
Structural Modifications of Deoxycholic Acid to Obtain Three Known Brassinosteroid Analogues and Full NMR Spectroscopic Characterization


Abstract
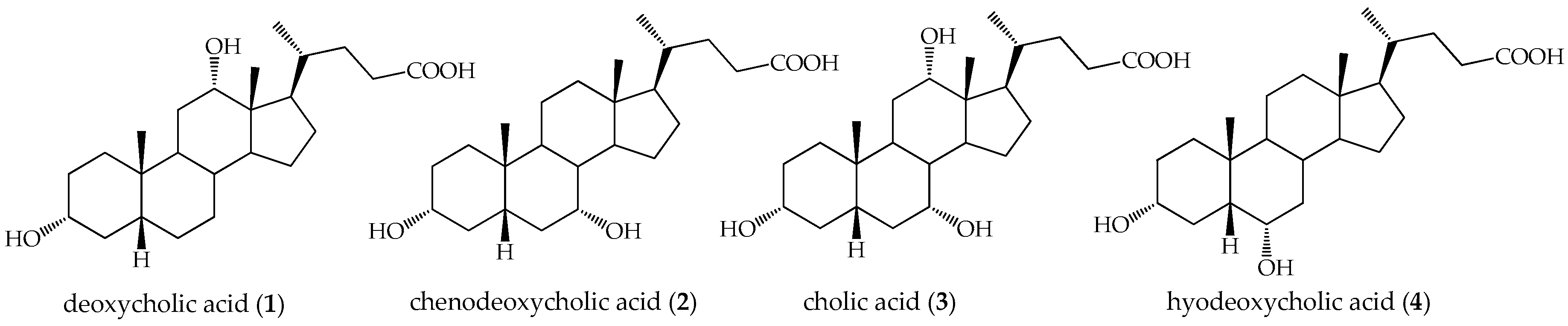
:1. Introduction
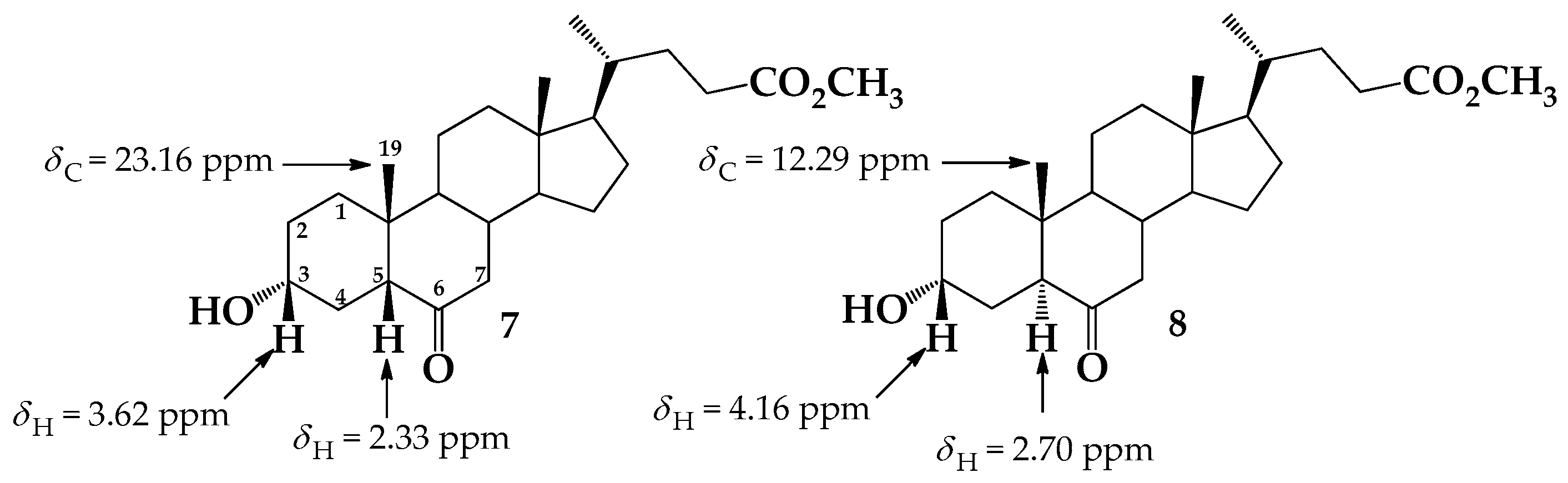
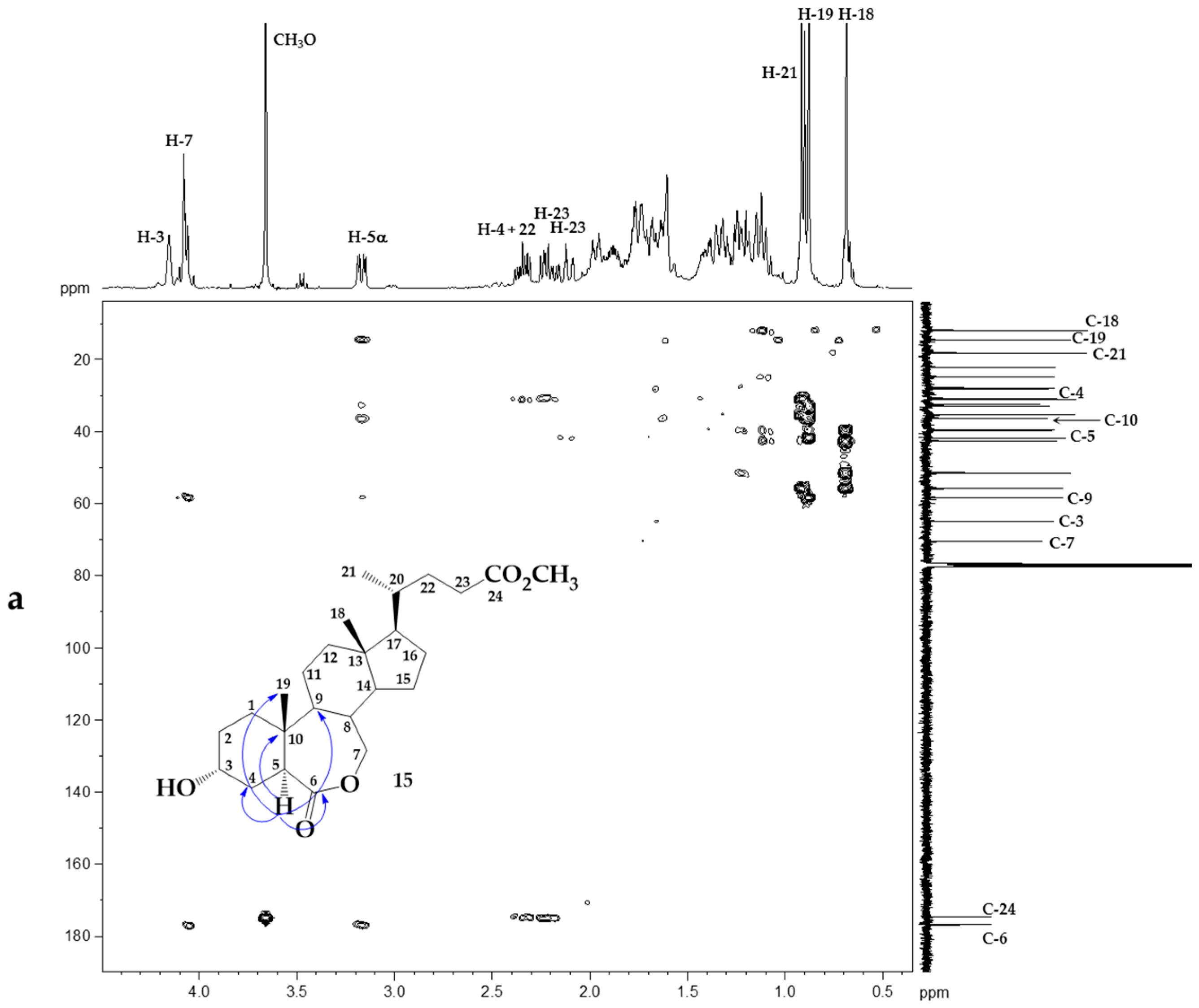
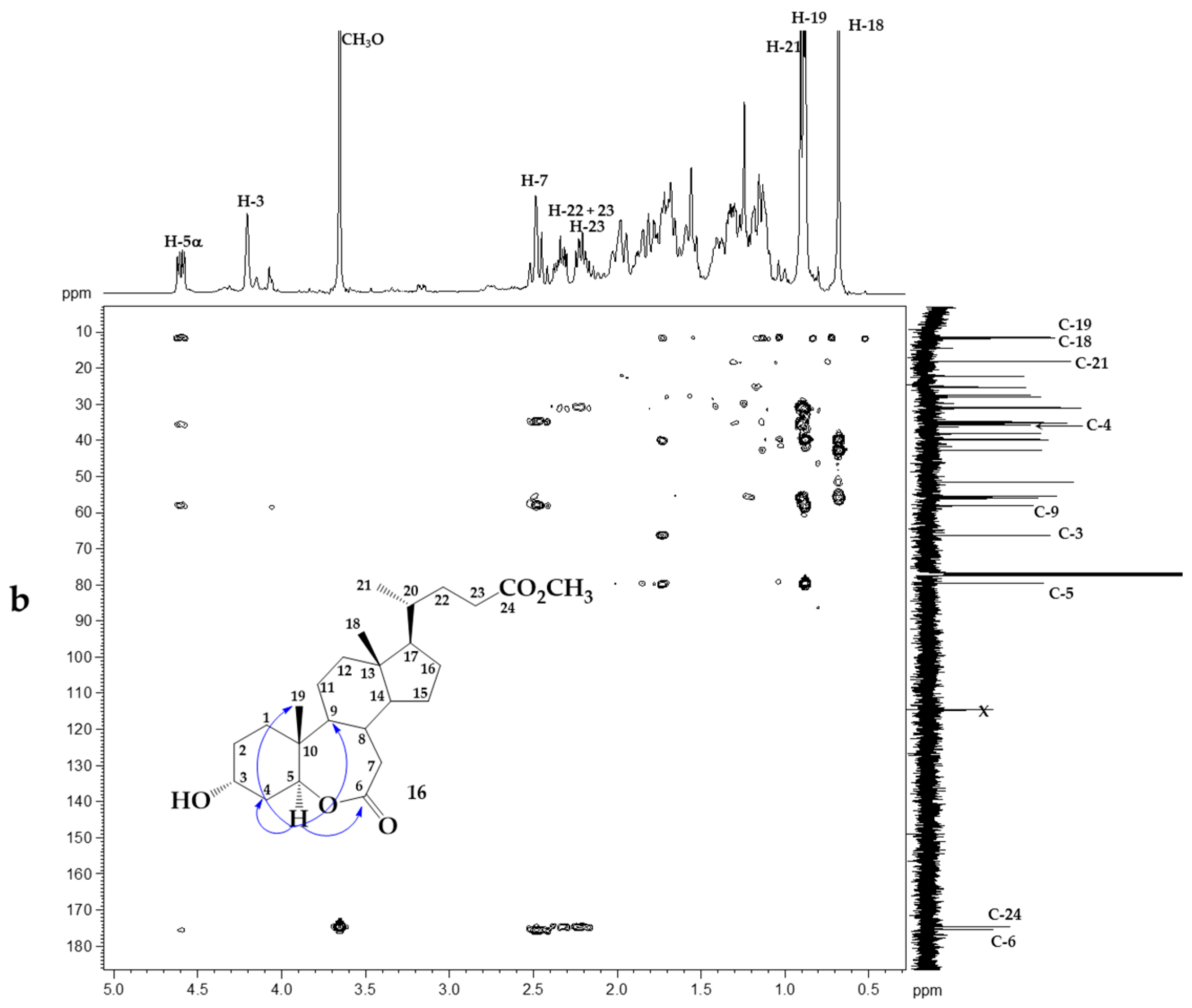
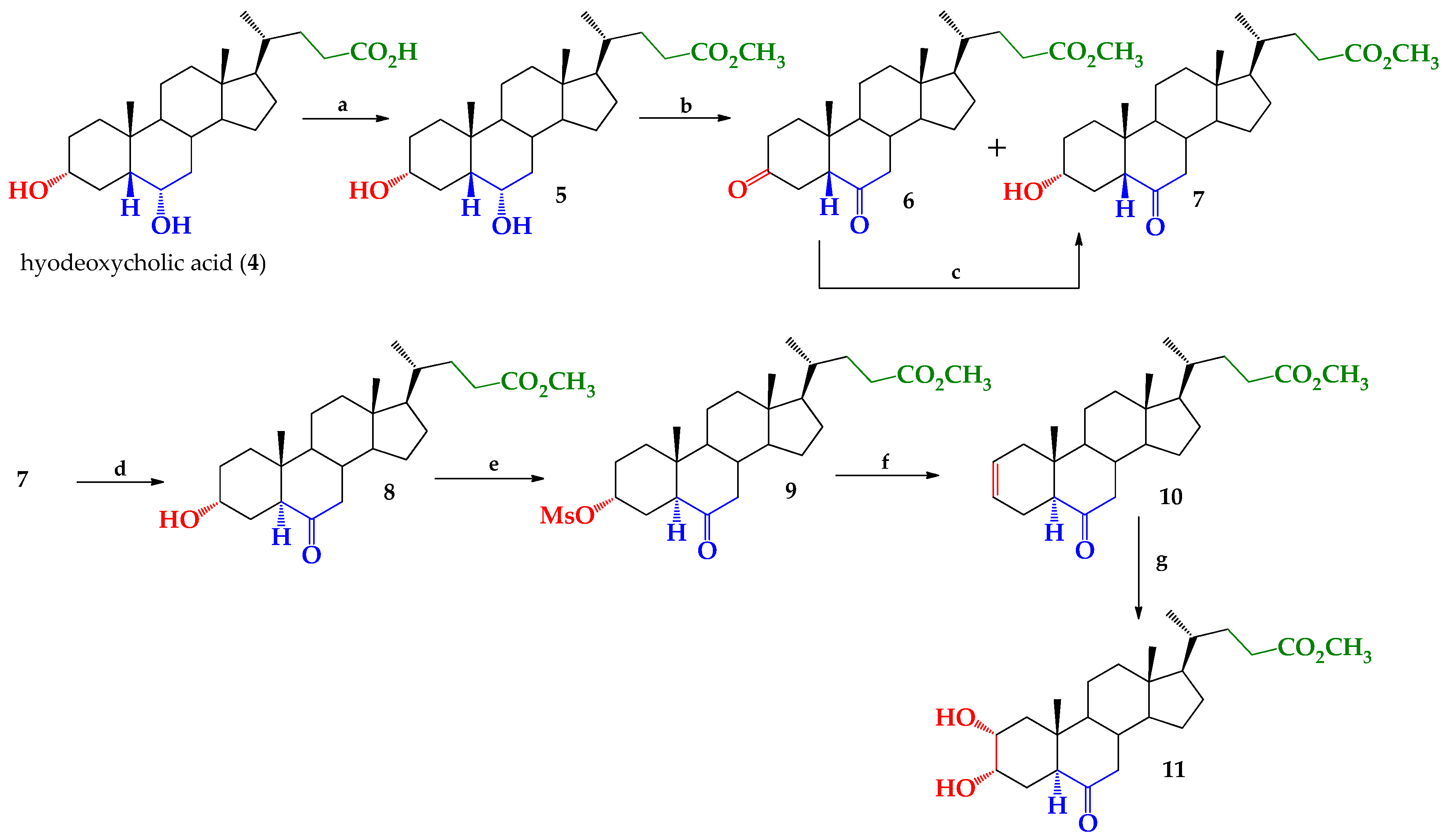
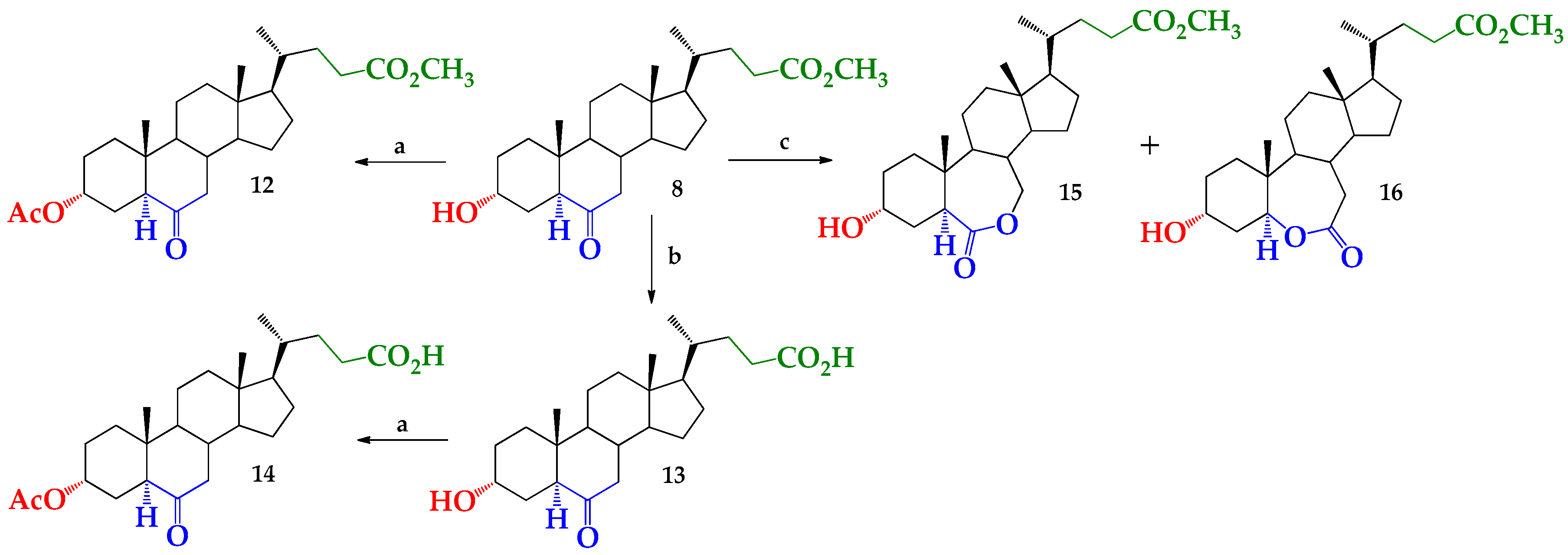
2. Results and Discussion
3. Materials and Methods
3.1. General
3.2. Synthesis
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| TLA | Three letter acronym |
| LD | linear dichroism |
| DCM | Dichloromethane |
| PCC | Pyridinium ChloroChromate |
| PDC | Pyridinium Dichromate |
| DMAP | 4-Dimethylaminopyridine |
| DMF | Dimethylformamide |
| NMO | N-Methylmorpholine-N-Oxide |
| m-CPBA | 3-Chloro or metha-chloroperoxybenzoic acid |
| NOESY | Nuclear Overhauser SpectroscopY |
| DEPT-135 | Distortionless Enhancement by Polarization Transfer with flip angle of 135° |
| gs | gradient selected |
| HSQC | Heteronuclear Single Quantum Coherence |
| HMBC | Heteronuclear Multiple Bond Correlation |
References
- Perez, R.; Iglesias, M.; Perez, C.; Coll, F.; Coll, D.; Rosado, A. Synthesis of analogues of brassinosteroids from chenodeoxycholic acid. Eur. J. Org. Chem. 1998, 2405–2407. [Google Scholar] [CrossRef]
- Perez, R.; Perez, C.; Coll, F. Synthesis of analogues of brassinosteroids with 5β-cholanic acid skeleton. Synth. Commun. 1998, 28, 3387–3396. [Google Scholar]
- Espinoza, L.; Bulat, F.; Coll, D.; Coll, F.; Preite, M.D.; Cortes, M. Synthesis and plant growth-activity of three new brassinosteroids analogues. Synth. Commun. 2000, 30, 1963–1974. [Google Scholar] [CrossRef]
