
Chemo-Enzymatic Synthesis of Chiral Epoxides Ethyl and Methyl (S)-3-(Oxiran-2-yl)propanoates from Renewable Levoglucosenone: An Access to Enantiopure (S)-Dairy Lactone

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results and Discussion
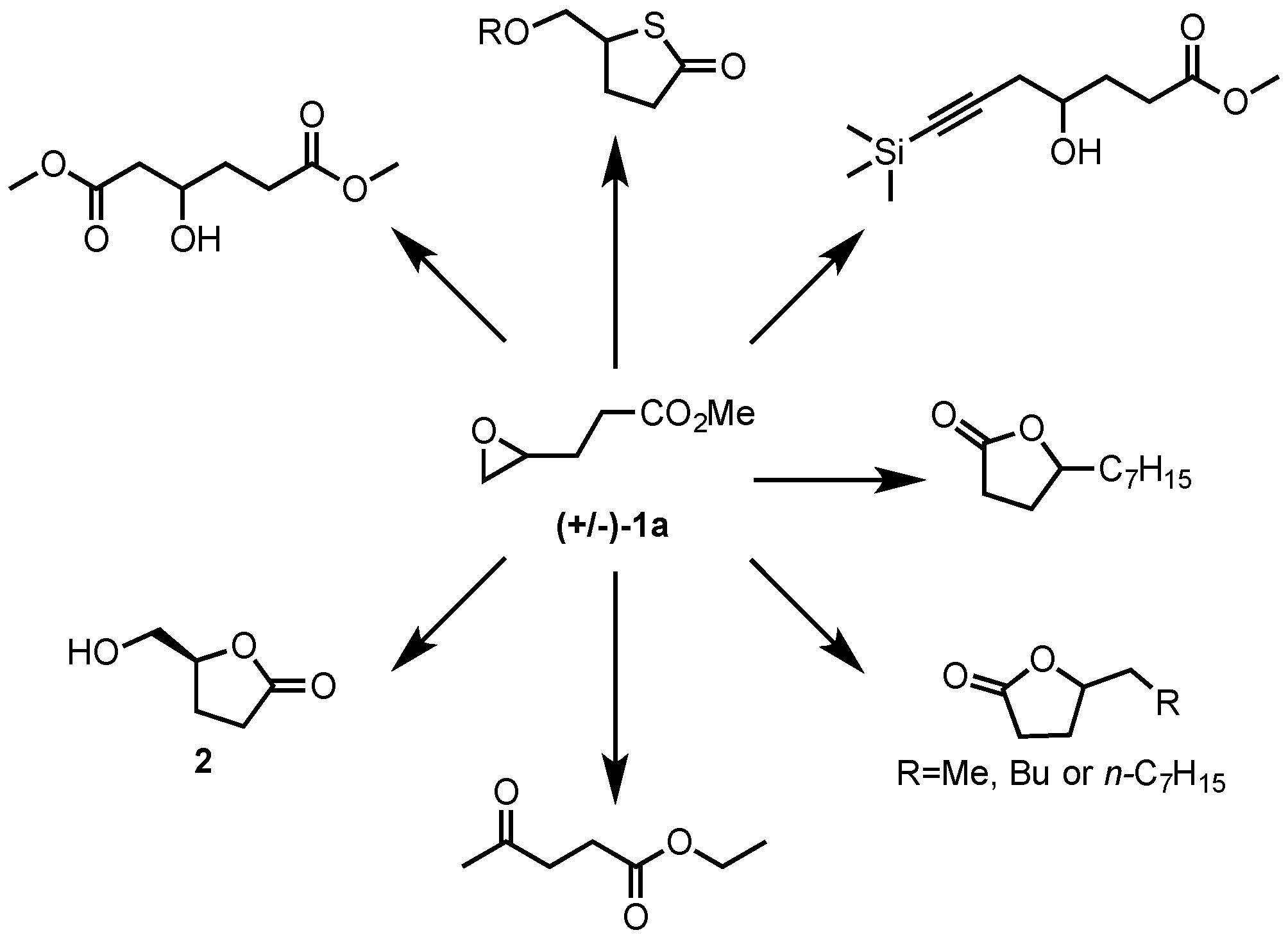
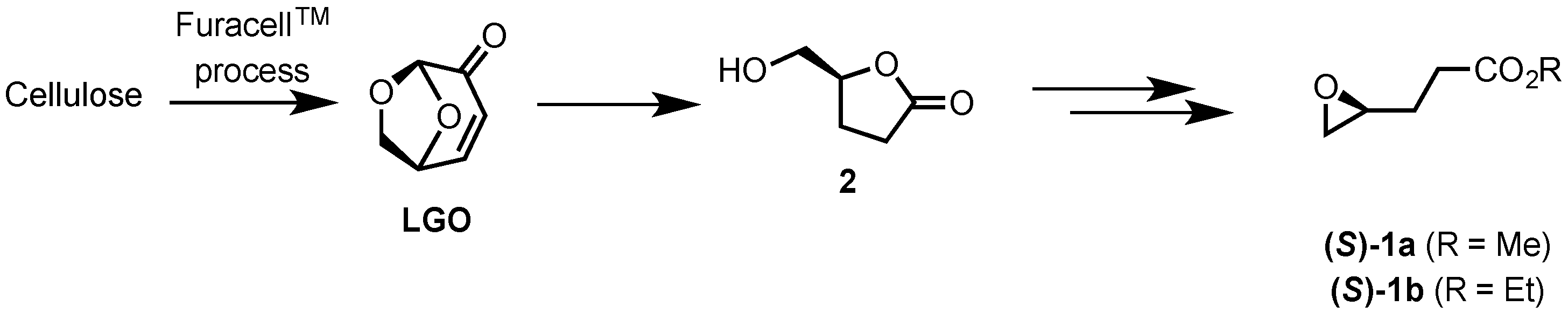
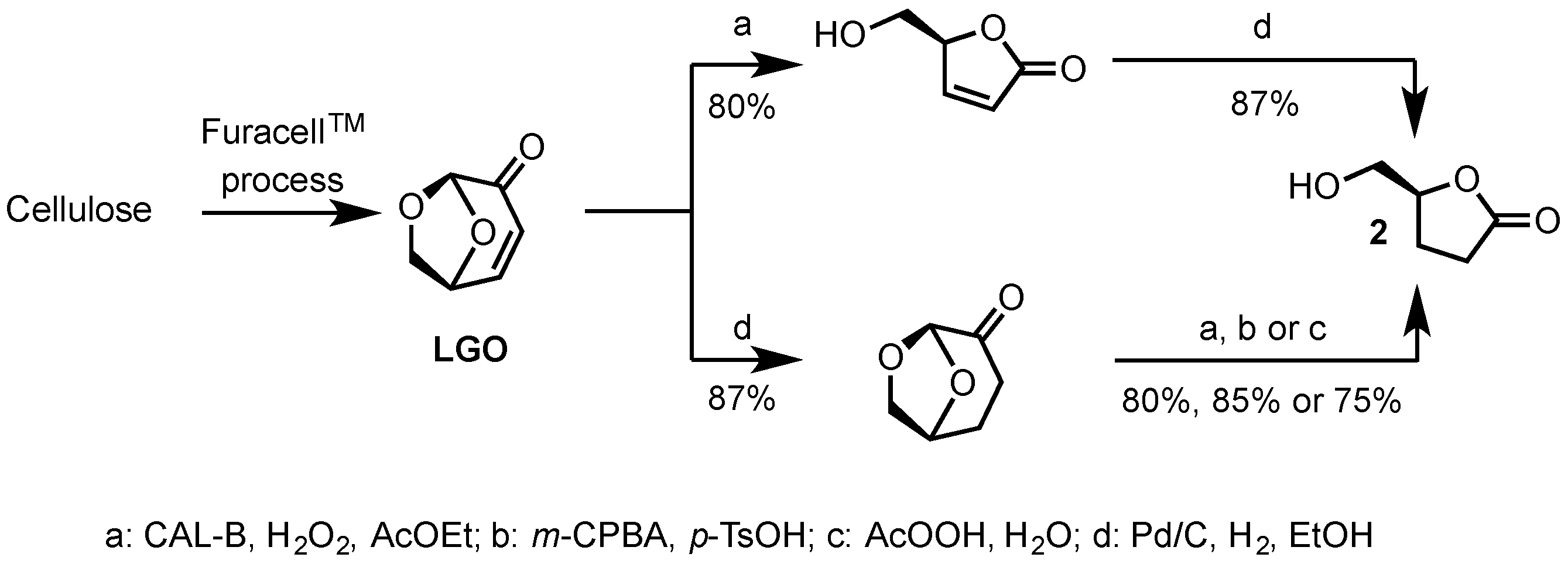
2.1. Synthesis of Epoxides from Levoglucosenone (LGO)
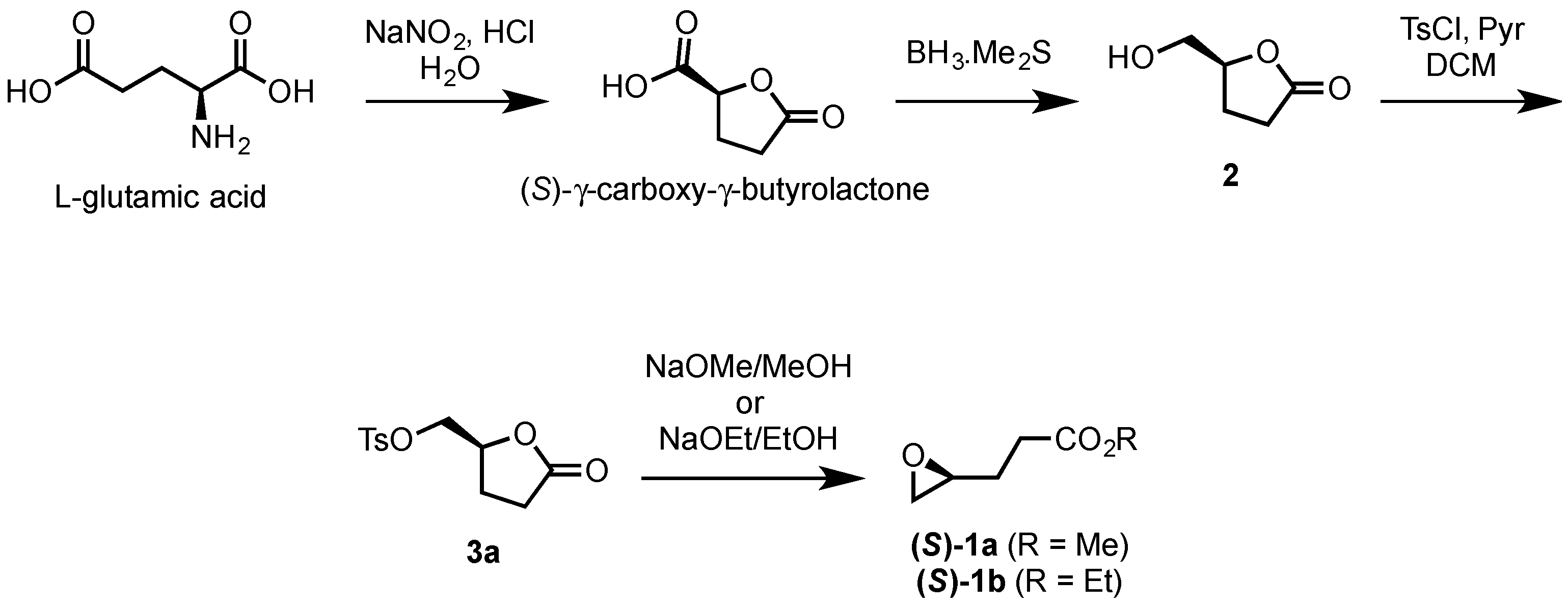
2.1.1. Synthesis of (S)-γ-Hydroxymethyl-γ-butyrolactone (2)
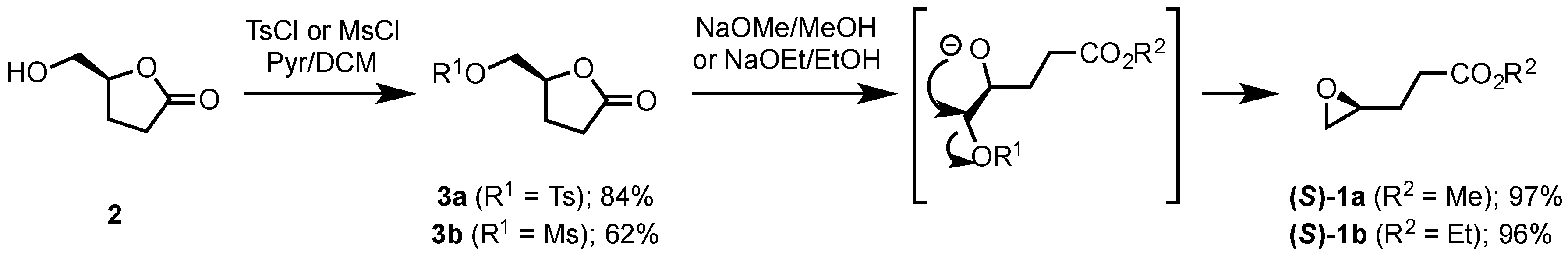
2.1.2. Synthesis of Ethyl and Methyl (S)-3-(Oxiran-2-yl)propanoates ((S)-1a and ((S)-1b)
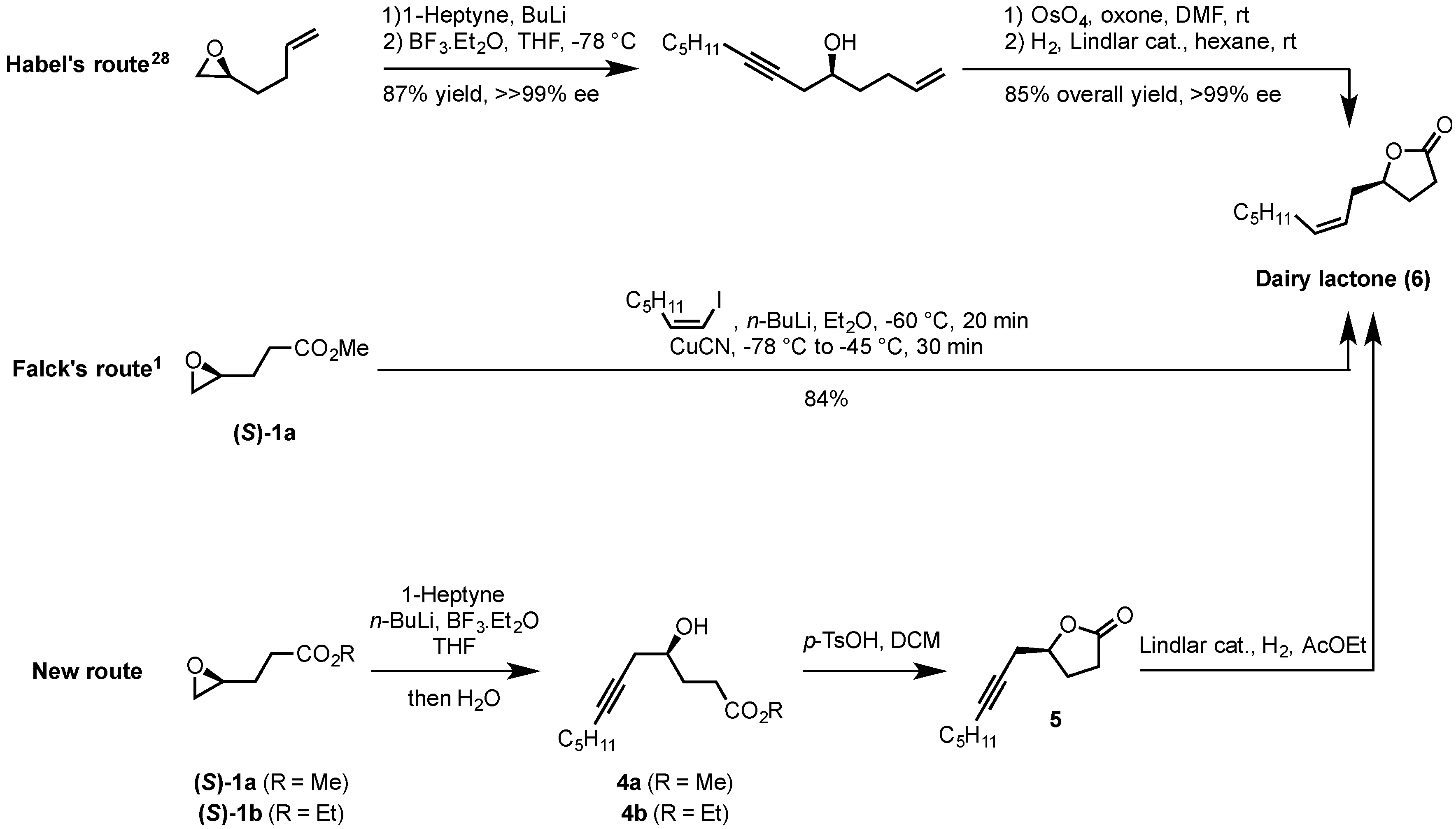
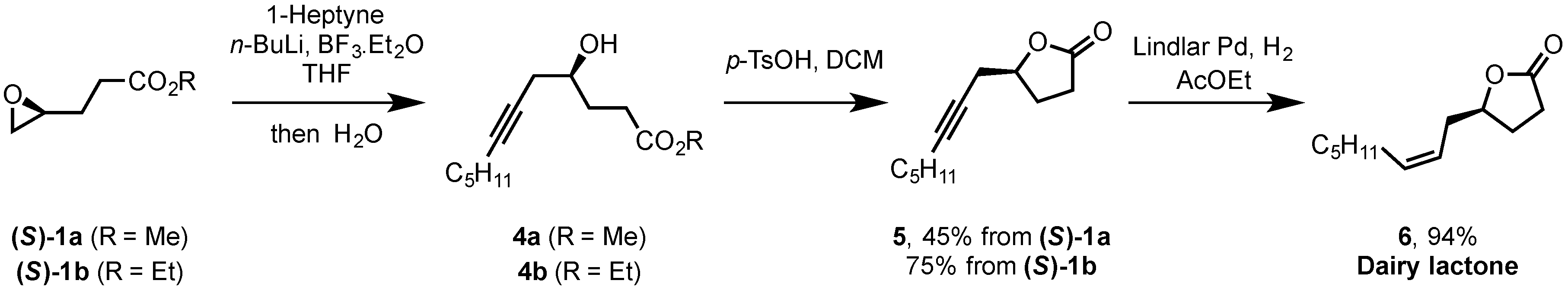
2.2. Synthesis of (S)-Dairy Lactone
3. Materials and Methods
3.1. Chemo-Enzymatic Synthesis of (S)-γ-Hydroxymethyl-γ-butyrolactone (2)
3.2. Synthesis of 2 Using m-CPBA
3.3. Synthesis of (S)-γ-Tosyloxymethyl-γ-butyrolactone (3a)
3.4. Synthesis of (S)-γ-Mesyloxymethyl-γ-butyrolactone (3b)
