
(+)-Podocarpic Acid as Chiral Template in the Synthesis of Aphidicolane, Stemodane and Stemarane Diterpenoids † †


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
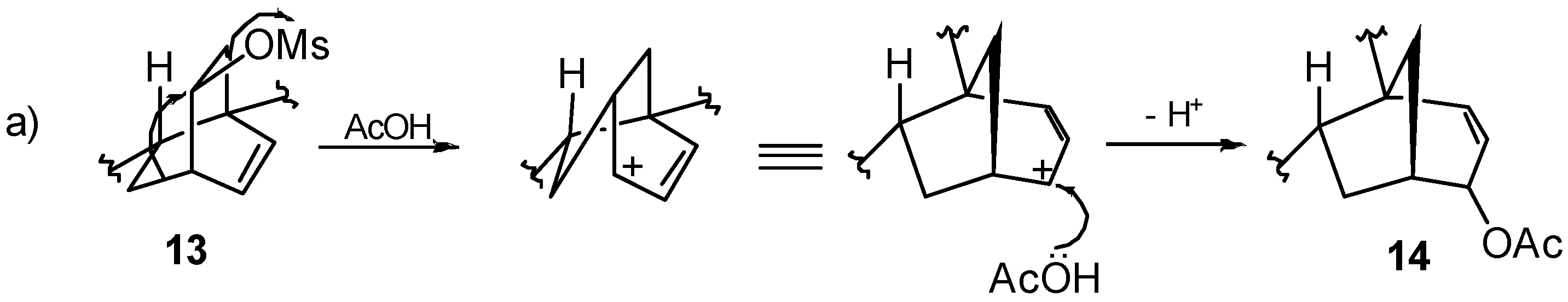{kind=link}
{kind=link}
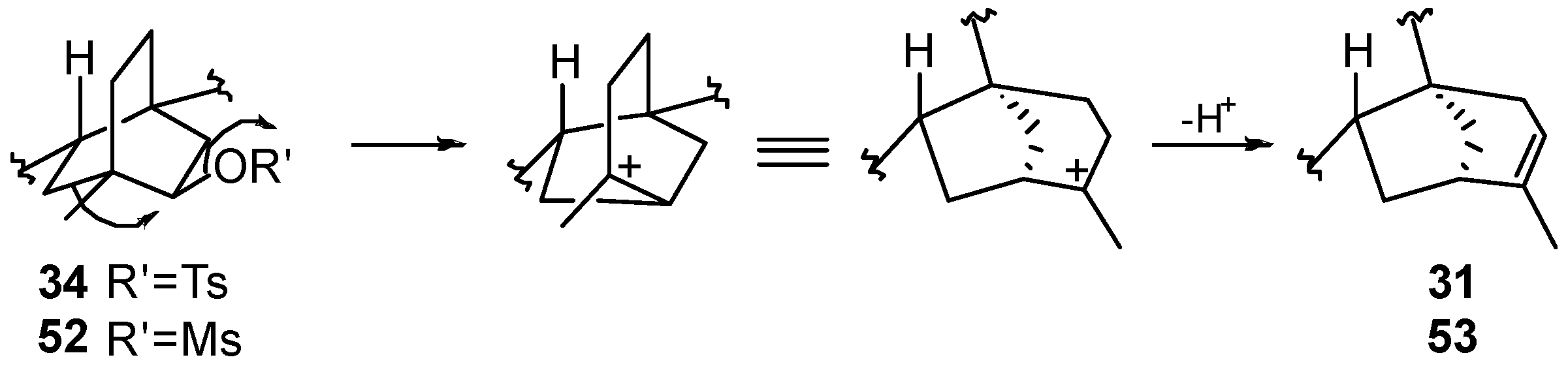{kind=link}
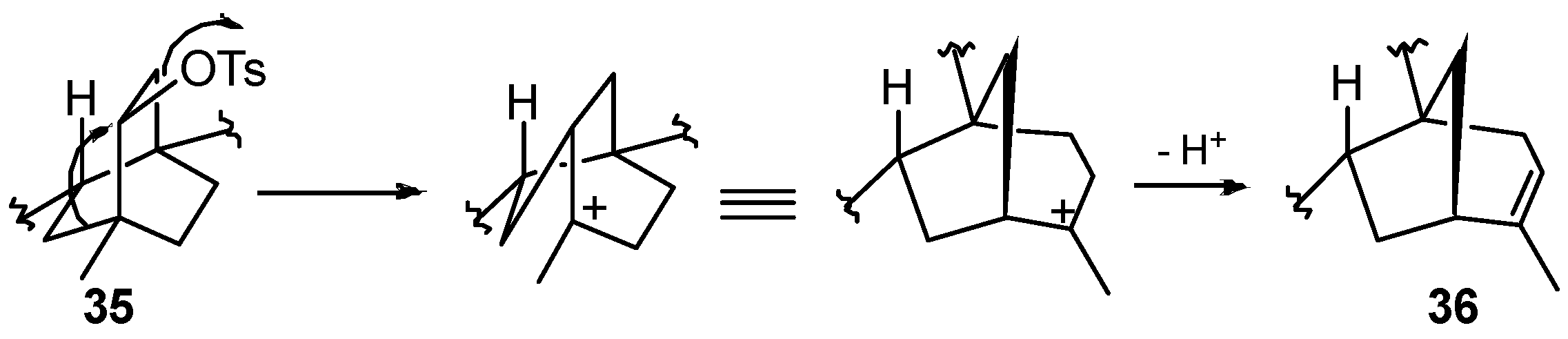{kind=link}
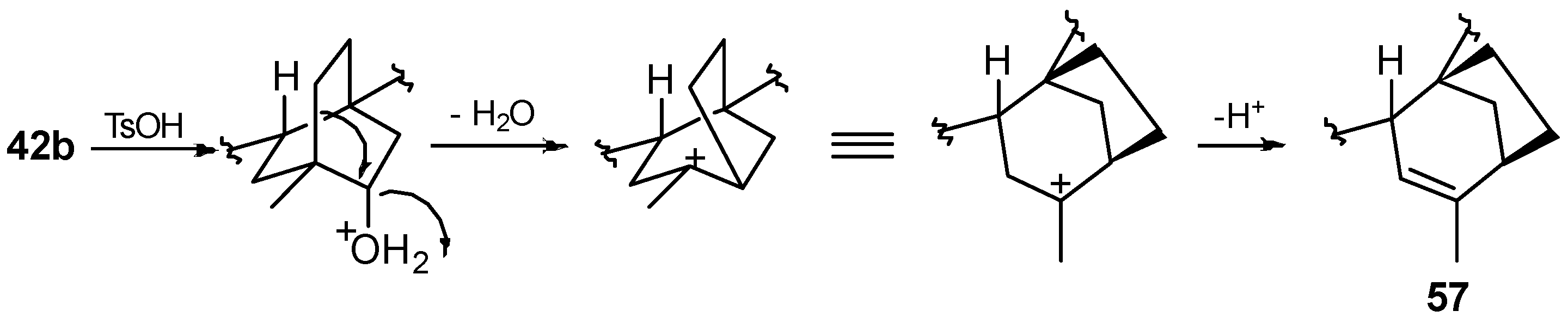{kind=link}
{kind=link}
{kind=link}
Abstract
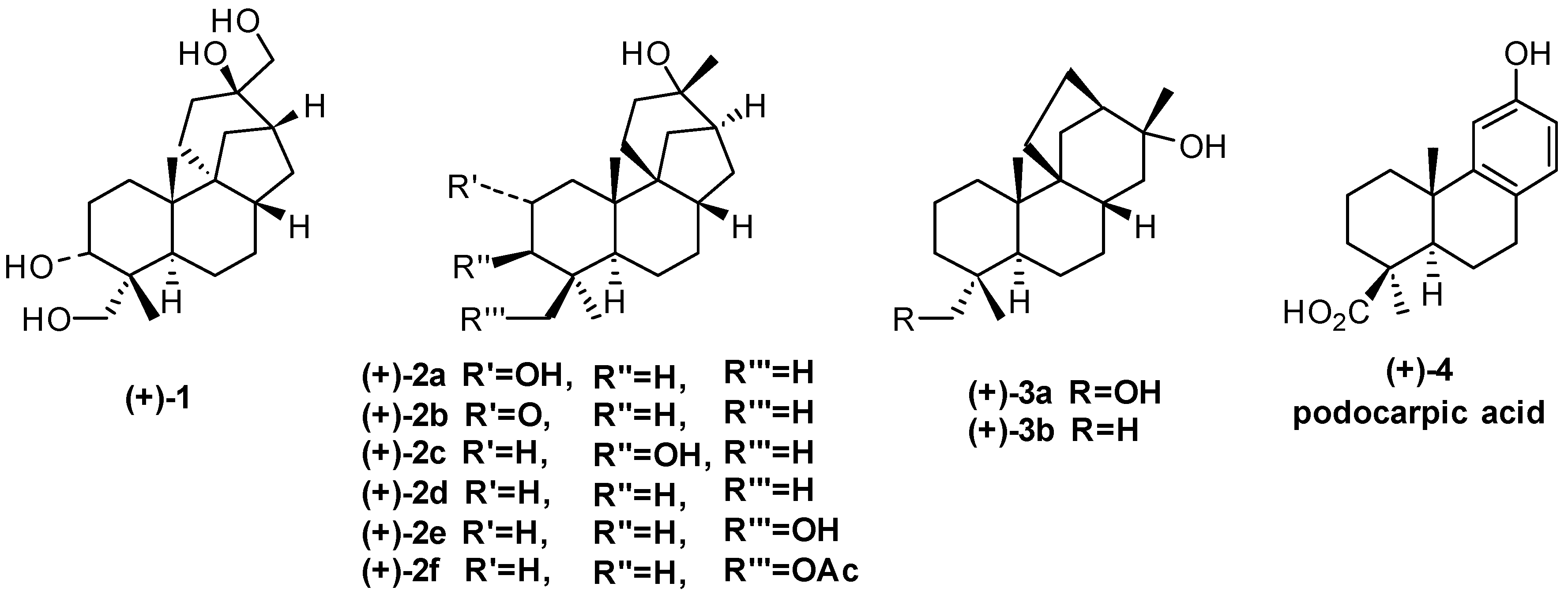
:1. Aphidicolane, Stemodane and Stemarane Diterpenoids
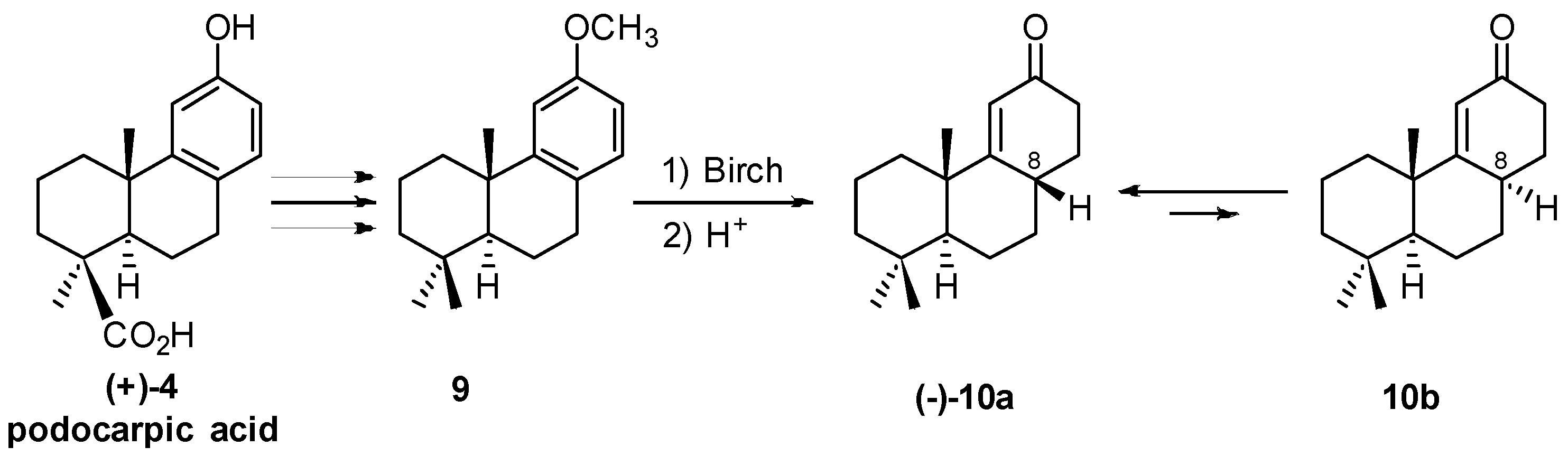
2. 1982 Synthesis of 17-Noraphidicolan-16-one and 17-Norstemodan-16-one from (+)-Podocarpic Acid via (−)-9(11)-Podocarpen-12-one (1982)
