
Molecular-Based Fluorescent Nanoparticles Built from Dedicated Dipolar Thienothiophene Dyes as Ultra-Bright Green to NIR Nanoemitters


Abstract
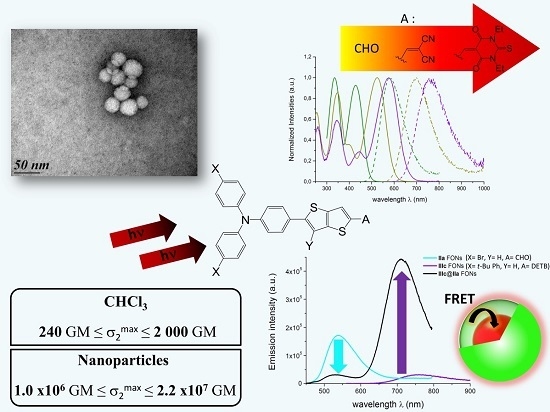
:
1. Introduction
2. Results and Discussion
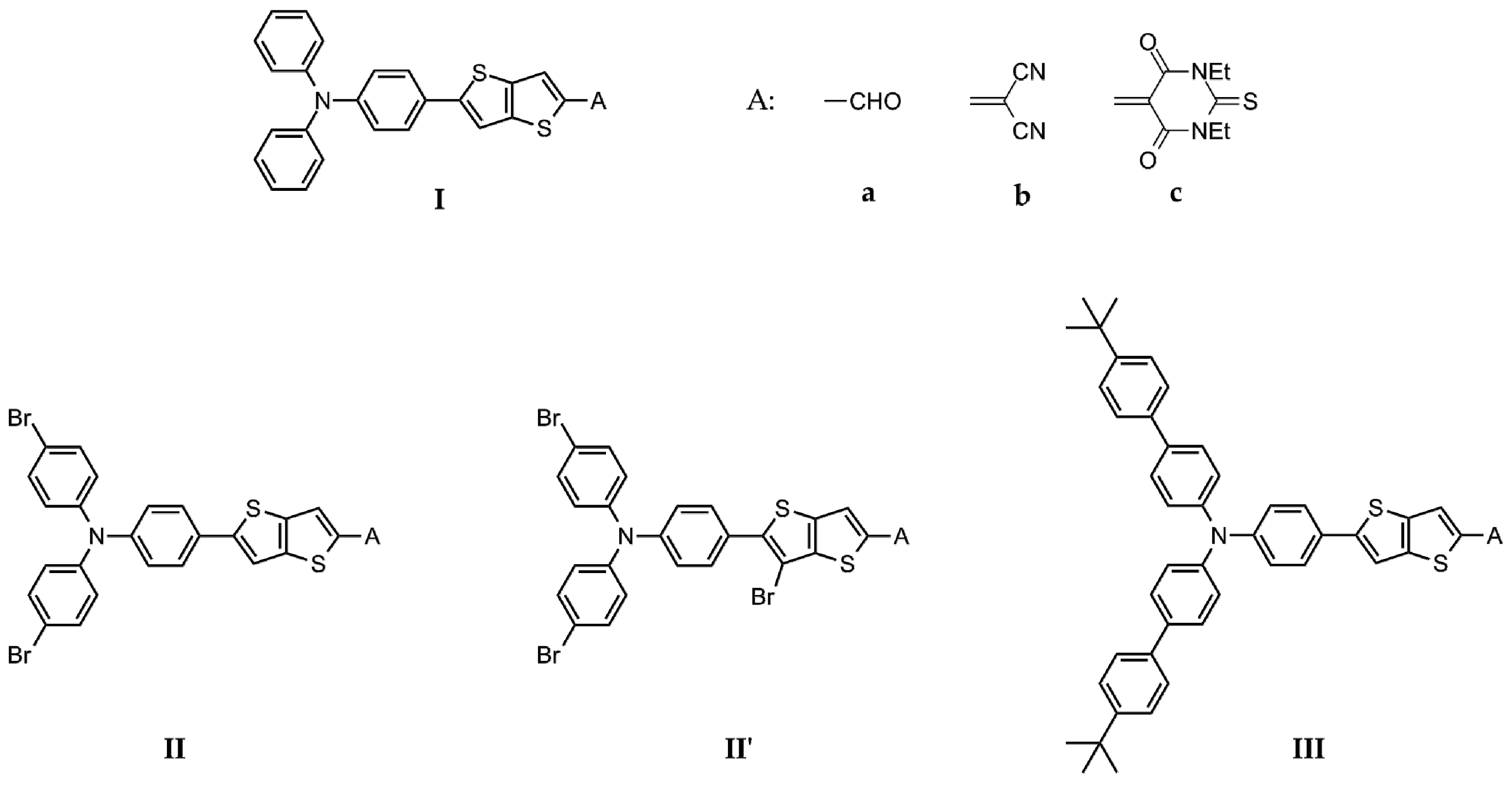
2.1. Design
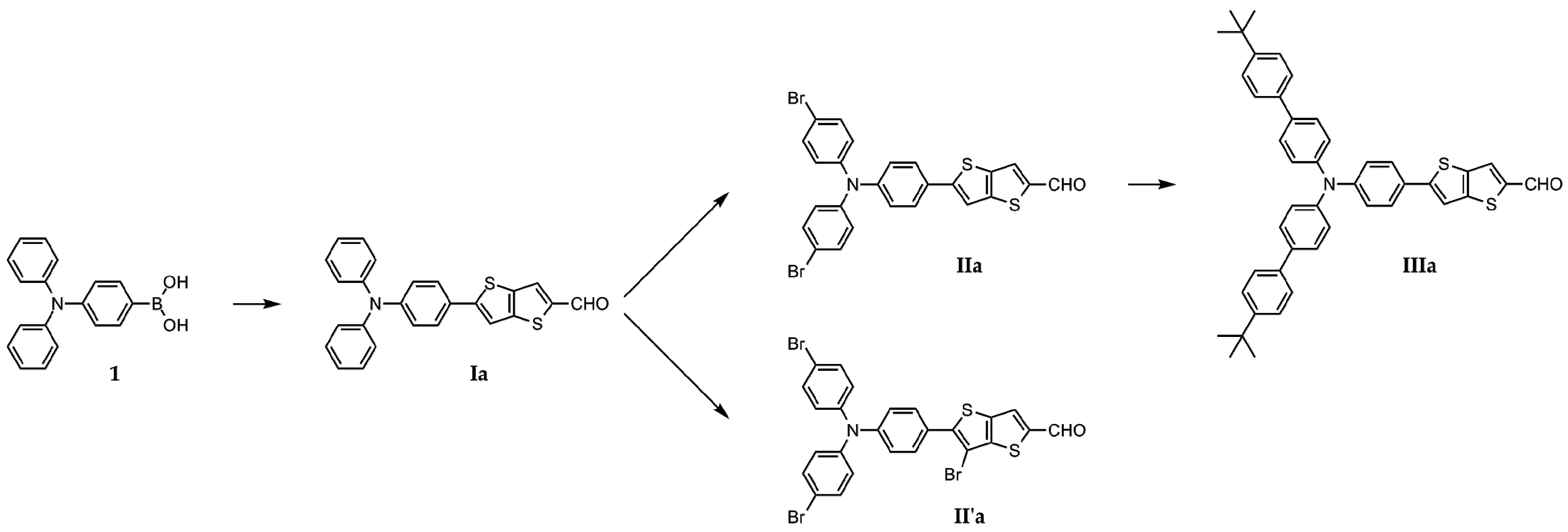