3.5. Synthesis of (S)-Methyl 4,5-epoxypentanoate ((S)-1a) and (S)-Ethyl 4,5-epoxypentanoate ((S)-1b)
3.6. Synthesis of (S)-5-(Oct-2-yn-1-yl)-γ-butyrolactone (5)
3.7. Synthesis of (S)-Dairy Lactone (6)
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Shin, D.-S.; Yadagiri, P.; Falck, J.R. Synthesis and Structure of Compound D, A Proinflammatory Arachidonate Metabolite. Tetrahedron Lett. 1989, 30, 3923–3926. [Google Scholar] [CrossRef]
- Haynes, S.W.; Sydor, P.K.; Corre, C.; Song, L.; Challis, G.L. Stereochemical Elucidation of Streptorubin B. J. Am. Chem. Soc. 2011, 133, 1793–1798. [Google Scholar] [CrossRef] [PubMed]
- Montagnat, O.D.; Lessene, G.; Hugues, A.B. Synthesis of Azide-Alkyne Fragments for “Click” Chemical Applications. Part 2. Formation of Oligomers from Orthogonally Protected Chiral Trialkylsilylhomopropargyl Azides and Homopropargyl Alcohols. J. Org. Chem. 2010, 75, 390–398. [Google Scholar] [CrossRef] [PubMed]
- Zunszain, P.A.; Varela, O. Two approaches to the enantioselective synthesis of (4R)-(−)-4-hydroxymethyl-4-thiobutyro-1,4-lactone. Tet. Asymmetry 2000, 11, 765–771. [Google Scholar] [CrossRef]