- Robaina, C.; Zullo, M.A.; Coll, F. Síntesis y Caracterización Espectroscópica de Análogos de Brasinoesteroides a partir de Ácido Cólico. Rev. Cuba. Quím. XIII 2001, 2, 396. [Google Scholar]
- Espinoza, L.; Cortes, M. Synthesis and biological activities of two new brassinosteroids functionalized in ring C. Bol. Soc. Chil. Quim. 2002, 47, 335–347. [Google Scholar]
- Espinoza, L.; Cortes, M. Synthesis and biological activity of brassinosteroids analogues. Bol. Soc. Chil. Quim. 2002, 47, 511–516. [Google Scholar]
- Espinoza, L. Synthesis of Four New Brassinosteroids Analogues 11-Oxo-Functionalizedon C Ring, with 24-Nor Side Chain and Containing 5β-Cholanic Acid Skeleton. Org. Chem. Curr. Res. 2015, 4, 156. [Google Scholar] [CrossRef]
- Zhou, W.S.; Tian, W.S. The Synthesis of Steroids Containing Structural Unit of A, B Ring of Brassinolide and Ecdysone from Hyodesoxycholic Acid. Acta Chim. Sin. 1984, 42, 1173–1177. [Google Scholar]
- Zhou, W.S.; Tian, W.S. Studies on Steroidal Plant-Growth Hormones. II. Stereoselective Synthesis of (22S,23S)-Typhasterol from Hyodeoxycholic Acid. Acta Chim. Sin. 1985, 43, 1060–1067. [Google Scholar]
- Zhou, W.S.; Tian, W.S. Study on the Synthesis of Brassinolide and Related-Compounds. III. Stereoselective Synthesis of Typhasterol from Hyodeoxycholic Acid. Tetrahedron 1987, 43, 3705–3712. [Google Scholar] [CrossRef]
- Zhou, W.S.; Jiang, L.Z.; Tian, W.S.; Zhao, X.Y.; Zheng, H. Studies on Steroidal Plant-Growth Hormone VII: Synthesis of 2-α,3-α-Dihydroxy-7-Oxa-6-Oxo-23, 24-Bisnor-B-Homo-5-α-Cholanic Acid and 2-α,3-α,-Dihydroxy-7-Oxa-6-Oxo-24-Nor-B-Homo-5-α-Cholanic Acid. Acta Chim. Sin. 1988, 46, 1212–1218. [Google Scholar]