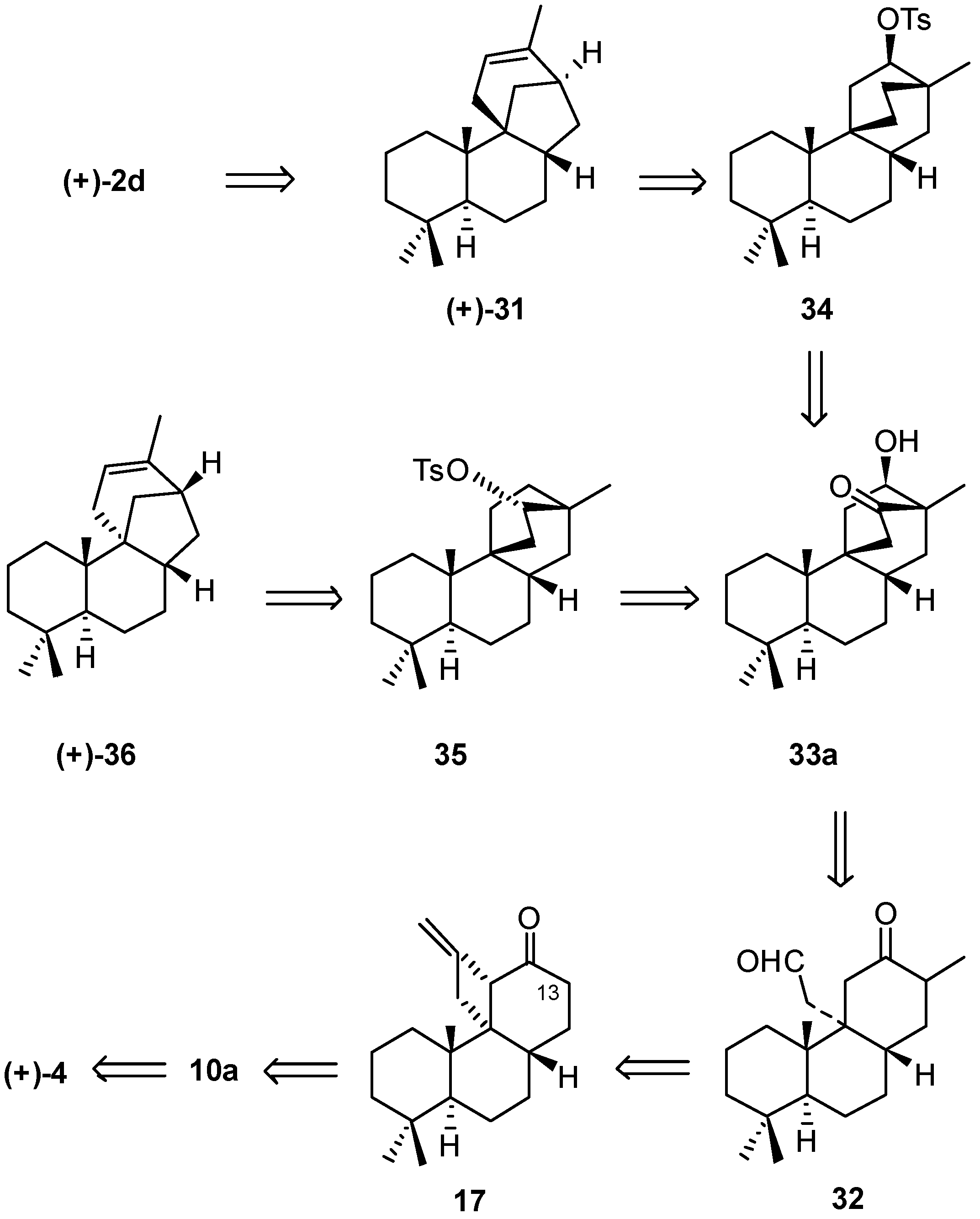
2.1. Retrosynthetic Analysis and Strategy
2.2. (+)-Podocarpic Acid as Chiral Template and (−)-9(11)Podocarpen-12-one as Suitable Chiron for Obtaining of 17-Noraphidicolan-16-one and 17-Norstemodan-16-one
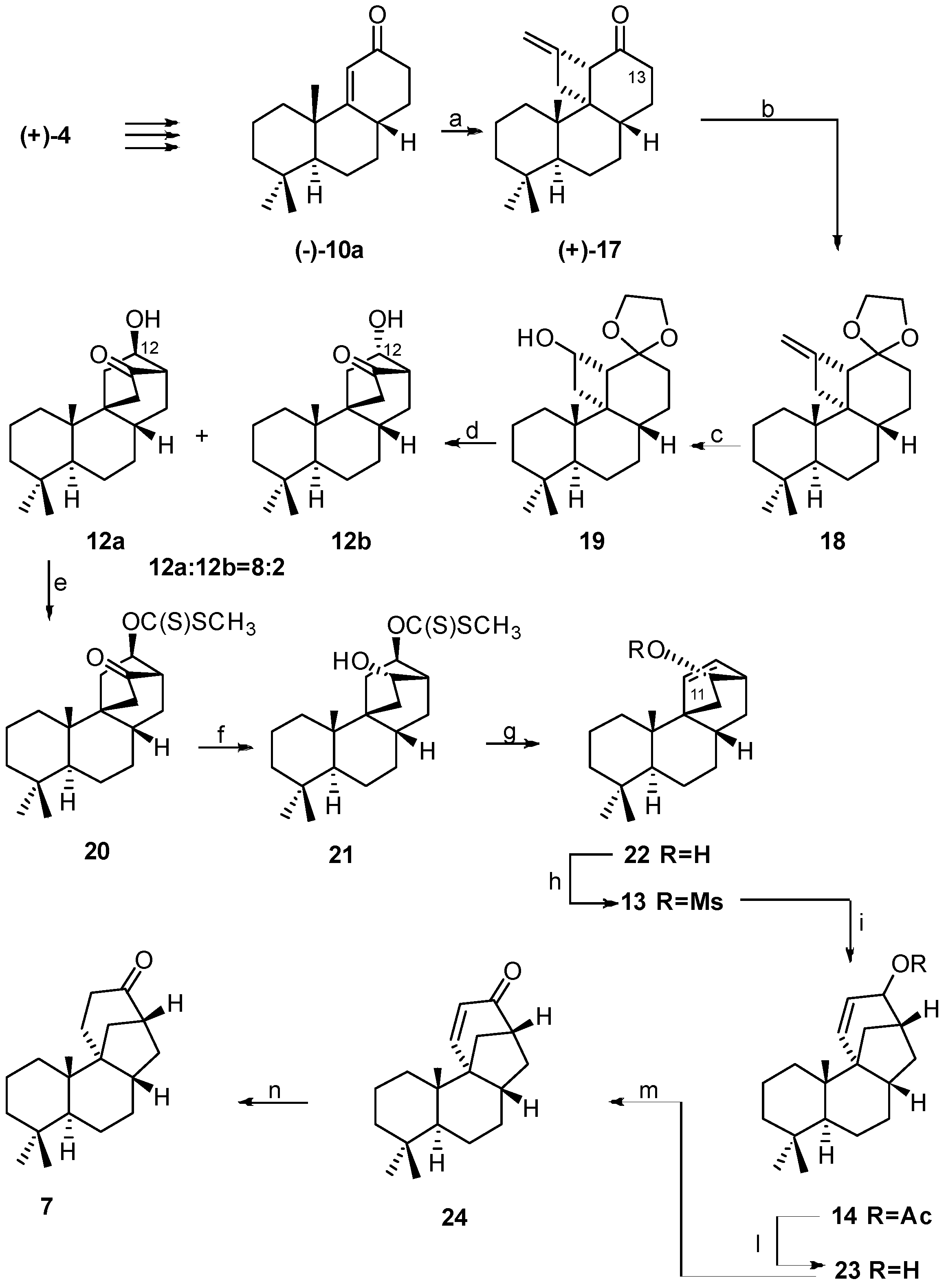
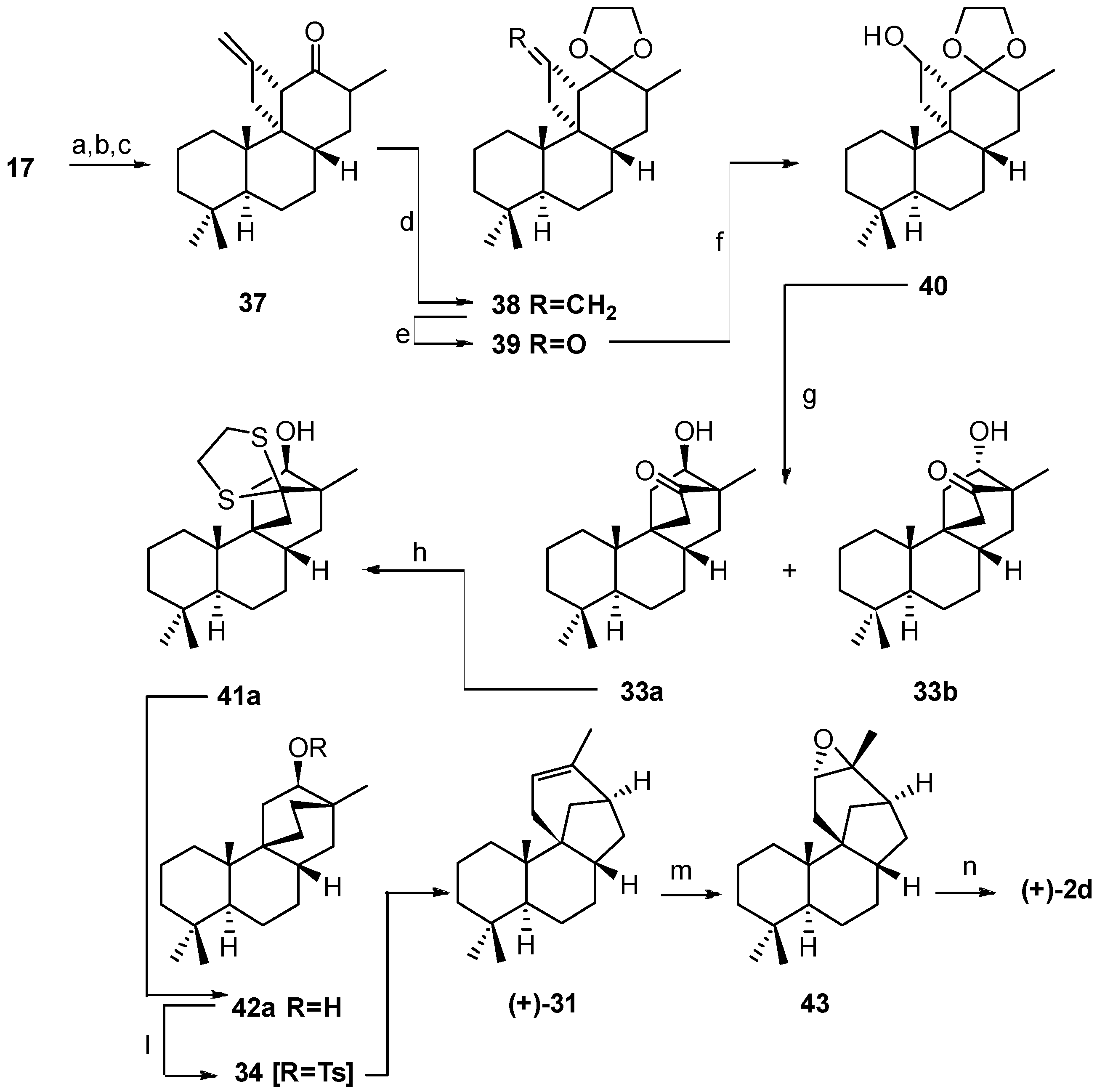
2.3. Synthesis of 17-Noraphidicolan-16-one (1982)
- protection of the HO-C(12);
- reduction of the carbonyl group to the corresponding 15α-alcohol;
- introduction of a double bond at C(11) in order to make the rearrangement quantitative.