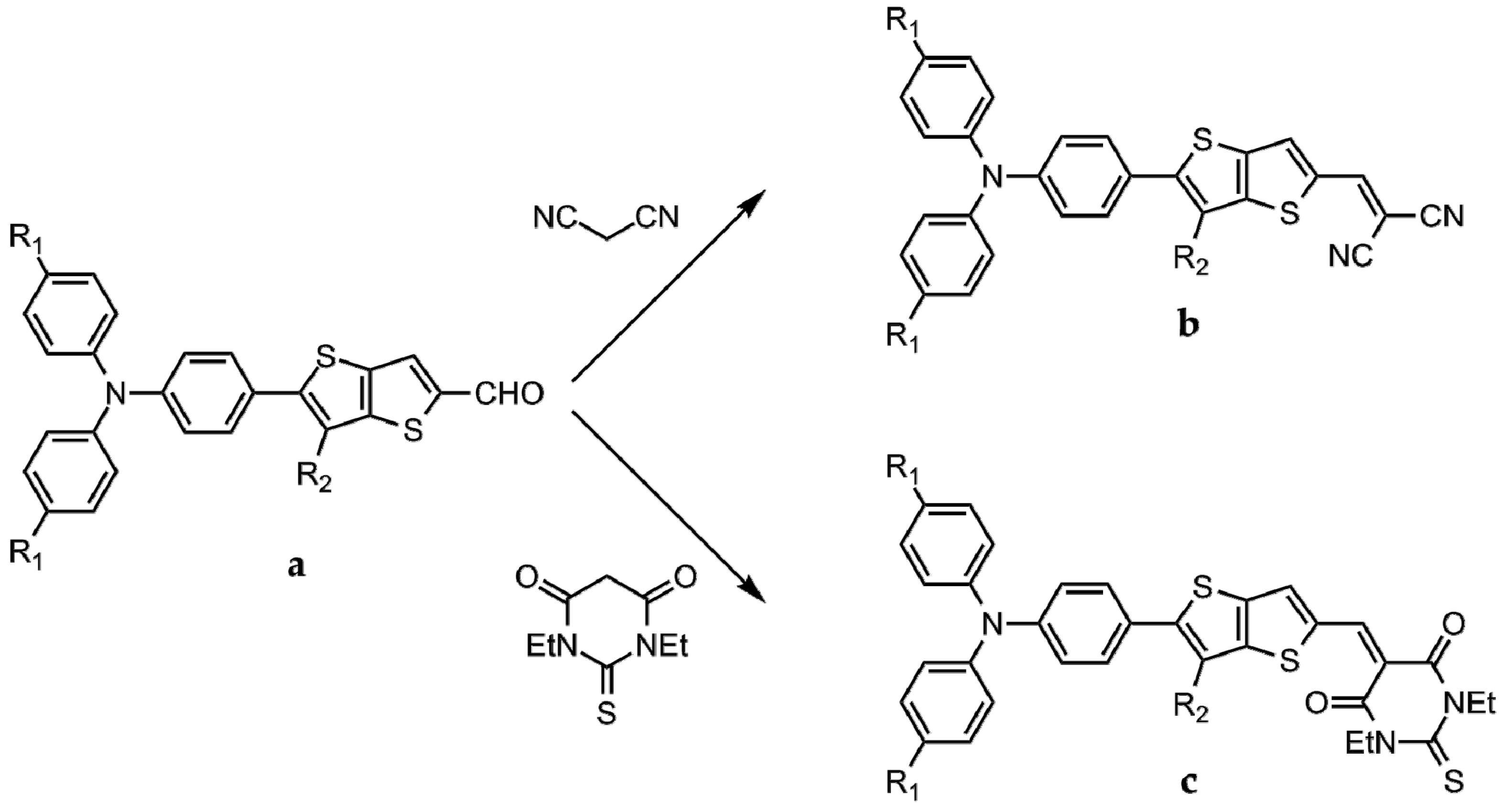
2.2. Synthesis of the Chromophores
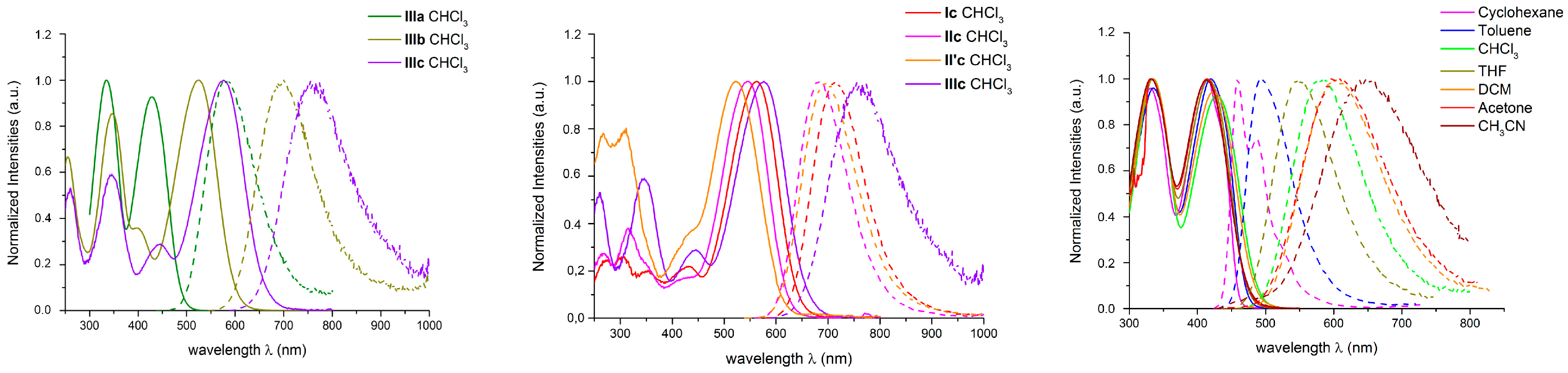
2.3. Photophysical Properties of the New Dyes in