethyl 2,6-dihydro-6-(prop-2-ynyl)furo(2,3-c)pyridine-3-carboxylate (6): Yellow oil (0.20 g, 11.0%), IR ν = 2924 (CH), 1723 (CO ester), 1616 (C=C-N), 1348 cm−1 (furan ring). C13H13NO3 (231.24), [M+] at m/z = 231, base peak at m/z = 180.
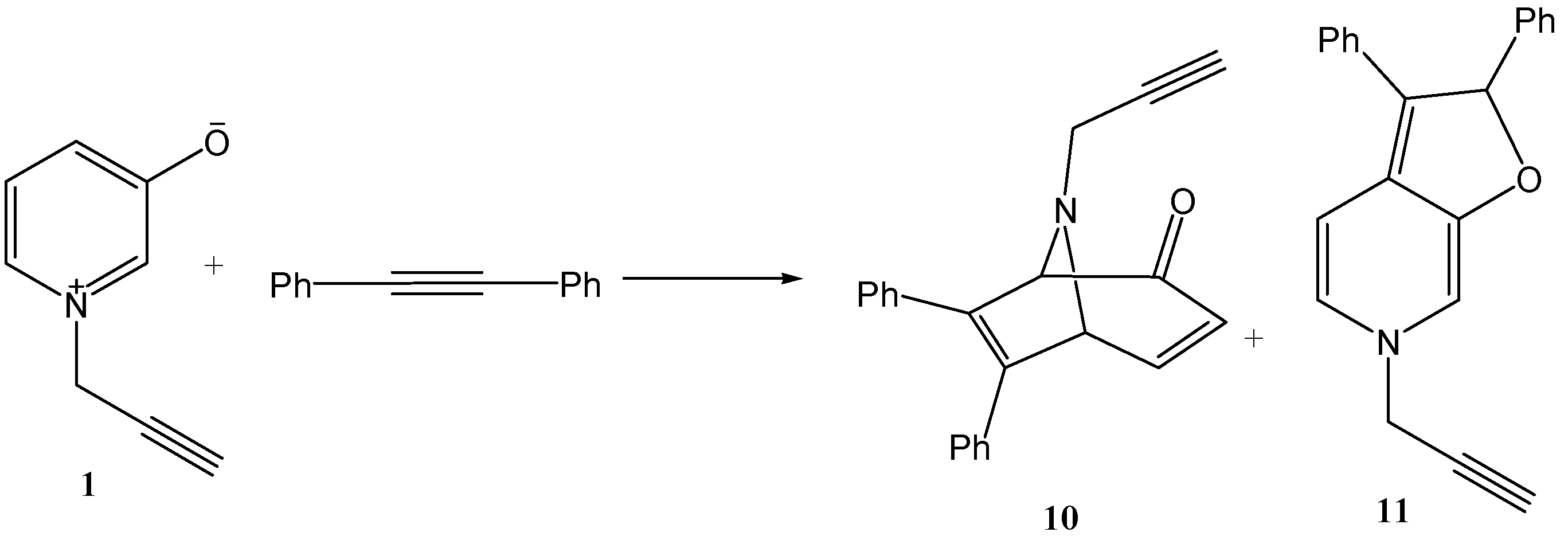
3.1. Reaction of 3-Hydroxy-1-prop-2-yne-1-ylpyridinium Chloride with Diphenyl Acetylene
To a mixture of 1-(propergyl)-pyridinium-3-olate (1) (1.69 g, 0.01 mol) and diphenyl acetylene (3.56 mL, 0.02 mol) in the presence of hydroquinone (0.01 g), triethylamine (1 mL) was added dropwise. The reaction mixture was heated in ultrasonic bath, for 6 h at 40 °C. At the end of the reaction, the brown residue obtained was triturated with distilled water and then extracted with chloroform that was separated, dried over anhydrous sodium sulfate, filtered off, and evaporated under reduced pressure to give a brown sticky residue. The residue obtained was eluted through a column of alumina with a mixture of chloroform–light petroleum (3:1). The solvent was removed under reduced pressure to give two products, 10–11 (2.54 g, overall yield 82%).
6,7-Diphenyl-8-(prop-2-ynyl)-8-aza-bicyclo(3.2.1)octa-3,6-dien-2-one (10): A brown oil, (1.52 g , 60%), IR ν = 2920 (CH), 1690 cm−1 (conjugated CO). C22H17NO (311.37), [M+] at m/z = 311 a.m.u., base peak at m/z = 95 a.m.u 1H-NMR (DMSO) at δ = 2.18 (s, 1H, acetylenic-H); 4.05 (s, 2H, CH2); 4.93 (d, J4,5 = 5 Hz, 1H, H-5); 5.22 (s, 1H, H-1); 6.00 (d, J3,4 = 5 Hz, 1H, H-3); 6.58 (dd, J4,5 = 5 Hz, J4,3 = 5 Hz, 1H, H-4); 7.20–7.91 (m, 10H, arom-H).
2,6-Dihydro-2,3-diphenyl-6-(prop-2-ynyl)furo(2,3-c)pyridine (11): orange oil, (0.42 g, 14%), IR ν = 2924 (CH), 1628–1348 (furan ring), 1525 cm−1 (C=C-N). C22H17NO (311.37) C (84.84%), H (5.50%), N (4.50%) required C (84.65%), H (5.33%), N (4.42%). 1H-NMR (DMSO) at δ = 2.1 (s, 1H, acetylenic-H); 3.55 (s, 2H, CH2 methylene-H); 4.92(s, 1H, H-1); 5.2(d, 1H, H-2); 6.35 (d, 1H, H-3); 7.90–8.07 (m, 10H, arom-H and 1H, H-4).



















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}