2.4. Synthesis of 17-Norstemodan-16-one (1982)
3. Synthesis of (+)-Stemod-12-ene, (+)-2-Deoxystemodinone and (+)-Aphidicol-15-ene from (+)-Podocarpic Acid via (−)-9(11)-Podocarpen-12-one (1983–1984)
3.1. Retrosynthetic Analysis and Strategy
3.2. Synthesis of (+)-Stemod-12-ene and (+)-2-Deoxystemodinone (1983)
3.3. Synthesis of (+)-Aphidicol-15-ene (1984)
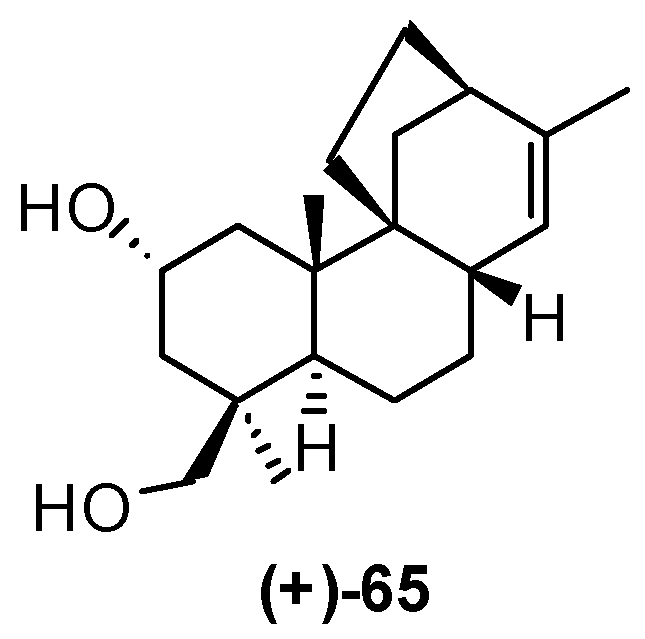
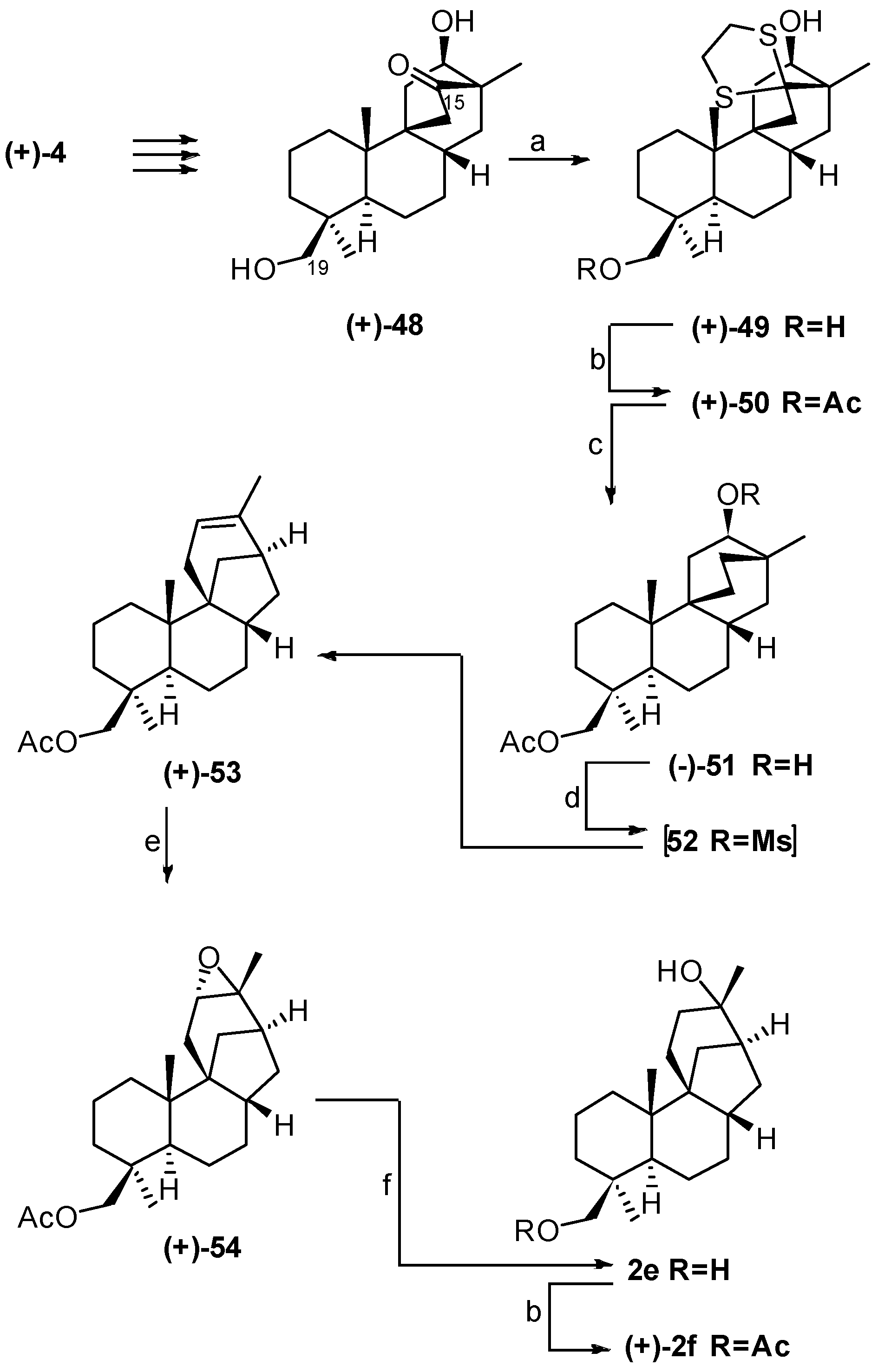
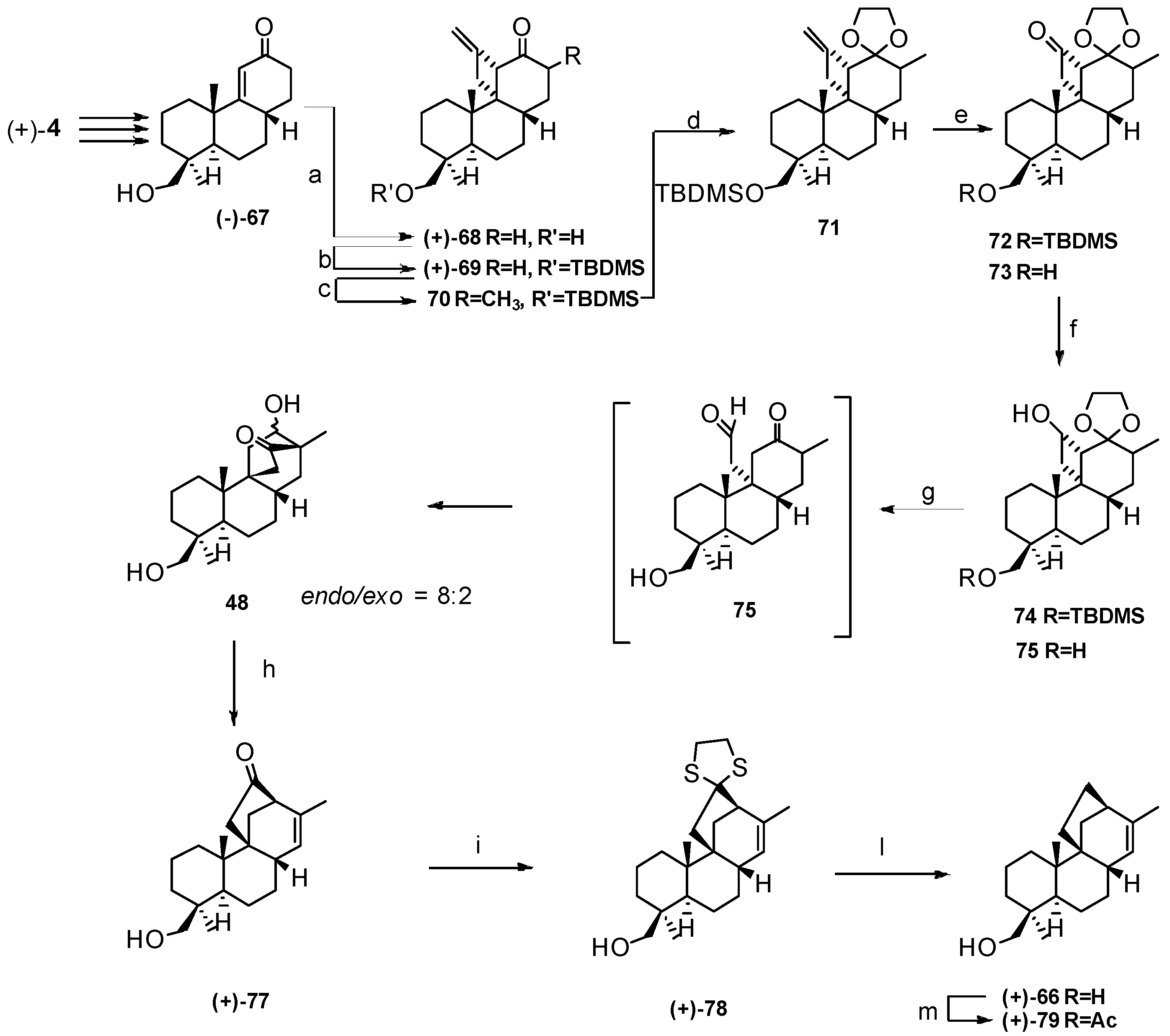
4. Proof of the Structure of the Stemodia chilensis Tetracyclic Diterpenoid (+)-19-Acetoxy-stemodan-12-ol by Synthesis from (+)-Podocarpic Acid (2016)
5. Synthesis of (+)-Stemar-13-ene, (+)-18-Deoxystemarin from (+)-Podocarpic Acid via 9(11)-Podocarpen-12-one (1991–2012)
5.1. Retrosynthetic Analysis and Strategy
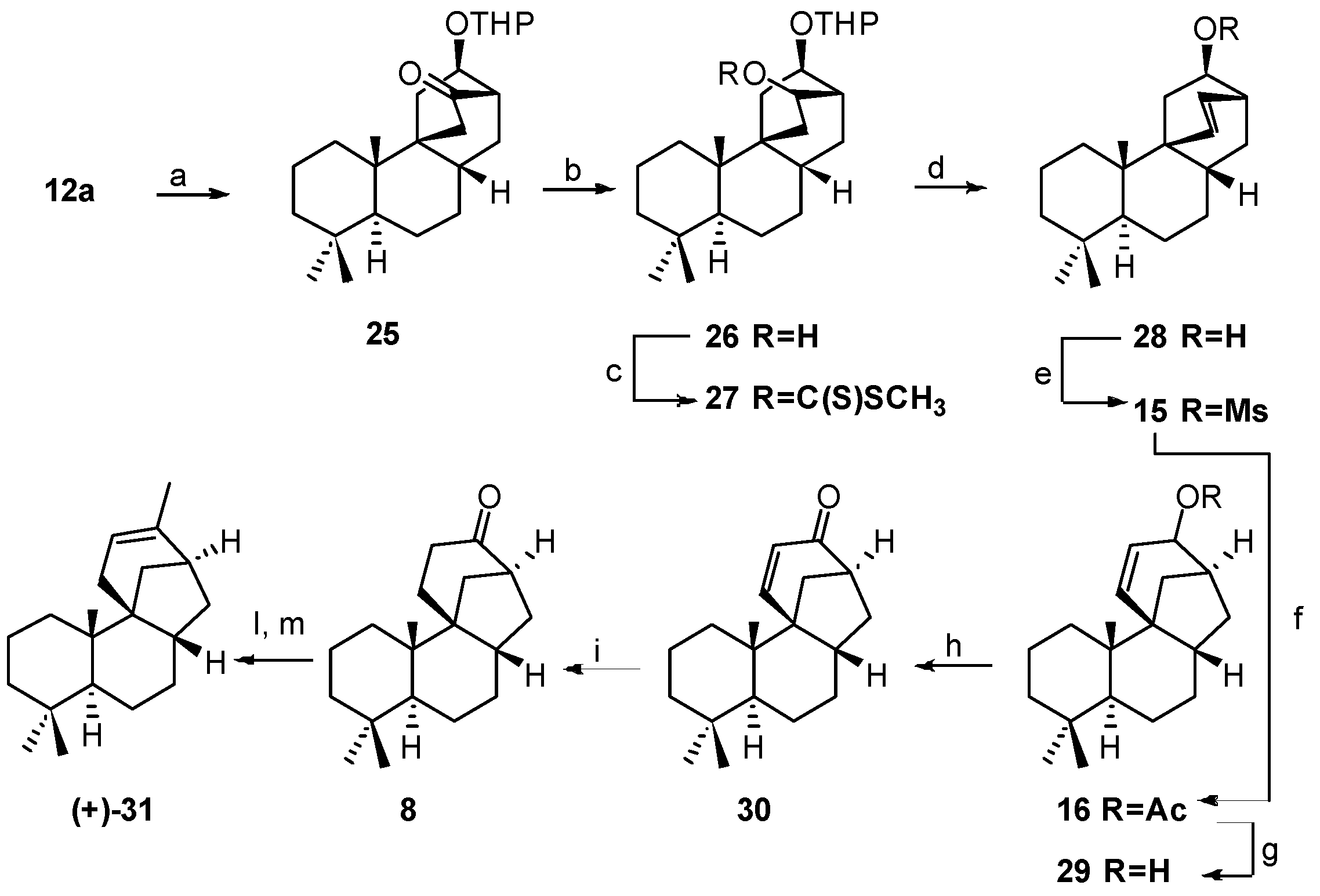
5.2. Diastereoselective Synthesis of (+)-Stemar-13-ene and (+)-18-Deoxystemarin by Inversion of Configuration at C(12) of Suitable Derivatives of the 13-Methyl-12-endo-hydroxybicyclo[2.2.2]octan-15-one Intermediate (1991)
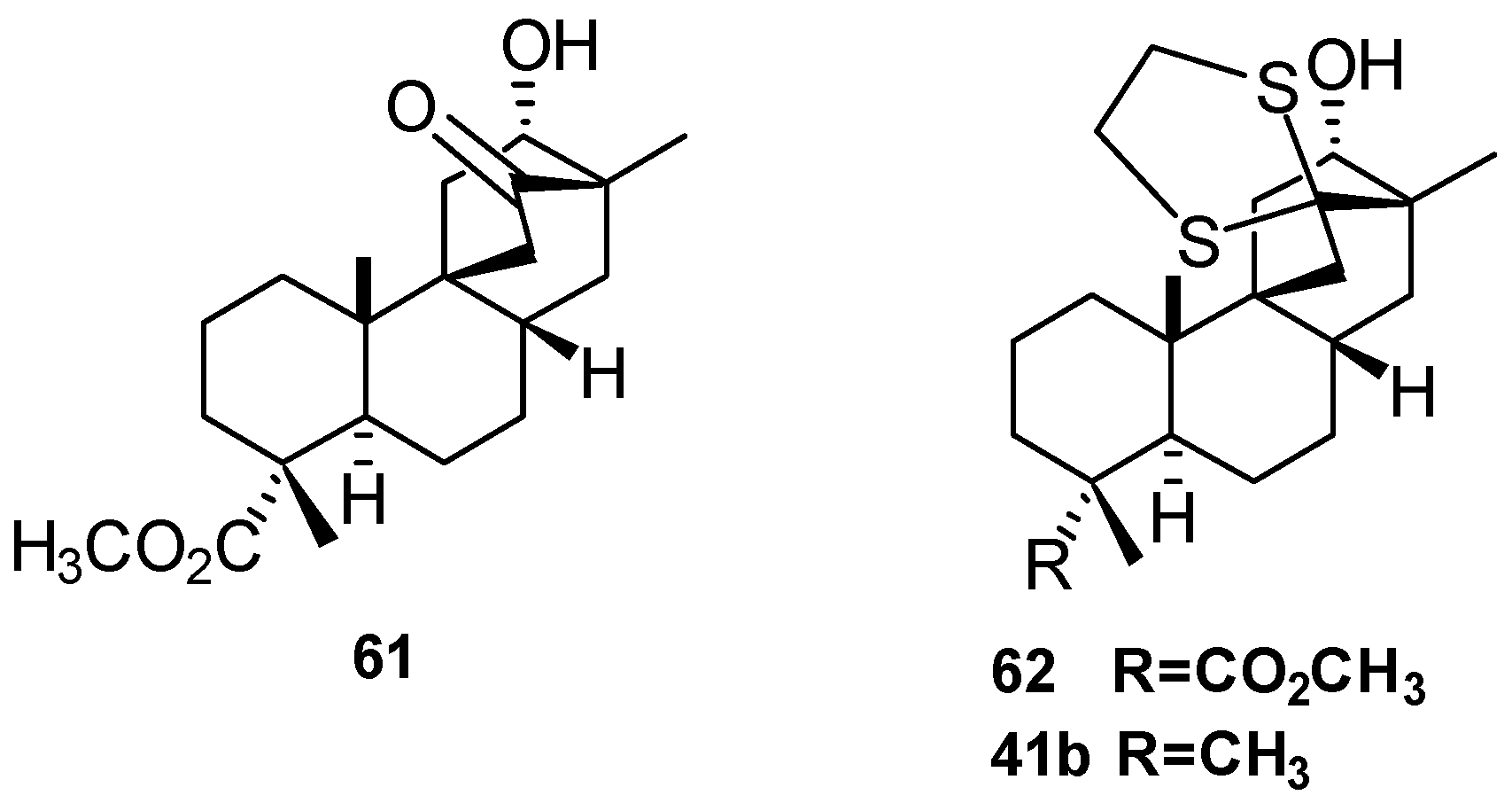
5.3. Synthesis of (+)-Stemar-13-ene and (+)-18-Deoxystemarin: Expeditious Preparation of the Key 13-Methyl-12-exo-hydroxybicyclo[2.2.2]octan-15-one Ethylene Dithioacetal (2008)
6. Regio- and Diastereoselective Synthesis of (+)-Stemar-13-ene and (+)-18-Deoxystemarin by the 6-Hydroxy-1-methylbicyclo[2.2.2]octan-2-one → 4-Methylbicyclo[3.2.1]oct-3-en-6-one Skeletal Rearrangement (2011)
6.1. Retrosynthetic Analysis and Strategy
6.2. Synthesis of (+)-Stemar-13-ene
7. Regio- and Diastereoselective Synthesis of (+)-2-Deoxyoryzalexin S from (+)-Podocarpic Acid (2012)
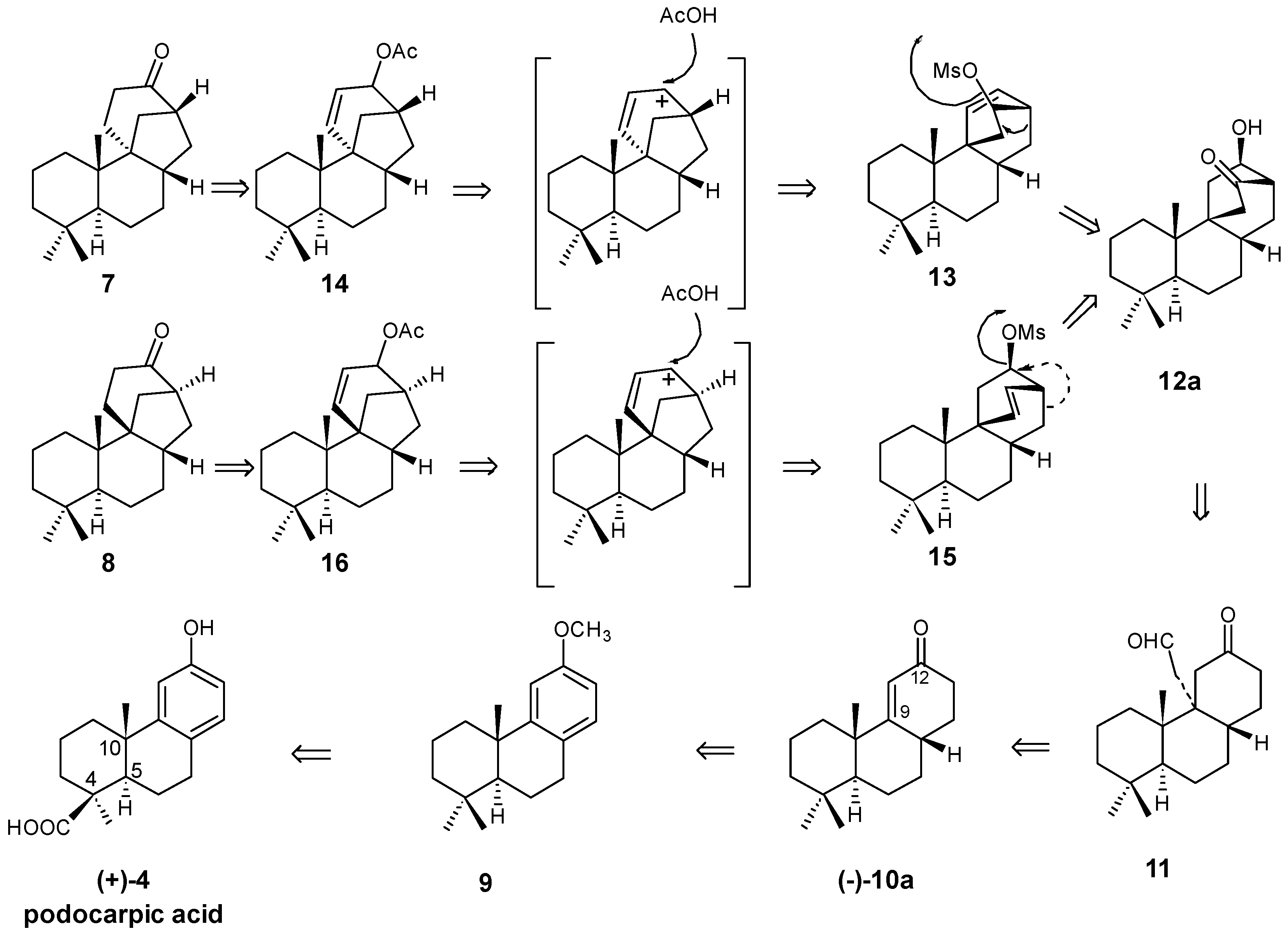
8. Main Skeletal Rearrangements: Mechanistic Aspects
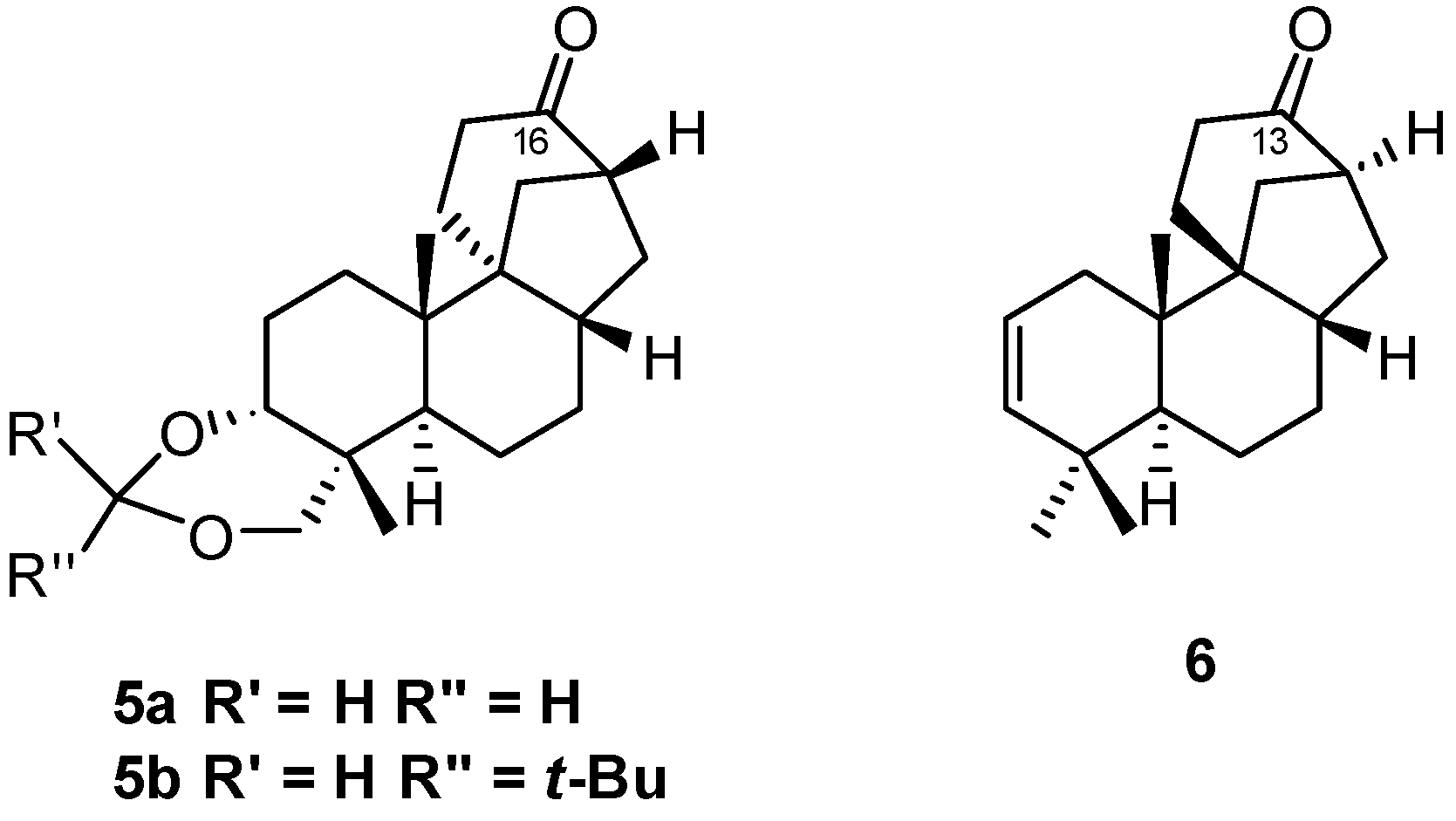
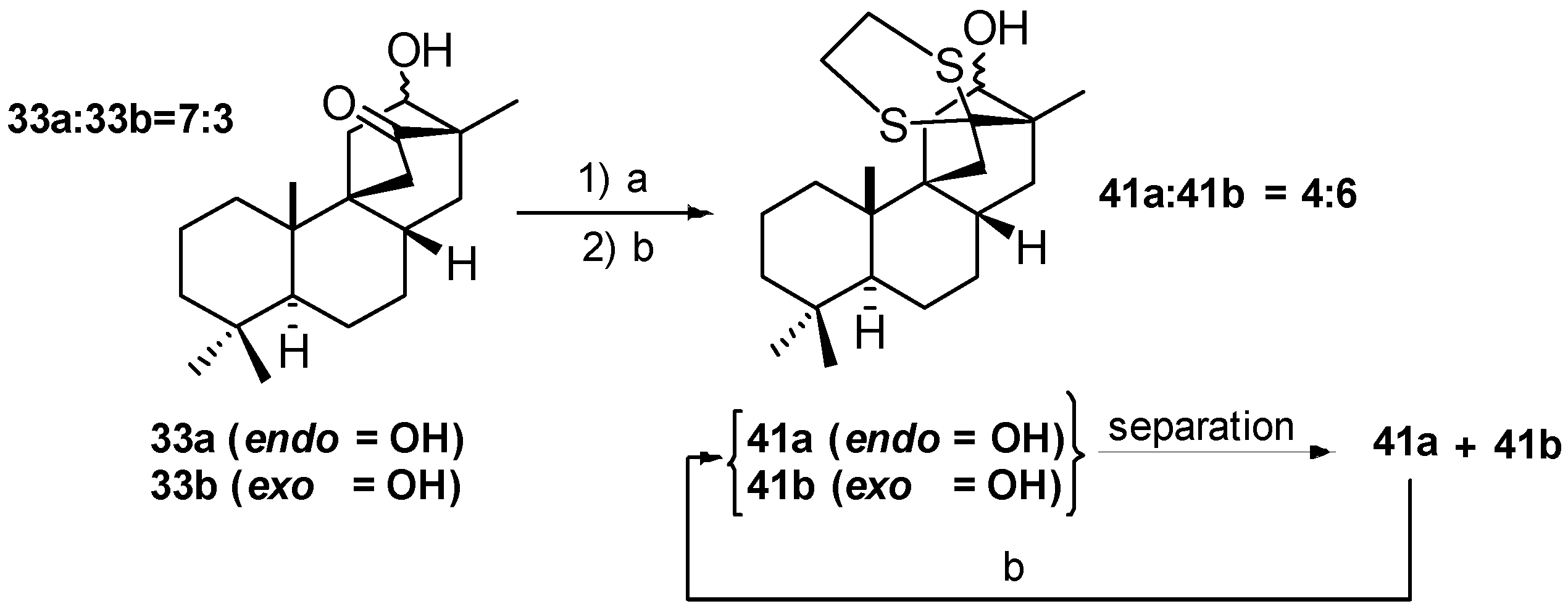
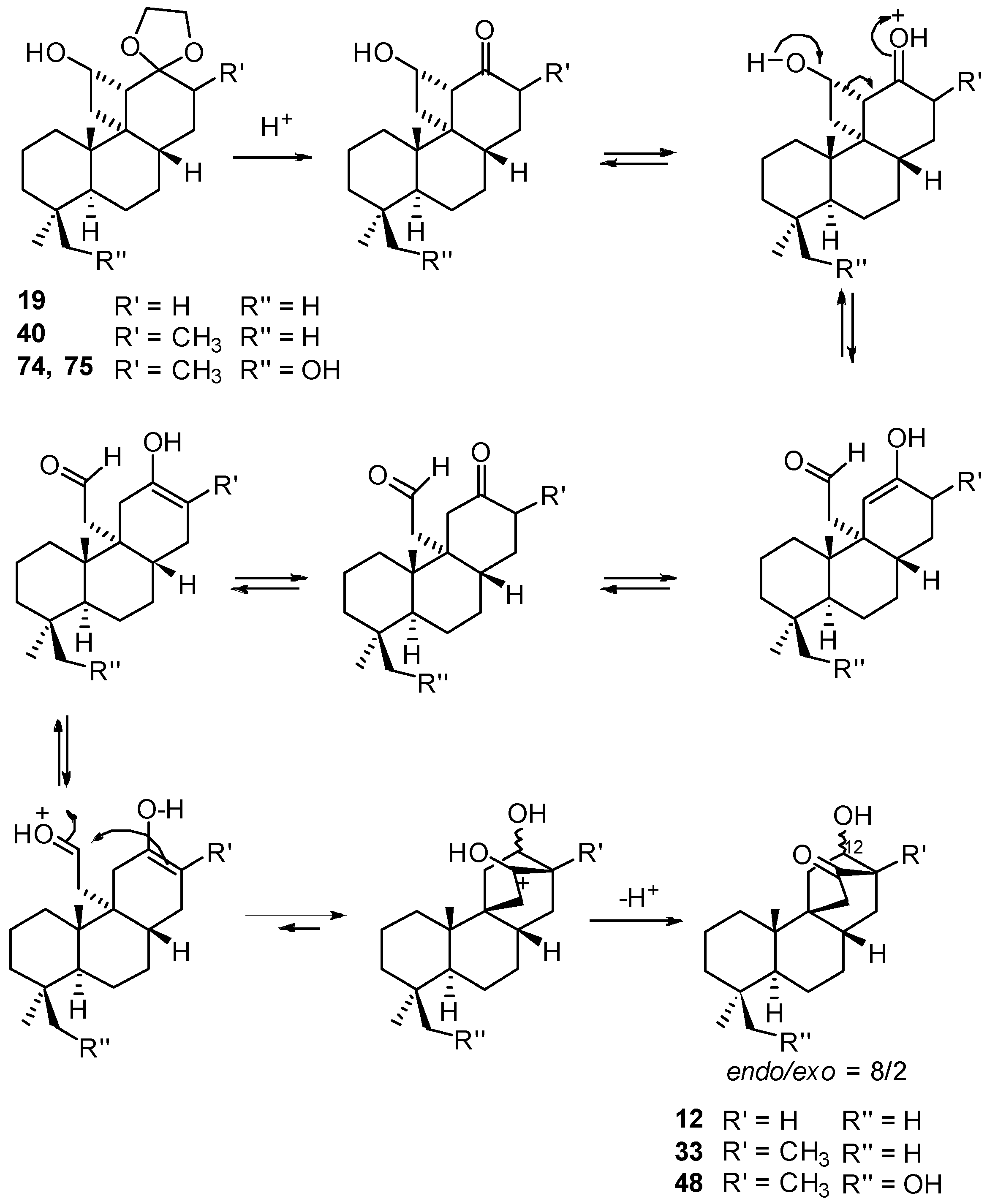
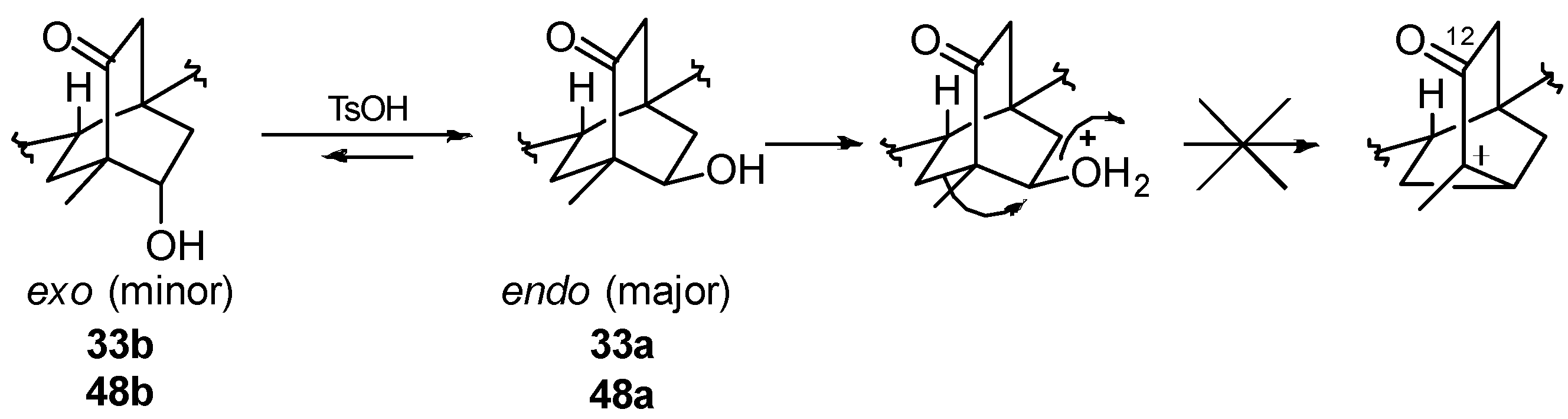
8.1. Conversion of Cyclobutanols 19, 40, 74 (and 75) into Hydroxybicyclo[2.2.2]octanones 12, 33, and 48, Respectively
8.2. Skeletal Rearrangements of Mesylates 13 and 15 into Allylic Acetates 14 and 16, Respectively
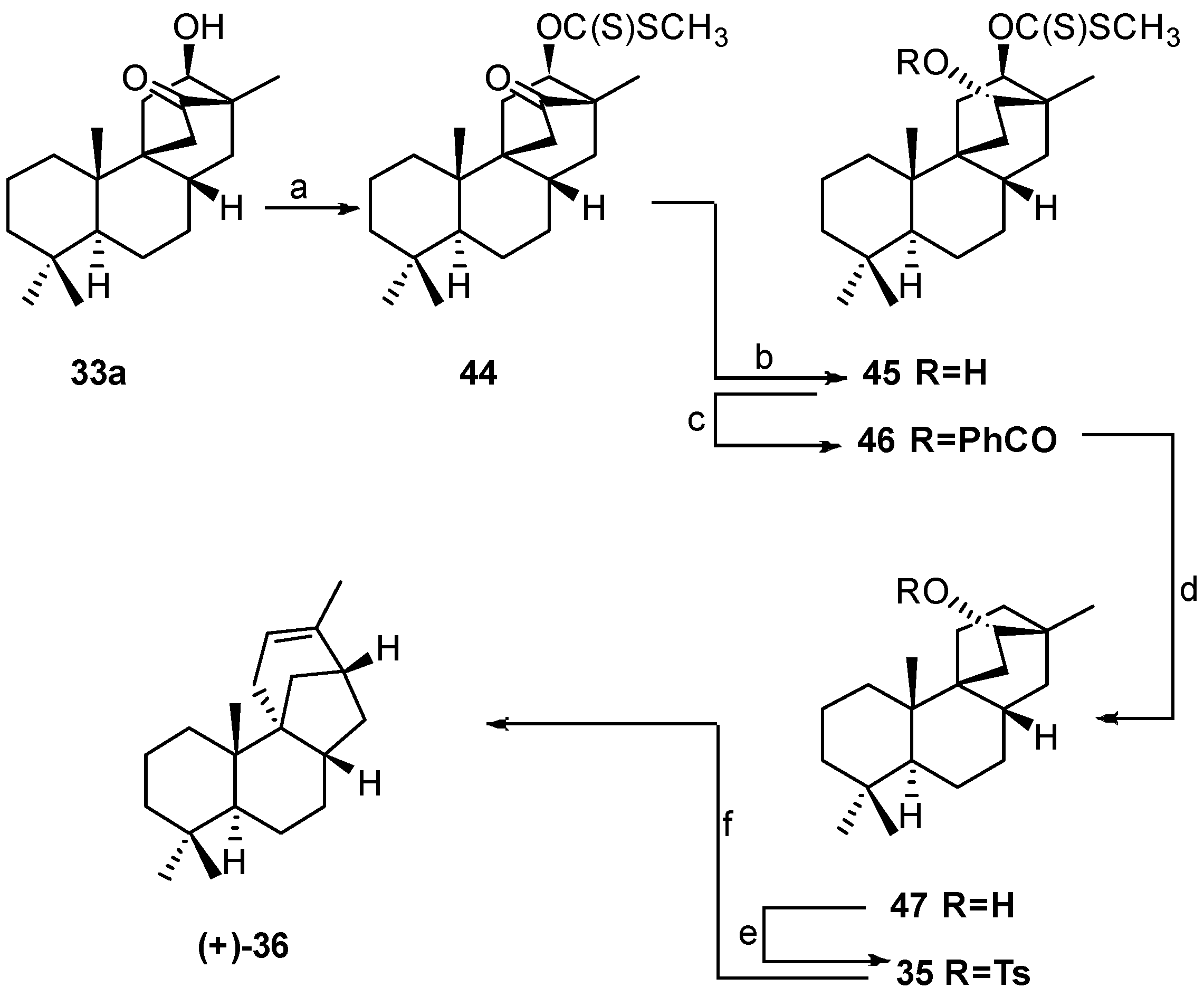
8.3. Skeletal Rearrangements of Compounds 34 and 52 into 31 and 53, Respectively
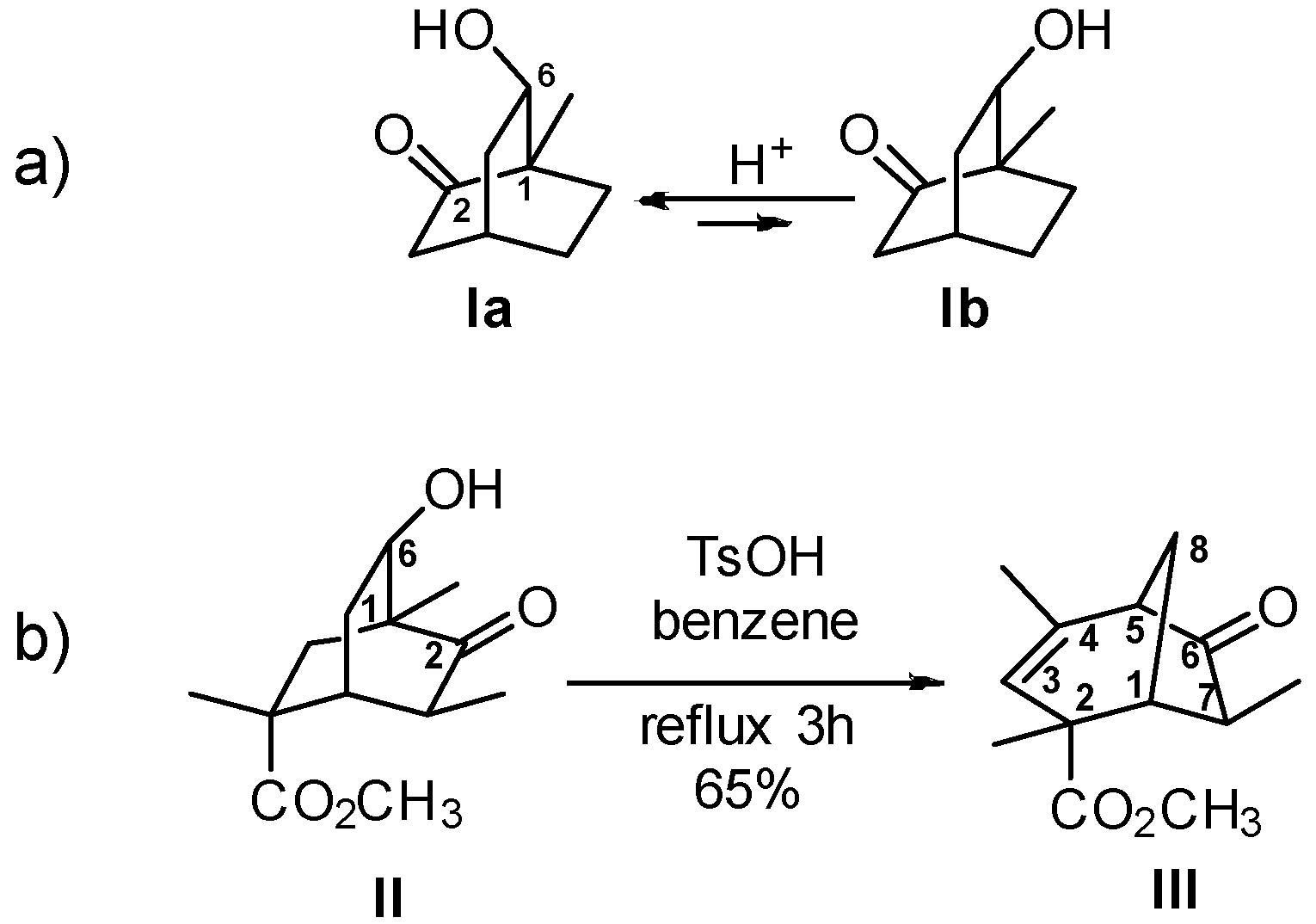
8.4. Skeletal Rearrangement of Tosylate 35 into 36
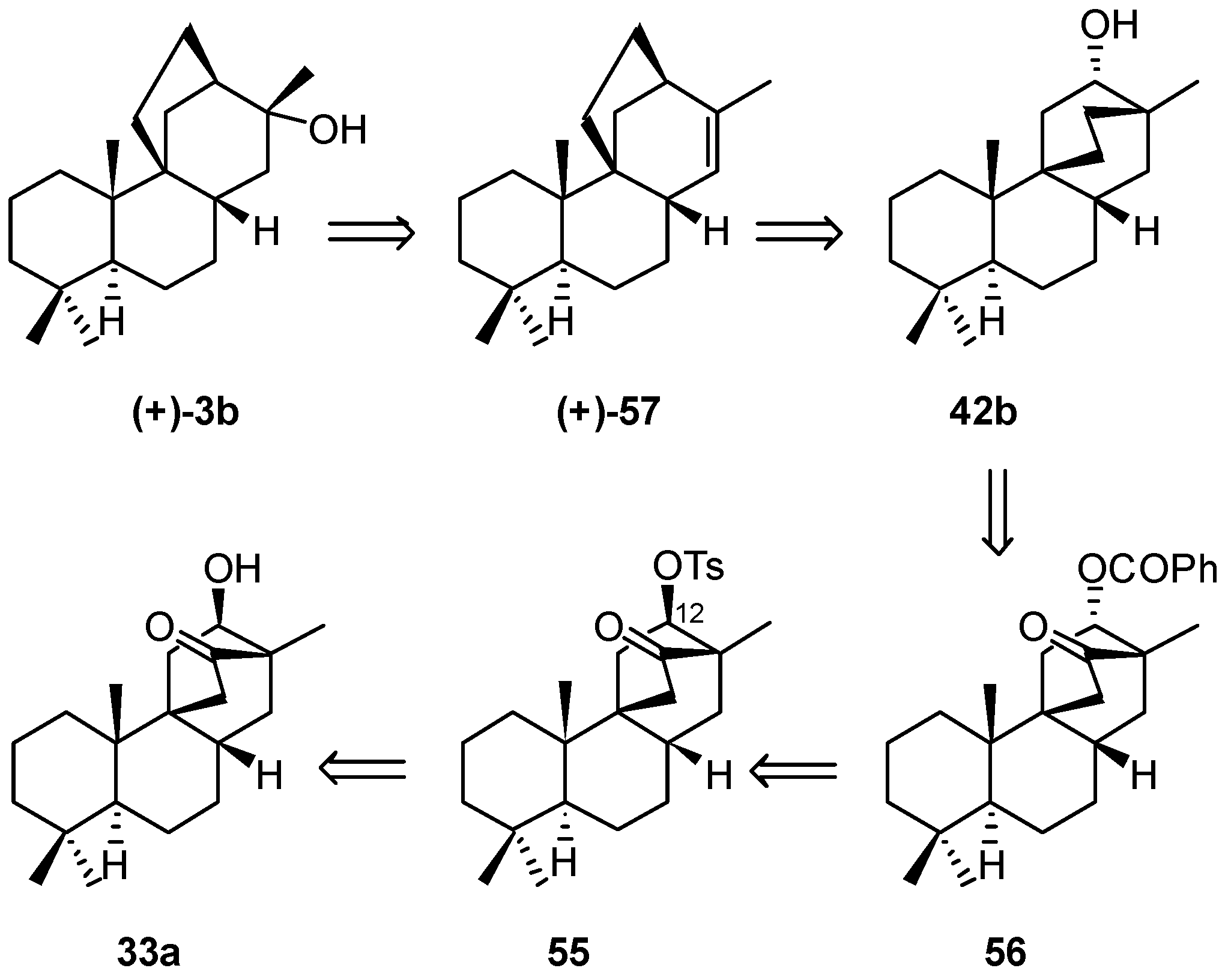
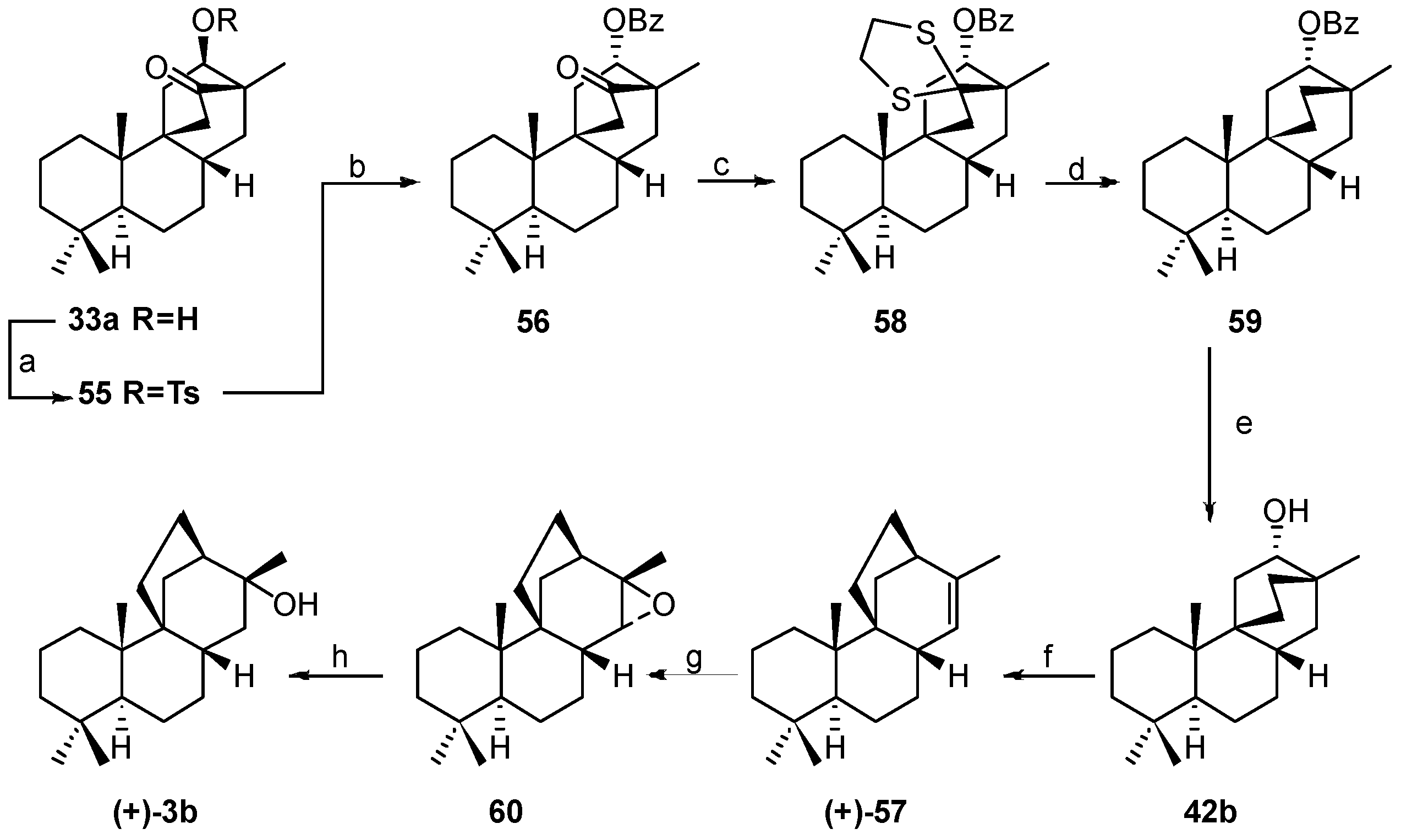
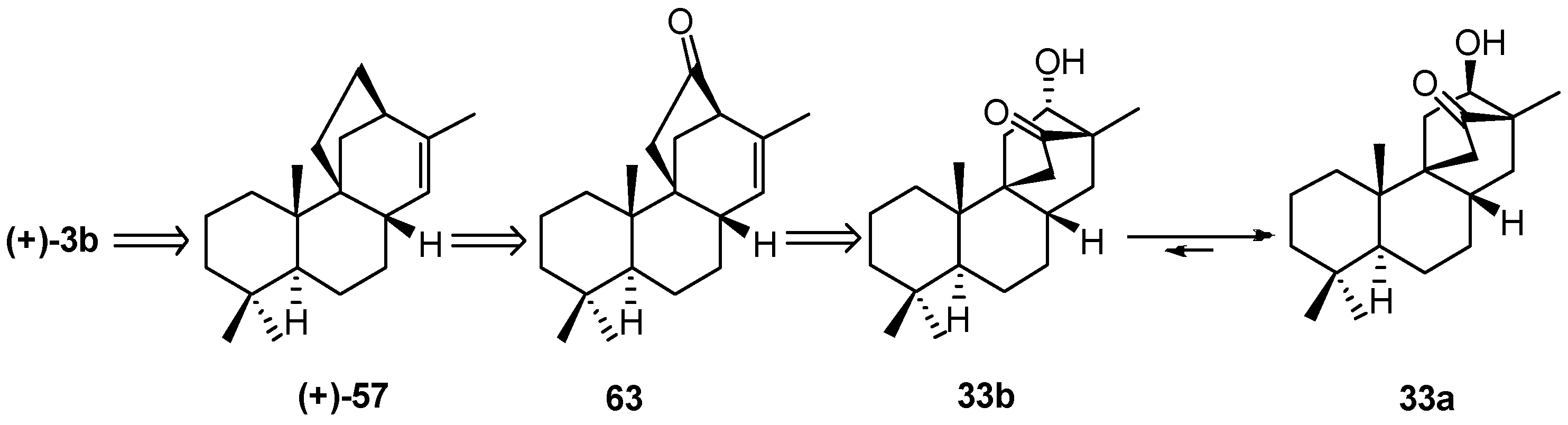
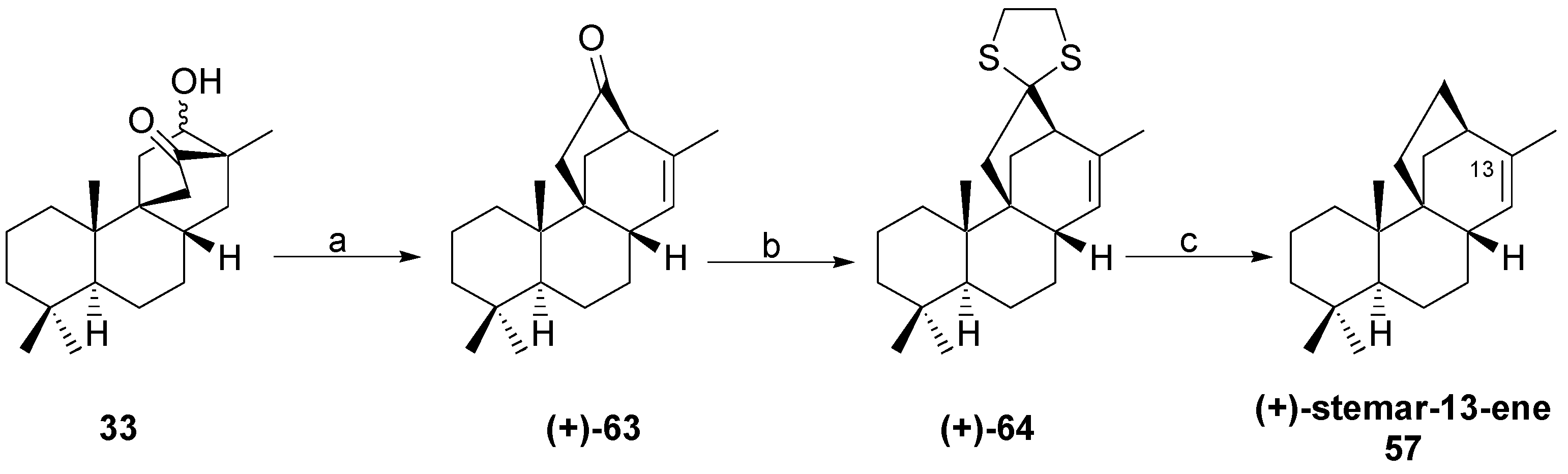
8.5. Skeletal Rearrangement of 42b into 57
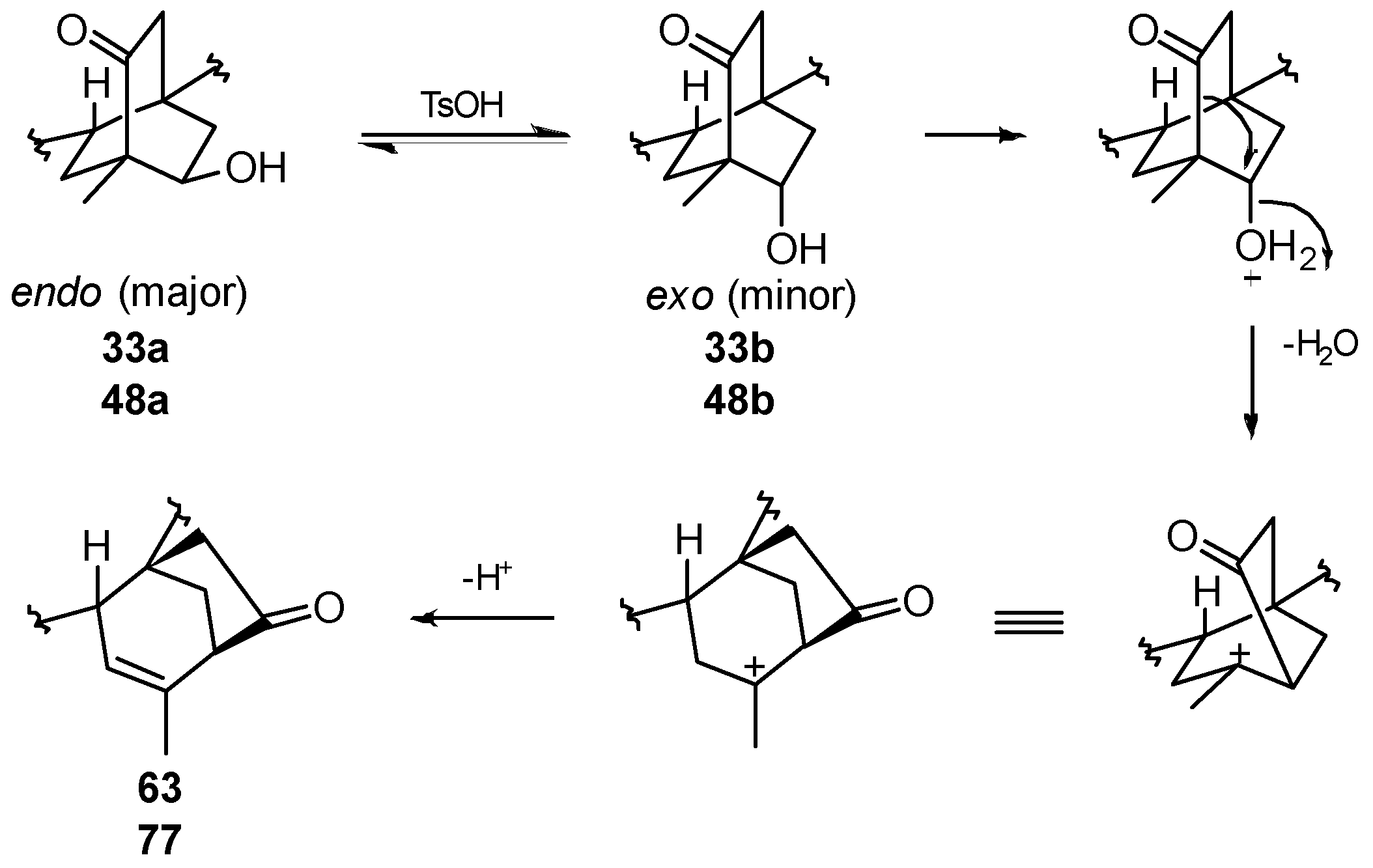
8.6. Skeletal Rearrangements of 33 and 48 into 63 and 77, Respectively
9. Conclusions
Acknowledgments
Conflicts of Interest
References
- Brundret, K.M.; Dalziel, W.; Hesp, B.; Jarvis, J.A.J.; Neidle, S. X-ray crystallographic determination of the structure of the antibiotic aphidicolin: A tetracyclic diterpenoid containing a new ring system. J. Chem. Soc. Chem. Commun. 1972, 18, 1027–1028. [Google Scholar] [CrossRef]
- Dalziel, W.; Hesp, B.; Stevenson, K.M.; Jarvis, J.A.J. The Structure and Absolute Configuration of the Antibiotic Aphidicolin: A Tetracyclic Diterpenoid Containing a New Ring System. Chem. Soc. Perkin Trans. 1 1973, 2841–2851. [Google Scholar] [CrossRef]