Solution in Organic Solvents
2.3.1. One-photon Photophysical Properties of Push-pull Derivatives I–IIIa–c in Solution
Absorption
Fluorescence
Effect of the Environment
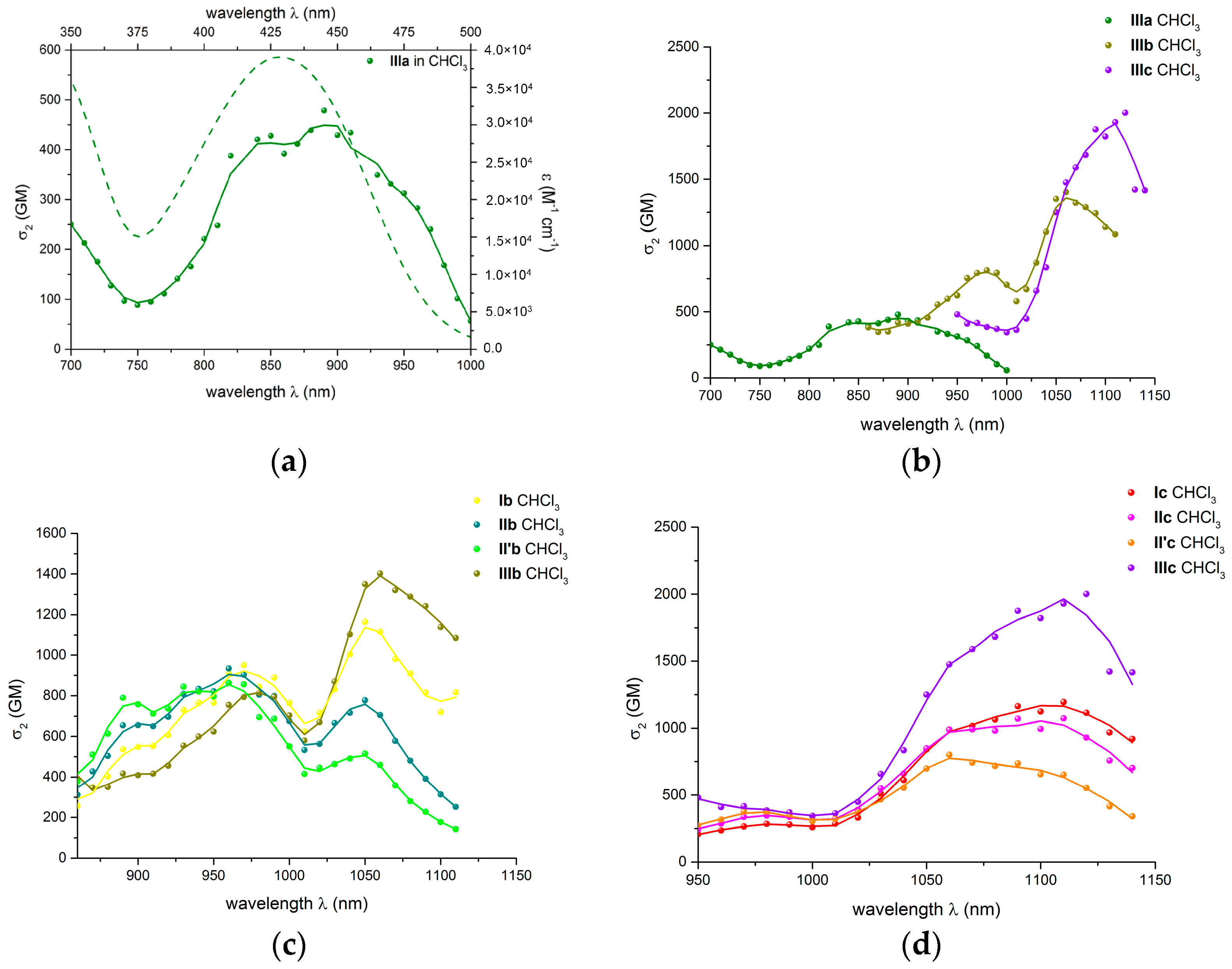
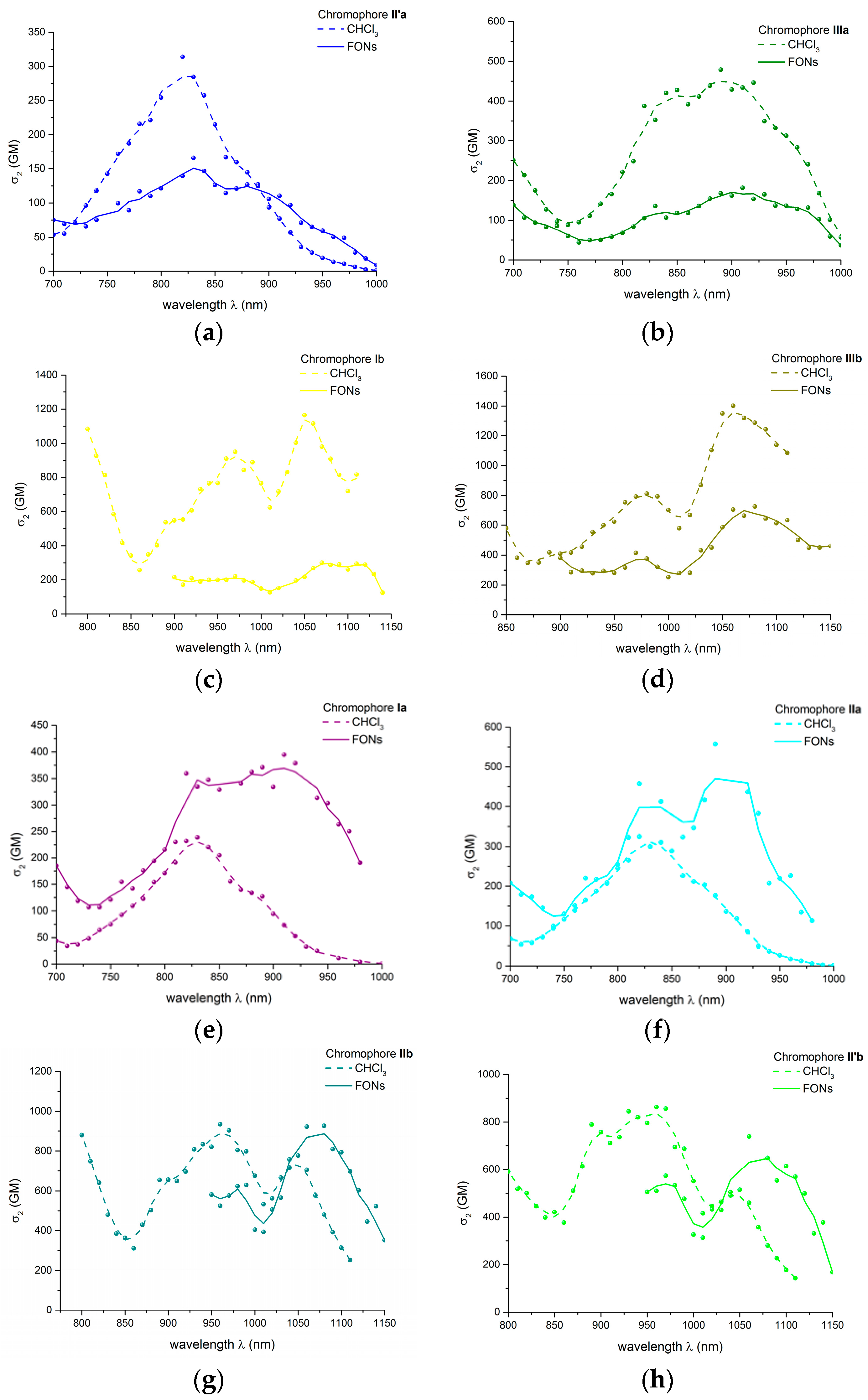
2.3.2. Two-Photon Absorption Properties of Push-pull Derivatives I–IIIa–c in Solution in Chloroform