- Tian, W.S.; Zhou, W.S.; Jiang, B.; Pan, X.F. Studies on Steroidal Plant-Growth Regulator IX: The Preparation of 22R-Penta-Nor-Brassinolides and 22S-24,25,26,27,28-Penta-Nor-Brassinolides. Acta Chim. Sin. 1989, 47, 1017–1021. [Google Scholar]
- Zhou, W.S. The Synthesis of Brassinosteroid. Pure Appl. Chem. 1989, 61, 431–434. [Google Scholar] [CrossRef]
- Zhou, W.S.; Jiang, B.; Pan, X.F. Studies on Steroidal Plant-Growth Hormones VIII: The Regioselective Synthesis of Methyl 3-α-Hydroxy-7-Oxa-6-Oxo-B-Homo-5-α-Cholanate. Acta Chim. Sin. 1989, 47, 182–185. [Google Scholar]
- Zhou, W.-S.; Biao, J.; Pan, X.-F. A Novel Synthesis of Brassinolide and Related Compounds. J. Chem. Soc. Chem. Commun. 1989, 10, 612–614. [Google Scholar]
- Wu, S.Z.; Zhou, W.S. Study on the Syntheses of Brassinolide and Related-Compounds. Part 14. Highly Stereoselective Construction of the Side-Chain of Brassinosteroids Utilizing the β-Alkylative 1,3-Carbonyl Transposition of the Steroidal 22-En-24-One. J. Chem. Soc. Perkin Trans. 1 1990, 1765–1767. [Google Scholar] [CrossRef]
- Zhou, W.S.; Zhou, H.Q.; Wang, Z.Q. Studies on Synthesis of Plant-Growth Hormone Steroids. Part 16. Stereoselective Synthesis of 26,27-Dinorbrassinolide. J. Chem. Soc. Perkin Trans. 1 1990, 2281–2286. [Google Scholar] [CrossRef]
- Zhou, W.S.; Jiang, B.; Pan, X.F. Stereoselective Synthesis of the Brassinolide Side-Chain—Novel Syntheses of Brassinolide and Related-Compounds. Tetrahedron 1990, 46, 3173–3188. [Google Scholar] [CrossRef]