- Manchand, P.S.; White, J.D.; Wright, H.; Clardy, J. Structures of Stemodin and Stemodinone. J. Am. Chem. Soc. 1973, 95, 2705–2706. [Google Scholar] [CrossRef]
- Manchand, P.S.; Blount, J.F. X-ray Structure and Absolute Stereochemistry of Stemarin, a Diterpene with a New Skeleton. J. Chem. Soc. Chem. Commun. 1975, 21, 894–895. [Google Scholar] [CrossRef]
- Hufford, C.D.; Guerrero, R.O.; Doorembos, N.J. Two new diterpenes from Stemodia maritima L. J. Pharm. Sci. 1976, 65, 778–780. [Google Scholar] [CrossRef] [PubMed]
- Sir, J.L.; Barton, D.H.R. The Terpenes, Vol. III, Simonsen; Cambridge University Press: Cambridge, UK, 1961; p. 472. [Google Scholar]
- Trost, B.M.; Nishimura, Y.; Yamamoto, K.; McElvain, S.S. A Total Synthesis of Aphidicolin. J. Am. Chem. Soc. 1979, 101, 1328–1330. [Google Scholar] [CrossRef]
- McMurry, J.E.; Andrus, A.; Ksander, G.M.; Musser, J.H.; Johnson, M.A. Stereospecific Total Synthesis of Aphidicolin. J. Am. Chem. Soc. 1979, 101, 1330–1332. [Google Scholar] [CrossRef]
- Rizzo, C.J.; Smith, A.B., III. A New End Game for Aphidicolin. Tetrahedron Lett. 1988, 29, 2793–2796. [Google Scholar] [CrossRef]
- Rizzo, C.J.; Smith, A.B., III. Aphidicolin synthetic studies: A stereocontrolled end game. J. Chem. Soc. Perkin Trans. 1 1991, 5, 969–979. [Google Scholar] [CrossRef]
- Corey, E.J.; Tius, M.A.; Das, J. Total Synthesis of (±)-Aphidicolin. J. Am. Chem. Soc. 1980, 102, 1742–1244. [Google Scholar] [CrossRef]
- Van Tamelen, E.E.; Zawacky, S.R.; Russell, R.K.; Carlson, J.G. Biogenetic-Type Total Synthesis of (±)-Aphidicolin. J. Am. Chem. Soc. 1983, 105, 142–143. [Google Scholar] [CrossRef]
- Marini Bettolo, R.; Tagliatesta, P.; Lupi, A.; Bravetti, D. A Total Synthesis of Aphidicolin: Stereospecific Synthesis of (±)-3α,18-Dihydroxy-17-noraphidicolan-16-one. Helv. Chim. Acta 1983, 66, 1922–1928. [Google Scholar] [CrossRef]
- Lupi, A.; Patamia, M.; Marini Bettolo, R. A Total Synthesis of (±)-Aphidicolin: Regio and Stereoselective Conversion of 3α,18-Di-O-benzyl-17-nor-14-aphidicolen-16-one into (±)-Aphidicolin. Helv. Chim. Acta 1988, 71, 872–875. [Google Scholar] [CrossRef]
- Toyota, M.; Nishikawa, Y.; Fukumoto, K. An Expeditious and Efficient Formal Synthesis of (±)-Aphidicolin. Tetrahedron Lett. 1994, 35, 6495–6498. [Google Scholar] [CrossRef]
- Toyota, M.; Nishikawa, Y.; Seishi, T.; Fukumoto, K. Aphidicolin Synthesis (I)—Formal Synthesis of (+)-Aphidicolin by the Successive Intramolecular Diels-Alder Ractions. Tetrahedron 1994, 50, 10183–10192. [Google Scholar] [CrossRef]
- Toyota, M.; Nishikawa, Y.; Seishi, T.; Fukumoto, K. Aphidicolin Synthesis (II)—An Expeditious and Efficient Formal Synthesis of (±)-Aphidicolin. Tetrahedron 1994, 50, 11153–11166. [Google Scholar] [CrossRef]
- Tanaka, T.; Murakami, K.; Okuda, O.; Inoue, T.; Kuroda, T.; Kamei, K.; Murata, T.; Yoshino, H.; Imanishi, T.; Kim, S.-W.; et al. Synthetic Studies on Aphidicolane and Stemodane Diterpenes. IV. A Stereoselective Formal Total Synthesis of (±)-Aphidicolin. Chem. Pharm. Bull. 1995, 43, 193–197. [Google Scholar] [CrossRef]