- Denmark, S.E.; Ahmad, M. Carbonylative ring opening of terminal epoxides at atmospheric pressure. J. Org. Chem. 2007, 72, 9630–9634. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.-Y.; Ji, J.-X.; Li, B.-G. The conversion of racemic terminal epoxides into either (+)- or (−)-diol-gamma and delta-lactones. J. Chem. Soc. Perkin Trans. 1 2000, 20, 3519–3521. [Google Scholar] [CrossRef]
- Lamb, J.R.; Jung, Y.; Coates, G.W. Meinwald-type rearrangement of monosubstituted epoxides to methyl ketones using an [Al porphyrin]+[Co(CO)4]− catalyst. Org. Chem. Front. 2015, 2, 346–349. [Google Scholar] [CrossRef]
- Ugurchieva, T.M.; Lozanova, A.V.; Zlokazov, M.V.; Veselovsky, V.V. Synthesis of (+/−)-4-alkanolides from pent-4-enoic acid. Russ. Chem. Bull. 2008, 57, 657–659. [Google Scholar] [CrossRef]
- Ho, P.-T.; Davies, N. A Practical synthesis of (R)-(−)-γ-Hydroxymethyl-γ-butyrolactone from Natural Glutamic Acid. Synthesis 1983, 462. [Google Scholar] [CrossRef]