- Zhou, W.S.; Huang, L.F.; Sun, L.Q.; Pan, X.F. Studies on A Steroidal Plant-Growth Regulator. Part 26. Stereoselective Construction of the Brassinolide Side-Chain—New Practical Syntheses of Brassinolide Analogs from Hyodeoxycholic Acid. J. Chem. Soc. Perkin Trans. 1 1992, 2039–2043. [Google Scholar] [CrossRef]
- Zhou, W.S.; Huang, L.F. Studies on Steroidal Plant-Growth Regulator. 25. Concise Stereoselective Construction of Side-Chain of Brassinosteroid from the Intact Side-Chain of Hyodeoxycholic Acid—Formal Syntheses of Brassinolide, 25-Methylbrassinolide, 26,27-Bisnorbrassinolide and Their Related-Compounds. Tetrahedron 1992, 48, 1837–1852. [Google Scholar]
- Zhou, W.S.; Sun, L.Q.; Pan, X.F. Synthesis of Steroidal Plant-Growth Regulators. Part 23. Stereoselective Synthesis of Crinosterol and Brassicasterol from Hyodeoxycholic Acid. Acta Chim. Sin. 1992, 50, 1192–1199. [Google Scholar]
- Zhou, W.S.; Huang, L.F.; Sun, L.Q.; Pan, X.F. Studies on Steroidal Plant-Growth Regulator. Part 21. A Stereoselective Construction of Brassinosteroid Side-Chain—A New Practical Synthesis of Brassinolide and Its Analogs. Tetrahedron Lett. 1991, 32, 6745–6748. [Google Scholar] [CrossRef]
- Zhou, W.S.; Shen, Z.W. Study on the Synthesis of Brassinolide and Related-Compounds. Part 15. Formal Synthesis of Brassinolide via Stereoselective Sulfenate Sulfoxide Transformation. J. Chem. Soc. Perkin Trans. 1 1991, 2827–2830. [Google Scholar] [CrossRef]
- Huang, L.F.; Zhou, W.S. Studies on Steroidal Plant-Growth Regulators. Part 33. Novel Method for Construction of the Side-Chain of 23-Arylbrassinosteroids via Heck Arylation and Asymmetric Dihydroxylation as Key Steps. J. Chem. Soc. Perkin Trans. 1 1994, 3579–3585. [Google Scholar] [CrossRef]