- Tanaka, T.; Okuda, O.; Murakami, K.; Yoshino, H.; Mikamiyama, H.; Kanda, A.; Kim, S.-W.; Iwata, C.C. Synthetic Studies on Aphidicolane and Stemodane Diterpenes. V. A Facile Formal Total Synthesis of (±)-Aphidicolin via a Lewis Acid-Mediated Stereoselective Spiroannelation. Chem. Pharm. Bull. 1995, 43, 1407–1411. [Google Scholar] [CrossRef]
- Holton, R.A.; Kennedy, R.M.; Kim, H.B.; Krafft, M.E. Enantioselective Total Synthesis of Aphidicolin. J. Am. Chem. Soc. 1987, 109, 1597–1600. [Google Scholar] [CrossRef]
- Tanis, S.P.; Chuang, Y.-H.; Head, D.B. Furans in Synthesis. Formal Total Syntheses of (±)- and (+)-Aphidicolin. J. Org. Chem. 1988, 53, 4929–4938. [Google Scholar] [CrossRef]
- Toyota, M.; Nishikawa, Y.; Fukumoto, K. Enantioselective total synthetic route to (+)-aphidicolin. Tetrahedron Lett. 1995, 36, 5379–5382. [Google Scholar] [CrossRef]
- Corey, E.J.; Tius, M.A.; Das, J. Total Synthesis of (±)-Stemodin and (±)-Stemodinone. J. Am. Chem. Soc. 1980, 102, 7612–7613. [Google Scholar] [CrossRef]
- Atwal, K.S.; Marini Bettolo, R.; Sanchez, I.H.; Tsai, T.Y.R.; Wiesner, K. On the construction of the C/D ring systems of chasmanine and napelline by diene addition. Can. J. Chem. 1978, 56, 1102–1113. [Google Scholar] [CrossRef]
- Tsai, T.Y.R.; Nambiar, K.P.; Krikorian, D.; Botta, M.; Marini Bettolo, R.; Wiesner, K. A new synthesis of chasmanine and 13-desoxydelphonine: A preferred route to the aromatic intermediate. Can. J. Chem. 1979, 57, 2124–2134. [Google Scholar] [CrossRef]
- Sethi, S.P.; Atwal, K.S.; Marini Bettolo, R.; Tsai, T.Y.R.; Wiesner, K. A stereospecific synthesis of napelline. Can. J. Chem. 1980, 58, 1889–1891. [Google Scholar] [CrossRef]
- Wiesner, K.; Tsai, T.Y.R.; Huber, K.; Bolton, S.E.; Vlahov, R. Total synthesis of talatisamine, a delphinine type alkaloid. J. Am. Chem. Soc. 1974, 96, 4990–4992. [Google Scholar] [CrossRef]
- Tsai, T.Y.R.; Tsai, C.S.J.; Sy, W.W.; Shanbag, M.N.; Liu, W.C.; Lee, S.F.; Wiesner, K. A Stereospecific Total Synthesis of Chasmanine. Heterocycles 1977, 7, 217–226. [Google Scholar]
- Wiesner, K. Centenary lecture. Systematic Development of Strategy in the Synthesis of Polycyclic Polysubstituted Natural Products: The Aconite Alkaloids. Chem. Soc. Rev. 1977, 6, 413–430. [Google Scholar] [CrossRef]
- Wiesner, K. Total Synthesis of Delphinine-Type Alkaloids by Simple, Fourth Generation Methods. Pure Appl. Chem. 1979, 51, 689–703. [Google Scholar] [CrossRef]
- Walborsky, H.M.; Baum, M.E.; Youssef, A.A. Acetolysis of Bicyclo[2.2.2]octyl-2 p-Bromobenzenesulfonate and the Absolute Configurations of Bicyclo[2.2.2]octanol-2 and cis- and trans-Bicyclo[3.2.1]octanol-2. J. Am. Chem. Soc. 1961, 83, 988–993. [Google Scholar] [CrossRef]
- Goering, H.L.; Sloan, M.F. Ionic Reactions in Bicyclic Systems. II. Carbonium Ion Reactions in Bicyclo[2.2.2]octane and Bicyclo[3.2.1]octane Derivatives. J. Am. Chem. Soc. 1961, 83, 1397–1401. [Google Scholar] [CrossRef]
- Goering, H.L.; Sloan, M.F. Ionic Reactions in Bicyclic Systems. III. Solvolysis of Bicycloöctanyl and Bicycloöctenyl p-Toluenesulfonates. J. Am. Chem. Soc. 1961, 83, 1992–1999. [Google Scholar] [CrossRef]