- Eguchi, C.; Kakuta, A. The novel synthesis of l-hydroxyproline from d-glutamic acid. Bull. Chem. Soc. Jpn. 1974, 47, 1704–1708. [Google Scholar] [CrossRef]
- Austin, A.T.; Howard, J. The Reaction of Nitrous Acid with Glutamine and Glutamic acid. J. Chem. Soc. 1961, 702, 3593–3603. [Google Scholar] [CrossRef]
- Ravid, U.; Silverstein, A.M.; Smith, L.R. Synthesis of the Enantiomers of 4-Susstituted γ-Lactones with Known Absolute Configuration. Tetrahedron 1978, 34, 1449–1452. [Google Scholar] [CrossRef]
- Ma, Z.; Wang, X.; Wang, X.; Rodriguez, R.A.; Moore, C.E.; Gao, S.; Tan, X.; Ma, Y.; Rheingold, A.L.; Baran, P.S.; et al. Asymmetric syntheses of sceptrin and massadine and evidence for biosynthetic enantiodivergence. Science 2014, 346, 219–224. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, M.; Yamada, S. Stereochemical studies—XXX: Stereoselective synthesis of d-ribose from l-glutamic acid. Tetrahedron 1974, 30, 3547–3552. [Google Scholar] [CrossRef]