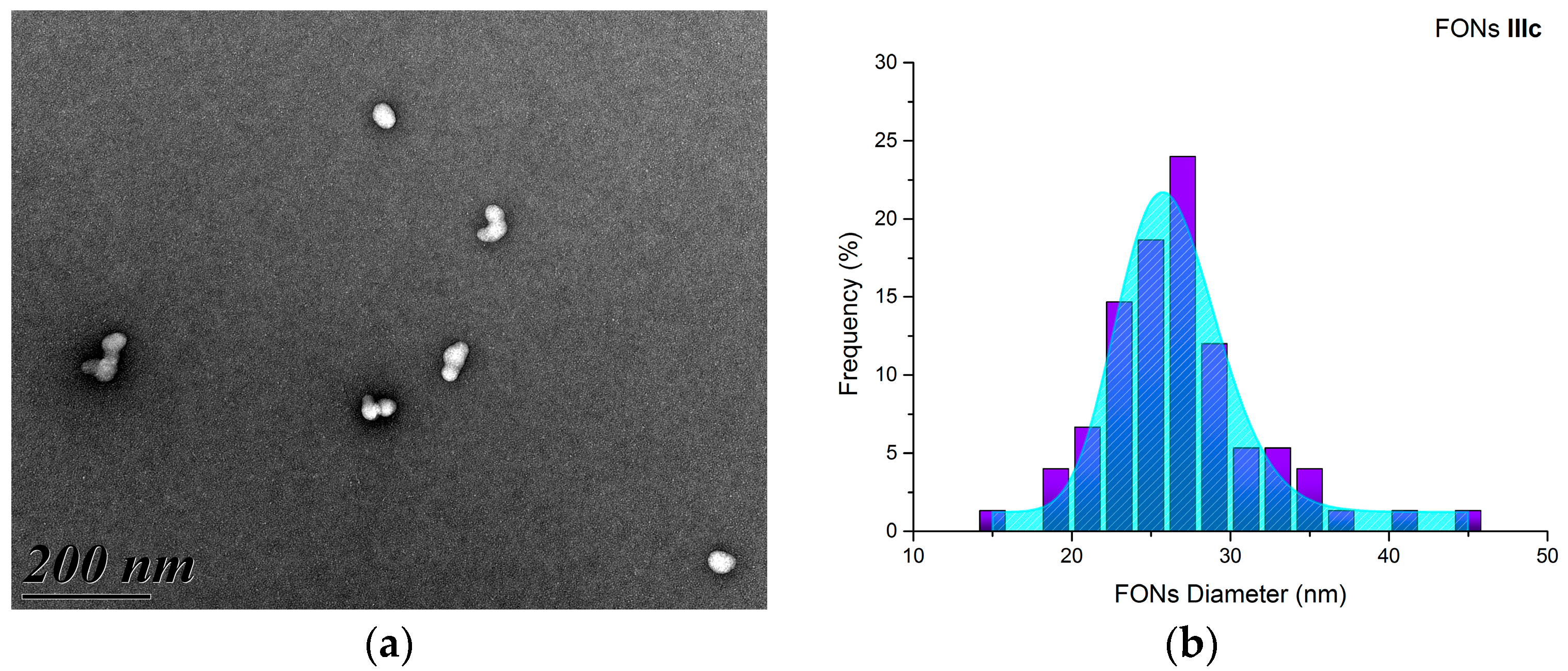
2.4. Preparation and Morphological Characterization of the FONs in Water
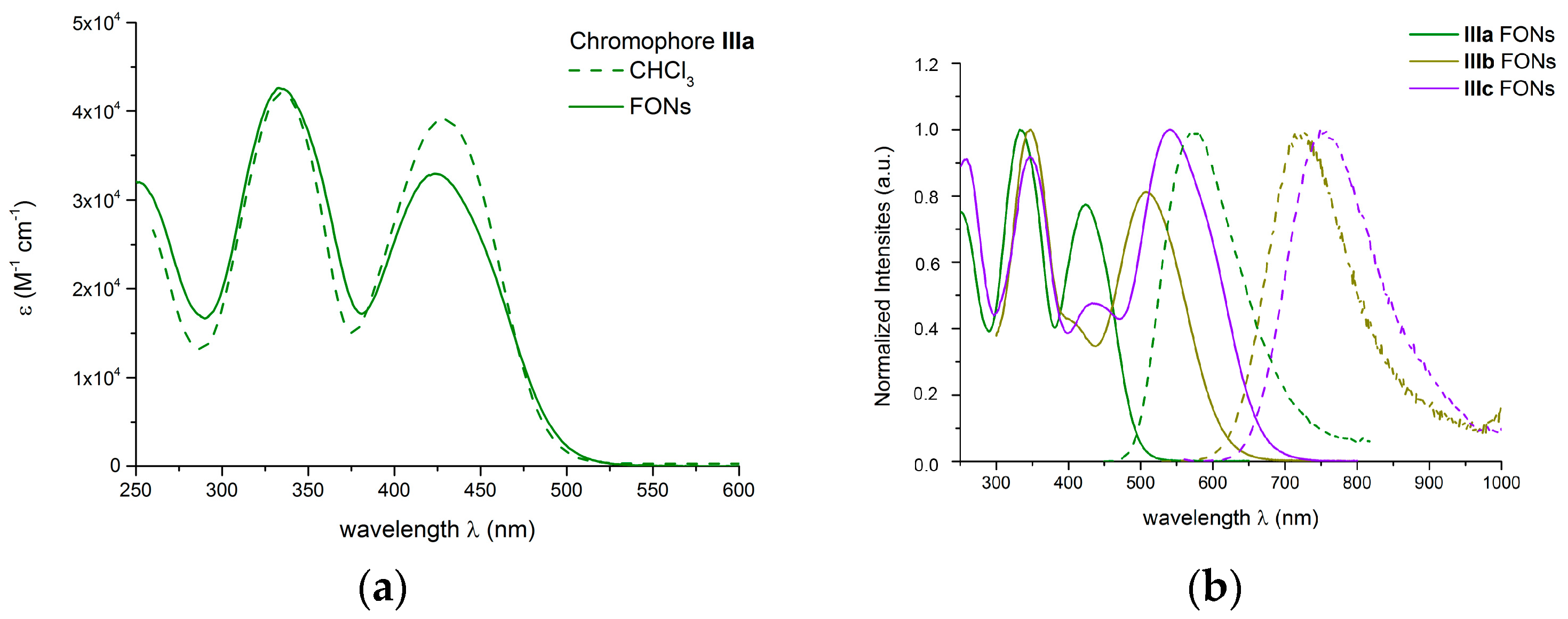