- Kraus, H.L.; Chassin, C.; Chassin, R.; Schmutte, P. Bicyclische Verbindungen, XVIII Solvolytische und reduktive Umlagerung von Bicyclo[2.2.2]octyltosylaten. Liebigs Ann. Chem. 1970, 738, 97–112. [Google Scholar] [CrossRef]
- Bell, R.A.; Ireland, R.E. The construction of the C/D ring system present in the diterpenoid alkaloids atisine and garryfoline. Tetrahedron Lett. 1963, 4, 269–273. [Google Scholar] [CrossRef]
- De Santis, B.; Iamiceli, A.L.; Marini Bettolo, R.; Migneco, L.M.; Scarpelli, R.; Cerichelli, G.; Fabrizi, G.; Lamba, D. On the Diastereoselectivity of the Aqueous-Acid Catalyzed Intramolecular Aldol Condensation of 3-Oxocyclohexaneacetaldehydes. Helv. Chim. Acta 1998, 81, 2375–2387. [Google Scholar] [CrossRef]
- Migneco, L.M.; Leonelli, F.; Marini Bettolo, R. The intramolecular aldol condensation of 3-oxocyclohexaneacetaldehydes: A useful tool in the synthesis of natural products. Arkivoc 2004, 7, 253–265. [Google Scholar]
- Van Tamelen, E.E.; Carlson, J.G.; Russell, R.K.; Zawacky, S.R. Total Synthesis of (±)-Maritimol. J. Am. Chem. Soc. 1981, 103, 4615–4616. [Google Scholar] [CrossRef]
- Hanessian, S. Total Synthesis of Natural Products: The “Chiron” Approach; Baldwin, J.E., Ed.; Pergamon: Oxford, UK, 1983. [Google Scholar]
- Wenkert, E.; Sternberg, V.; Beak, P. Podocarpic Acid Derivatives. Synthesis of Nimbiol. J. Am. Chem. Soc. 1961, 83, 2320–2325. [Google Scholar] [CrossRef]
- Leonelli, F.; Borocci, S.; Migneco, L.M.; Marini Bettolo, R.; Lamba, D. The Formation of 8-Epipodocarp-9(11)en-12-one in the Course of the Preparation of Podocarp-9(11)-en-12-one from O-Methylpodocarpane and Related Studies. Helv. Chim. Acta 2002, 85, 2817–2826. [Google Scholar] [CrossRef]
- Guthrie, R.W.; Philipp, A.; Valenta, Z.; Wiesner, K. Synthesis in the diterpene alkaloid series III. A novel synthesis of a keto lactam. A photochemical approach to the C,D-ring system of atisine. Tetrahedron Lett. 1965, 34, 2945–2950. [Google Scholar] [CrossRef]
- Wiesner, K. On the stereochemistry of photoaddition between α,β-unsaturated ketones and olefins. Tetrahedron 1975, 31, 1655–1658. [Google Scholar] [CrossRef]
- Marini Bettolo, G.; Sahoo, S.P.; Poulton, G.A.; Tsai, T.Y.R.; Wiesner, K. On the stereochemistry of photoaddition between α,β-unsaturated ketones and olefins—II. Tetrahedron 1980, 36, 719–721. [Google Scholar] [CrossRef]
- Blount, J.F.; Gray, G.D.; Atwal, K.S.; Tsai, T.Y.R.; Wiesner, K. On the stereochemistry of photoaddition between α,β-unsaturated ketones and olefins. III. Tetrahedron Lett. 1980, 21, 4413–4416. [Google Scholar] [CrossRef]
- Valenta, K.; Grein, F. Excited states of acrolein: Ab initio model studies on α,β-unsaturated carbonyl compounds. Can. J. Chem. 1982, 60, 601–606. [Google Scholar] [CrossRef]
- Bravetti, D.; Marini Bettolo, R.; Lupi, A. On the Construction of the C/D-Ring Systems of Aphidicolin and Stemodin. A regio and Stereospecific Synthesis of 17-Noraphidicolan-16-one and 17-Norstemodan-16-one. Helv. Chim. Acta 1982, 65, 371–376. [Google Scholar] [CrossRef]
- Marini Bettolo, R.; Tagliatesta, P.; Lupi, A.; Bravetti, D. A Stereoselective Total Synthesis of (±)-Maritimol, (±)-2-Deoxystemodinone, (±)-Stemodinone and (±)-Stemodin. Helv. Chim. Acta 1983, 66, 760–770. [Google Scholar] [CrossRef]