- Koga, K.; Taniguchi, M.; Yamada, S.-I. A new synthesis of d-ribose from l-glutamic acid. Tetrahedron Lett. 1971, 12, 263–266. [Google Scholar] [CrossRef]
- Figadère, B.; Franck, X.; Cavé, A. A facile and highly chemoselective protection of primary hydroxyl groups with 2-methyl-1-butene. Tetrahedron Lett. 1993, 34, 5893–5894. [Google Scholar] [CrossRef]
- Huh, N.; Thompson, C.M. Enantioenriched N-(2-chloroalkyl)-3-acetoxypiperidines as potential cholinotoxic agents. Synthesis and preliminary evidence for spirocyclic aziridinium formation. Tetrahedron 1995, 51, 5935–5950. [Google Scholar] [CrossRef]
- Guan, J.; Zou, Y.; Gao, P.; Wu, Y.; Yue, Z. A Total Synthesis of Natural Rhizobialide. Chin. J. Chem. 2010, 28, 1613–1617. [Google Scholar] [CrossRef]
- Yoon, N.M.; Pak, C.S.; Krishnamurthy, S.; Stocky, T.P. Selective reductions. XIX. Rapid reaction of carboxylic acids with borane-tetrahydrofuran. Remarkably convenient procedure for the selective conversion of carboxylic acids to the corresponding alcohols in the presence of other functional groups. J. Org. Chem. 1973, 88, 2786–2792. [Google Scholar] [CrossRef]