- Zhou, W.; Jiang, B.; Shen, J. Synthesis of Cholesteric Lactones and Analogs as Plant Growth Regulators Assignee: Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. China Patent CN 1184113 A, 10 June 1998. [Google Scholar]
- Zhou, W.-S. Studies on Steroidal Plan Growth Regulator. Part 27. Stereoselective Construction of the Side Chain of Brassinosteroid by Employin a Separated Dialkylation and a Tandem Dialkylation of the Pyranone moiety. Chin. J. Chem. 1993, 11, 376–384. [Google Scholar] [CrossRef]
- Schwarz, S.; Onken, D.; Schubert, A. The steroid story of Jenapharm: From the late 1940s to the early 1970s. Steroids 1999, 64, 439–445. [Google Scholar] [CrossRef]
- Singhal, A.K.; Cohen, B.I.; Mosbach, E.H.; Une, M.; Stenger, R.J.; Mcsherry, C.K.; Maydonath, P.; Palaia, T. Prevention of Cholesterol-Induced Gallstones by Hyodeoxycholic Acid in the Prairie Dog. J. Lipid Res. 1984, 25, 539–549. [Google Scholar] [PubMed]
- Cohen-Solal, C.; Parquet, M.; Ferezou, J.; Serougne, C.; Lutton, C. Effects of Hyodeoxycholic Acid and α-Hyocholic Acid, two 6-α-Hydroxylated Bile-Acids, on Cholesterol and Bile-Acid Metabolism in the Hamster. BBA Lipid Lipid Metab. 1995, 1257, 189–197. [Google Scholar] [CrossRef]
- Iida, T.; Momose, T.; Tamura, T.; Matsumoto, T.; Chang, F.C.; Goto, J.; Nambara, T. Potential Bile-Acid Metabolites. 13. Improved Routes to 3-β,6-β-Dihydroxy-5-β-Cholanoic and 3-β,6-α-Dihydroxy-5-β-Cholanoic Acids. J. Lipid Res. 1988, 29, 165–171. [Google Scholar] [PubMed]
- Iida, T.; Tamaru, T.; Chang, F.C.; Niwa, T.; Goto, J.; Nambara, T. Potential Bile-Acid Metabolites. 20. A New Synthetic Route to Stereoisomeric 3,6-Dihydroxy-5-α-Cholanoic and 6-Hydroxy-5-α-Cholanoic Acids. Steroids 1993, 58, 362–369. [Google Scholar] [CrossRef]