- Kelly, R.B.; Harley, M.L.; Alward, S.J. A total synthesis of (±)-stemarin, a diterpenoid with a unique bicyclic C/D ring System. Can. J. Chem. 1980, 58, 755–756. [Google Scholar] [CrossRef]
- Kelly, R.B.; Harley, M.L.; Alward, S.J.; Rej, R.N.; Gowda, G.; Mukhopadhyay, A.; Manchand, P.S. Total synthesis of the stemodane-type diterpenoids, (±)-2-desoxystemodinone, (+)-2-desoxystemodinone, and (±)-stemodinol. Can. J. Chem. 1983, 61, 269–275. [Google Scholar] [CrossRef]
- Kelly, R.B.; Lal, S.; Gowda, G.; Rej, R.N. A synthesis of (+)-aphidicol-15-ene. Can. J. Chem. 1984, 62, 1930–1932. [Google Scholar] [CrossRef]
- Chamy, M.C.; Piovano, M.; Garbarino, J.A.; Gambaro, V. Stemodane diterpenoids from Stemodia Chilensis. Phytochemistry 1991, 30, 1719–1721. [Google Scholar] [CrossRef]
- Leonelli, F.; Mostarda, A.; de Angelis, L.; Lamba, D.; Demitri, N.; la Bella, A.; Ceccacci, F.; Migneco, L.M.; Marini Bettolo, R. Proof of the Structure of the Stemodia chilensis Tetracyclic Diterpenoid (+)-19-Acetoxystemodan-12-ol by Synthesis from (+)-Podocarpic Acid: X-ray Structure Determination of a Key Intermediate. J. Nat. Prod. 2016, 79, 1155–1159. [Google Scholar] [CrossRef] [PubMed]
- Berettoni, M.; Marini Bettolo, R.; Montanari, V.; Prencipe, T.; Romeo, S. Studies for a Diastereoselective Synthesis of the Tetracyclic Diol Stemarin: A Model Study for a New Preparation of the Key Intermediate and the Synthesis of (+)-18-Deoxystemarin. Helv. Chim. Acta 1991, 74, 1671–1678. [Google Scholar] [CrossRef]
- Marini Bettolo, R.; Migneco, L.M.; Moretti, P.; Scarpelli, R. Improved Conversion of 6-endo-Tosyloxybicyclo[2.2.2]octan-2-ones into 6-exo-Acetoxy and 6-exo-Benzoyloxy-bicyclo[2.2.2]octan-2-ones. J. Prakt. Chem. 1999, 341, 687–690. [Google Scholar] [CrossRef]
- Steiweiswer, A., Jr.; Walsh, T.D.; Wolfe, J.R., Jr. Stereochemistry of Acetolysis of Alkyl Sulfonates. J. Am. Chem. Soc. 1965, 87, 3682–3685. [Google Scholar]
- Oikawa, H.; Ohashi, S.; König, W.A.; Kenmoku, H.; Sassa, T. Diversity of diterpene hydrocarbons in fungus Phoma betae. Tetrahedron Lett. 2001, 42, 2329–2332. [Google Scholar] [CrossRef]
- Neverov, A.A.; McDonald, T.; Gibson, G.; Brown, R.S. Catalysis of transesterification reactions by lanthanides—Unprecedented acceleration of methanolysis of aryl and alkyl esters promoted by La(OTf)3 at neutral pH and ambient temperatures. Can. J. Chem. 2001, 79, 1704–1710. [Google Scholar]
- Di Stefano, S.; Leonelli, F.; Garofalo, B.; Mandolini, L.; Marini Bettolo, R.; Migneco, L.M. Elusive 6-exo-Hydroxybicyclo[2.2.2]octan-2-ones from the Corresponding Acetates by Methanolysis in the Presence of CH3ONa/La(OTf)3. Org. Lett. 2002, 4, 2783–2785. [Google Scholar] [CrossRef] [PubMed]
- Leonelli, F.; Caschera, B.; Silvestri, L.; Prastaro, A.; Corso, G.; Ceccacci, F.; la Bella, A.; Migneco, L.M.; Marini Bettolo, R. Synthesis of (+)-13-Stemarene and (+)-18-Deoxystemarin: Expeditious Preparation of the Key 6-exo-Hydroxybicyclo[2.2.2]octan-2-one Ethylene Dithioacetal. Helv. Chim. Acta 2008, 91, 598–607. [Google Scholar] [CrossRef]