- Kinushita, S. l-Glutamic acid production. In Encyclopedia of Industrial Biotechnology; Wiley: New York, NY, USA, 2010; pp. 1–25. [Google Scholar]
- Witczak, Z.J. Levoglucosenone and Levoglucosans: Chemistry and Applications; ATL Press, Inc.: Mount Prospect, IL, USA, 1994. [Google Scholar]
- Witczak, Z.J. Levoglucosenone: Chiral Building Block with New Perspectives. In Chemicals and Materials from Renewable Resources; Bozell, J.J., Ed.; ACS Books: Washington, DC, USA, 2001; pp. 81–97. [Google Scholar]
- Koshi, K.; Takashi, E.; Kawakami, H.; Matsushita, H.; Naoi, Y.; Itoh, K. A Method for Easy Preparation of Optically Pure (S)-5-Hydroxy-2-penten-4-olide and (S)-5-Hydroxypentan-4-olide. Heterocycles 1990, 31, 423–426. [Google Scholar]
- Flourat, A.L.; Peru, A.A.M.; Teixeira, A.R.S.; Brunissen, F.; Allais, F. Chemo-enzymatic synthesis of key intermediates (S)-γ-hydroxymethyl-α,β-butenolide and (S)-γ-hydroxymethyl-γ-butyrolactone via lipase-mediated Baeyer-Villiger oxidation of Levoglucosenone. Green Chem. 2015, 17, 404–412. [Google Scholar] [CrossRef]
- Teixeira, A.R.S.; Flourat, A.L.; Peru, A.A.M.; Brunissen, F.; Allais, F. Lipase-catalyzed Baeyer-Villiger Oxidation of cellulose-derived Levoglucosenone into (S)-γ-hydroxymethyl-α,β-butenolide: Optimization by Response Surface Methodology. Front. Chem. 2016, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Court, G.R.; Lawrence, C.H.; Raverty, W.D.; Duncan, A.J. Method for Converting Lignocellulosic Materials into Useful Chemicals. U.S. Patent US20120111714 A1, 6 January 2011. [Google Scholar]
- Wilson, R.D. The Flavour of New Zealand Whole Milk Powder. Ph.D. Thesis, Massey University, Palmerston North, New Zealand, 1992. [Google Scholar]
- Latrasse, A.; Guichard, E.; Piffaut, C.; Fournier, N.; Dufossé, L. Chirality of the γ-lactones formed by Fusarium poae INRA 45. Chirality 1993, 5, 379–384. [Google Scholar] [CrossRef] [PubMed]
- (Z)-Dairy Lactone. Available online: http://www.thegoodscentscompany.com/data/rw1038301.html (accessed on 6 May 2016).