- Yang, Y.X.; Zheng, L.T.; Shi, J.J.; Gao, B.; Chen, Y.K.; Yang, H.C.; Chen, H.L.; Li, Y.C.; Zhen, X.C. Synthesis of 5α-cholestan-6-one derivatives and their inhibitory activities of NO production in activated microglia: Discovery of a novel neuroinflammation inhibitor. Bioorg. Med. Chem. Lett. 2014, 24, 1222–1227. [Google Scholar] [CrossRef] [PubMed]
- Takatsuto, S.; Yazawa, N.; Ishiguro, M.; Morisaki, M.; Ikekawa, N. Stereoselective Synthesis of Plant Growth-Promoting Steroids, Brassinolide, Castasterone, Typhasterol, and Their 28-Nor Analogs. J. Chem. Soc. Perkin Trans. 1 1984, 139–146. [Google Scholar] [CrossRef]
- Sample Availability: Samples of compounds 5–16 are available from the authors.







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| C | 12 | 13 * | 14 | 15 | 16 |
|---|---|---|---|---|---|
| 1 | 32.38 | 32.87 | 32.36 | 32.88 | 27.46 |
| 2 | 27.89 | 28.96 | 27.88 | 22.13 | 30.75 |
| 3 | 68.87 | 66.01 | 68.85 | 64.88 | 66.31 |
| 4 | 25.26 | 28.70 | 25.28 | 30.81 | 35.63 |
| 5 | 52.58 | 52.83 | 52.56 | 41.78 | 79.65 |
| 6 | 211.86 | 215.65 | 211.84 | 176.76 | 175.35 |
| 7 | 46.71 | 47.59 | 46.73 | 70.44 | 38.07 |
| 8 | 37.91 | 39.41 | 37.93 | 39.45 | 35.27 |
| 9 | 53.73 | 55.03 | 53.72 | 58.35 | 57.97 |
| 10 | 41.26 | 43.71 | 41.24 | 36.29 | 39.85 |
| 11 | 21.06 | 22.17 | 21.09 | 24.80 | 22.17 |
| 12 | 39.44 | 40.78 | 39.47 | 39.68 | 39.66 |
| 13 | 43.02 | 44.16 | 43.07 | 42.66 | 42.71 |
| 14 | 55.79 | 57.18 | 55.76 | 55.72 | 56.00 |
| 15 | 25.00 | 28.42 | 25.00 | 28.23 | 25.24 |
| 16 | 23.88 | 24.92 | 23.87 | 27.82 | 27.93 |