- Srikrishna, A.; Satyanarayana, G.; Ravi Kumar, P. Enantiospecific synthesis of tricyclo[5.2.1.04,8]decanes via acid catalysed rearrangement of isotwistanes. Tetrahedron Lett. 2005, 47, 363–366. [Google Scholar] [CrossRef]
- Leonelli, F.; Latini, V.; Trombetta, A.; Bartoli, G.; Ceccacci, F.; la Bella, A.; Migneco, L.M.; Marini Bettolo, R. Diastereoselective Total Synthesis of (+)-13-Stemarene by Fourth Generation Methods: A Formal Total Synthesis of (+)-18-Deoxystemarin. J. Org. Chem. 2011, 76, 6871–6876. [Google Scholar] [CrossRef] [PubMed]
- Kodama, O.; Li, W.X.; Tamogami, S.; Akatsuka, T. Oryzalexin S a novel stemarane-type diterpene rice phytoalexin. Biosci. Biotechnol. Biochem. 1992, 56, 1002–1003. [Google Scholar] [CrossRef] [PubMed]
- Tamogami, S.; Mitani, M.; Kodama, O.; Akatsuka, T. Oryzalexin structure: A new stemarane-type diterpene rice phytoalexin and its biogenesis. Tetrahedron 1993, 49, 2025–2032. [Google Scholar] [CrossRef]
- Kodama, O.; Suzuki, T.; Miyakawa, J.; Akatsuka, T. Ultraviolet-induced accumulation of phytoalexins in rice leaves. Agric. Biol. Chem. 1988, 52, 2469–2473. [Google Scholar]
- Chamy, M.C.; Piovano, M.; Garbarino, J.A.; Miranda, C.; Gambaro, V. Diterpenes from Calceolaria lepida. Phytochemistry 1990, 29, 2943–2946. [Google Scholar] [CrossRef]
- Garbarino, J.A.; Molinari, A. Diterpenes from Calceolaria latifolia. Phytochemistry 1990, 29, 3037–3039. [Google Scholar] [CrossRef]
- Chamy, M.C.; Piovano, M.; Garbarino, J.A.; Gambaro, V. Diterpenes from Calceolaria polifolia. Phytochemistry 1991, 30, 3365–3368. [Google Scholar] [CrossRef]
- Leonelli, F.; Blesi, F.; Dirito, P.; Trombetta, A.; Ceccacci, F.; la Bella, A.; Sferrazza, A.; Lamba, D.; Migneco, L.M.; Marini Bettolo, R. Regio- and Diastereoselective Synthesis and X-ray Structure Determination of (+)-2-Deoxyoryzalexin S from (+)-Podocarpic Acid. Structural Non-identity with Its Nominal Natural Isolated Enantiomer. J. Nat. Prod. 2012, 75, 1944–1950. [Google Scholar] [CrossRef] [PubMed]
- Cambie, R.C.; Rutledge, P.S.; Woodgate, P.D. Transformations of podocarpic acid. Aust. J. Chem. 1993, 46, 1447–1471. [Google Scholar] [CrossRef]



























© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
La Bella, A.; Leonelli, F.; Migneco, L.M.; Marini Bettolo, R. (+)-Podocarpic Acid as Chiral Template in the Synthesis of Aphidicolane, Stemodane and Stemarane Diterpenoids †. Molecules 2016, 21, 1197. https://doi.org/10.3390/molecules21091197
La Bella A, Leonelli F, Migneco LM, Marini Bettolo R. (+)-Podocarpic Acid as Chiral Template in the Synthesis of Aphidicolane, Stemodane and Stemarane Diterpenoids †. Molecules. 2016; 21(9):1197. https://doi.org/10.3390/molecules21091197
Chicago/Turabian StyleLa Bella, Angela, Francesca Leonelli, Luisa Maria Migneco, and Rinaldo Marini Bettolo. 2016. "(+)-Podocarpic Acid as Chiral Template in the Synthesis of Aphidicolane, Stemodane and Stemarane Diterpenoids †" Molecules 21, no. 9: 1197. https://doi.org/10.3390/molecules21091197
APA StyleLa Bella, A., Leonelli, F., Migneco, L. M., & Marini Bettolo, R. (2016). (+)-Podocarpic Acid as Chiral Template in the Synthesis of Aphidicolane, Stemodane and Stemarane Diterpenoids †. Molecules, 21(9), 1197. https://doi.org/10.3390/molecules21091197