- Farbood, M.I.; Mclean, L.B.; Morris, J.A. Fermentation Process for Preparing 10-Hydroxy-C18-carboxylic Acid and Gamma-Dodelactone Derivatives. U.S. Patent EP0578388 B1, 12 January 1998. [Google Scholar]
- Habel, A.; Boland, W. Efficient and flexible synthesis of chiral γ- and δ-lactones. Org. Biomol. Chem. 2008, 6, 1601–1604. [Google Scholar] [CrossRef] [PubMed]
- Yadagiri, P.; Lumin, S.; Falck, J.R. Total Synthesis of 5(S),12(S)- and 5(S),12(R)-Dihydroxyeicosa-6(Z),8(E),14(Z)-Trienoic acids, metabolites of leukotriene B4. Tetrahedron Lett. 1989, 30, 429–432. [Google Scholar] [CrossRef]
- Schwartzman, M.L.; Falck, J.R. 12-HETrE Analogs and Methods of Use Thereof. U.S. Patent US2002151734 A1, 2 February 2002. [Google Scholar]
- Koseki, K.; Ebata, T.; Kawakami, H.; Matsushita, H.; Itoh, K.; Naoi, Y. Method of Producing (S)-4-Hydroxymethyl-γ-lactone. U.S. Patent US5112994, 12 May 1992. [Google Scholar]
- Koseki, K.; Ebata, T.; Kawakami, H.; Matsushita, H.; Itoh, K.; Naoi, Y. Method of Preparing (S)-γ-Hydroxymethyl-α,β-butenolide. U.S. Patent US4994585, 19 February 1991. [Google Scholar]
- Paris, C.; Moliner, M.; Corma, A. Metal-containing zeolites as efficient catalysts for the transformation of highly valuable chiral biomass-derived products. Green Chem. 2013, 15, 2101–2109. [Google Scholar] [CrossRef]
- Sample Availability: Samples of all compounds are available from the authors.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}





© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peru, A.A.M.; Flourat, A.L.; Gunawan, C.; Raverty, W.; Jevric, M.; Greatrex, B.W.; Allais, F. Chemo-Enzymatic Synthesis of Chiral Epoxides Ethyl and Methyl (S)-3-(Oxiran-2-yl)propanoates from Renewable Levoglucosenone: An Access to Enantiopure (S)-Dairy Lactone. Molecules 2016, 21, 988. https://doi.org/10.3390/molecules21080988
Peru AAM, Flourat AL, Gunawan C, Raverty W, Jevric M, Greatrex BW, Allais F. Chemo-Enzymatic Synthesis of Chiral Epoxides Ethyl and Methyl (S)-3-(Oxiran-2-yl)propanoates from Renewable Levoglucosenone: An Access to Enantiopure (S)-Dairy Lactone. Molecules. 2016; 21(8):988. https://doi.org/10.3390/molecules21080988
Chicago/Turabian StylePeru, Aurélien A. M., Amandine L. Flourat, Christian Gunawan, Warwick Raverty, Martyn Jevric, Ben W. Greatrex, and Florent Allais. 2016. "Chemo-Enzymatic Synthesis of Chiral Epoxides Ethyl and Methyl (S)-3-(Oxiran-2-yl)propanoates from Renewable Levoglucosenone: An Access to Enantiopure (S)-Dairy Lactone" Molecules 21, no. 8: 988. https://doi.org/10.3390/molecules21080988
APA StylePeru, A. A. M., Flourat, A. L., Gunawan, C., Raverty, W., Jevric, M., Greatrex, B. W., & Allais, F. (2016). Chemo-Enzymatic Synthesis of Chiral Epoxides Ethyl and Methyl (S)-3-(Oxiran-2-yl)propanoates from Renewable Levoglucosenone: An Access to Enantiopure (S)-Dairy Lactone. Molecules, 21(8), 988. https://doi.org/10.3390/molecules21080988