| 17 | 56.74 | 57.89 | 56.75 | 51.45 | 55.48 |
| 18 | 12.01 | 12.41 | 12.01 | 11.79 | 11.78 |
| 19 | 12.39 | 12.68 | 12.40 | 14.51 | 11.53 |
| 20 | 35.24 | 36.60 | 35.25 | 35.31 | 34.83 |
| 21 | 18.19 | 18.72 | 18.17 | 18.14 | 18.12 |
| 22 | 30.67 | 32.01 | 30.65 | 31.01 | 30.99 |
| 23 | 30.84 | 32.24 | 30.83 | 32.49 | 31.15 |
| 24 | 174.67 | 178.19 | 179.20 | 174.60 | 174.64 |
| CH3O | 51.45 | - | - | 51.50 | 51.52 |
| CH3CO- | 170.31 | - | 170.33 | - | - |
| CH3CO- | 21.39 | - | 21.41 | - | - |
| C | 5 | 6 | 7 | 8 | 9 | 10 | 11 |
|---|---|---|---|---|---|---|---|
| 1 | 35.56 | 35.71 | 34.37 | 31.65 | 27.80 | 39.34 | 40.18 |
| 2 | 29.20 | 36.42 | 34.86 | 28.16 | 31.64 | 124.49 | 68.29 |
| 3 | 71.48 | 208.58 | 70.15 | 65.41 | 78.71 | 124.95 | 68.39 |
| 4 | 34.86 | 39.83 | 29.85 | 27.90 | 26.56 | 27.88 | 26.28 |
| 5 | 48.40 | 59.67 | 59.40 | 51.65 | 51.78 | 53.35 | 50.69 |
| 6 | 67.98 | 210.75 | 213.88 | 212.70 | 211.08 | 211.96 | 214.15 |
| 7 | 30.11 | 42.08 | 37.97 | 46.83 | 46.52 | 46.95 | 46.73 |
| 8 | 34.79 | 36.61 | 37.06 | 37.94 | 37.79 | 37.67 | 35.65 |
| 9 | 39.81 | 40.83 | 39.60 | 53.77 | 53.35 | 53.82 | 53.66 |
| 10 | 35.90 | 38.22 | 39.99 | 41.53 | 40.99 | 40.00 | 42.98 |
| 11 | 20.72 | 21.23 | 20.83 | 21.03 | 20.94 | 21.08 | 21.17 |
| 12 | 39.92 | 39.41 | 42.91 | 39.45 | 39.25 | 39.44 | 39.35 |
| 13 | 42.80 | 43.02 | 43.09 | 42.89 | 42.90 | 42.84 | 42.55 |
| 14 | 55.89 | 55.74 | 55.79 | 55.71 | 56.58 | 56.69 | 56.61 |
| 15 | 28.10 | 27.88 | 27.98 | 27.67 | 51.79 | 53.35 | 23.91 |
| 16 | 24.17 | 23.85 | 23.96 | 23.88 | 23.79 | 21.70 | 27.91 |
| 17 | 56.14 | 56.70 | 56.82 | 56.73 | 55.62 | 55.75 | 55.69 |
| 18 | 11.98 | 11.92 | 11.96 | 12.00 | 11.92 | 11.92 | 12.01 |
| 19 | 23.48 | 22.40 | 23.16 | 12.29 | 12.39 | 13.48 | 13.55 |
| 20 | 35.32 | 35.19 | 35.28 | 35.29 | 35.19 | 35.29 | 35.30 |
| 21 | 18.21 | 18.18 | 18.23 | 18.21 | 18.13 | 18.23 | 18.22 |
| 22 | 30.91 | 30.80 | 30.90 | 30.89 | 30.79 | 30.90 | 31.04 |
| 23 | 31.02 | 30.95 | 31.05 | 31.03 | 30.94 | 31.03 | 30.89 |
| 24 | 174.71 | 174.50 | 174.67 | 174.67 | 174.56 | 174.64 | 174.72 |
| CH3O- | 51.48 | 51.44 | 51.52 | 51.50 | 51.42 | 51.49 | 51.54 |
| MsO- | - | - | - | - | 38.38 | - | - |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Herrera, H.; Carvajal, R.; Olea, A.F.; Espinoza, L. Structural Modifications of Deoxycholic Acid to Obtain Three Known Brassinosteroid Analogues and Full NMR Spectroscopic Characterization. Molecules 2016, 21, 1139. https://doi.org/10.3390/molecules21091139
Herrera H, Carvajal R, Olea AF, Espinoza L. Structural Modifications of Deoxycholic Acid to Obtain Three Known Brassinosteroid Analogues and Full NMR Spectroscopic Characterization. Molecules. 2016; 21(9):1139. https://doi.org/10.3390/molecules21091139
Chicago/Turabian StyleHerrera, Heidy, Rodrigo Carvajal, Andrés F. Olea, and Luis Espinoza. 2016. "Structural Modifications of Deoxycholic Acid to Obtain Three Known Brassinosteroid Analogues and Full NMR Spectroscopic Characterization" Molecules 21, no. 9: 1139. https://doi.org/10.3390/molecules21091139
APA StyleHerrera, H., Carvajal, R., Olea, A. F., & Espinoza, L. (2016). Structural Modifications of Deoxycholic Acid to Obtain Three Known Brassinosteroid Analogues and Full NMR Spectroscopic Characterization. Molecules, 21(9), 1139. https://doi.org/10.3390/molecules21091139