2.5. Photophysical Properties of Fluorescent Organic Nanoparticles in Water
2.5.1. One-photon Photophysical Properties of FONs in Water
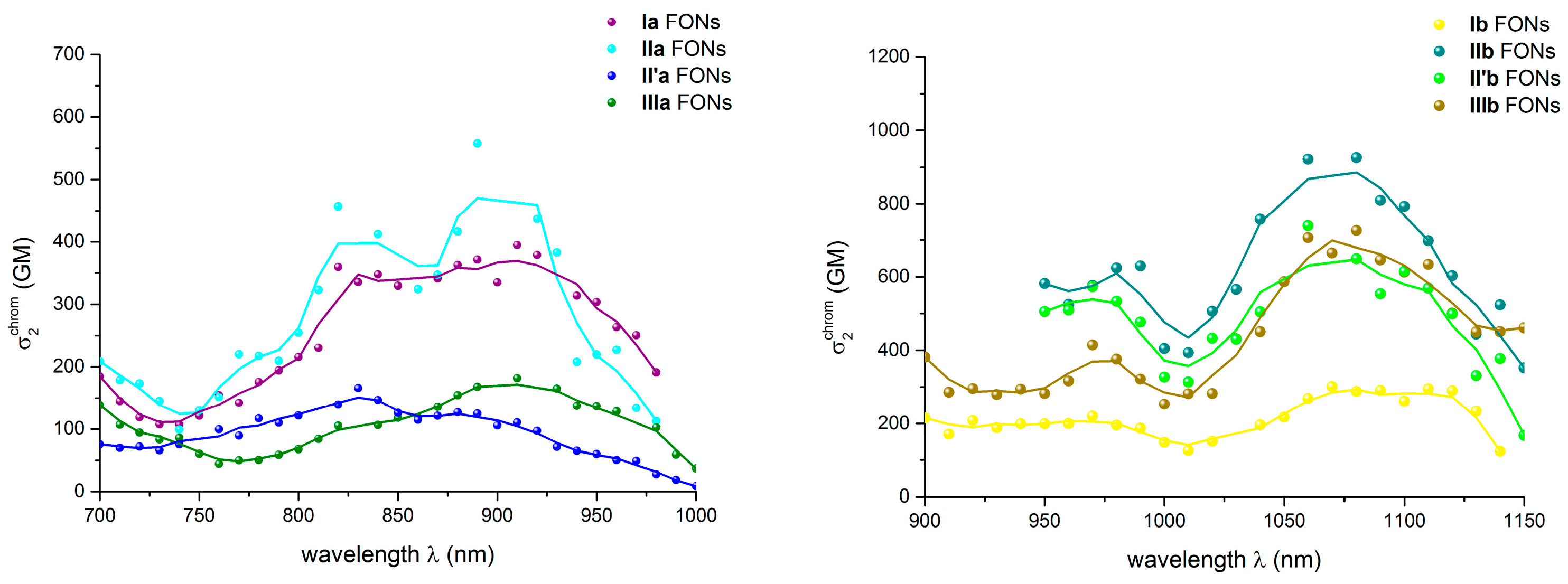
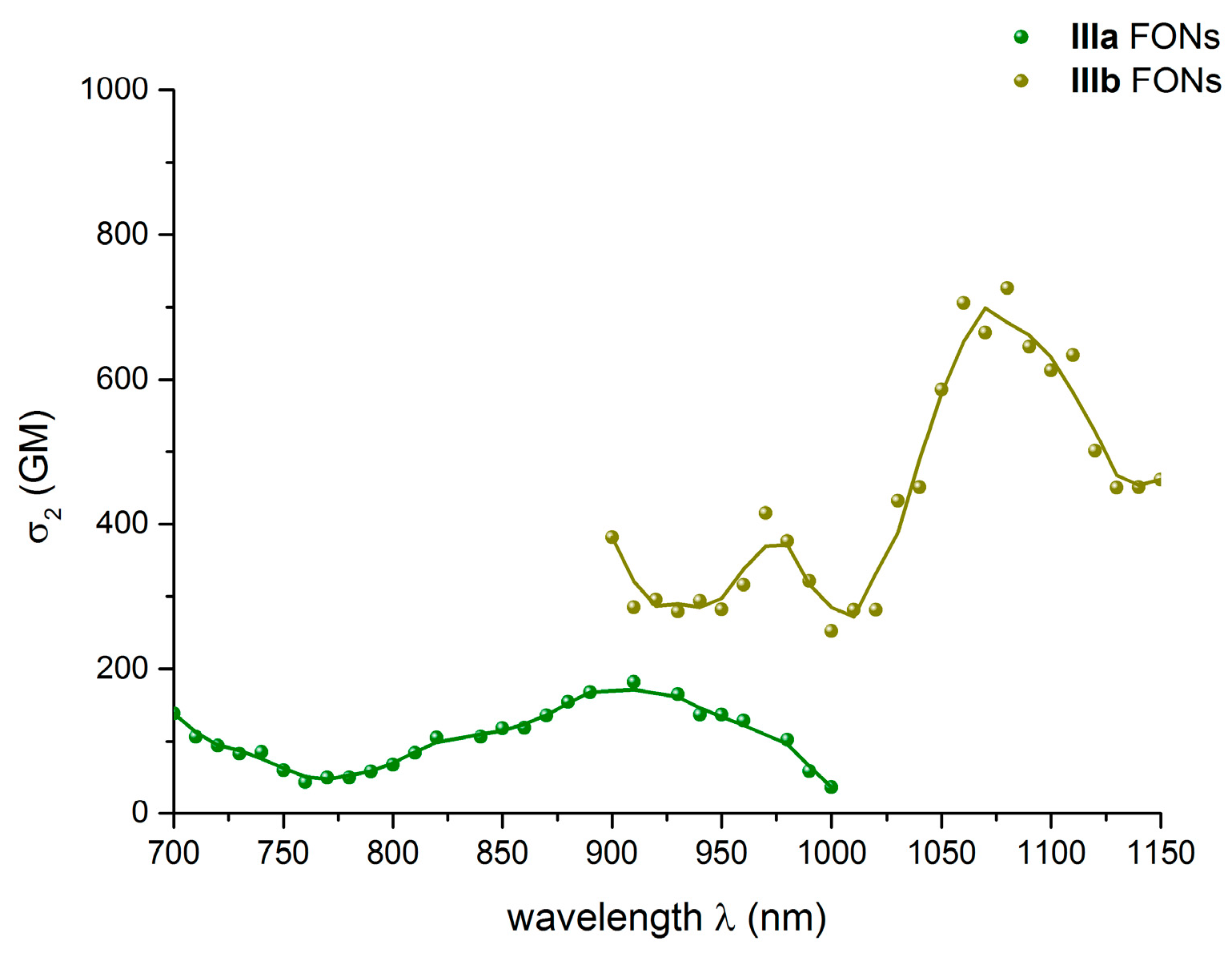
2.5.2. Two-Photon Absorption Properties of the FONs in Water
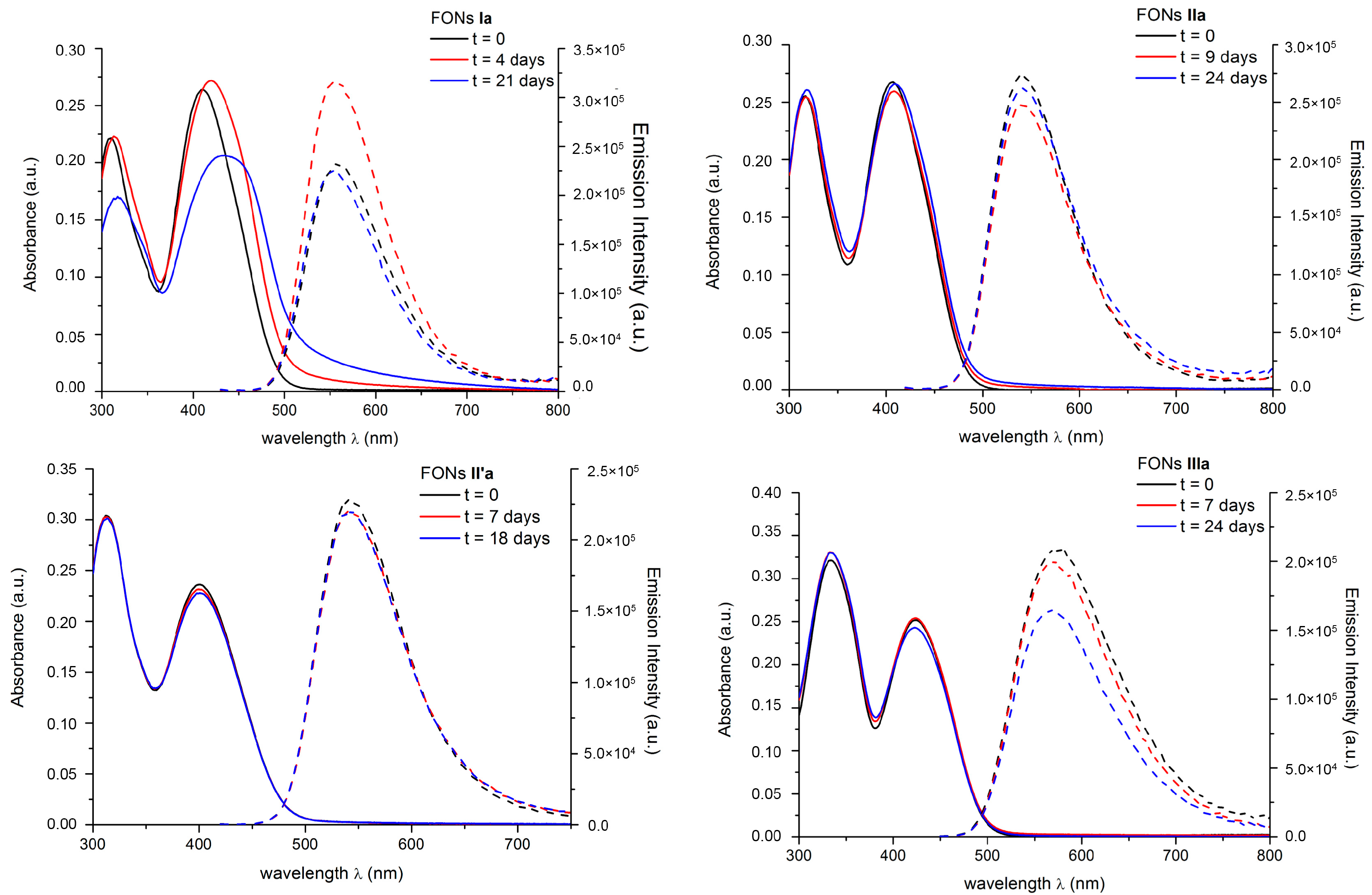
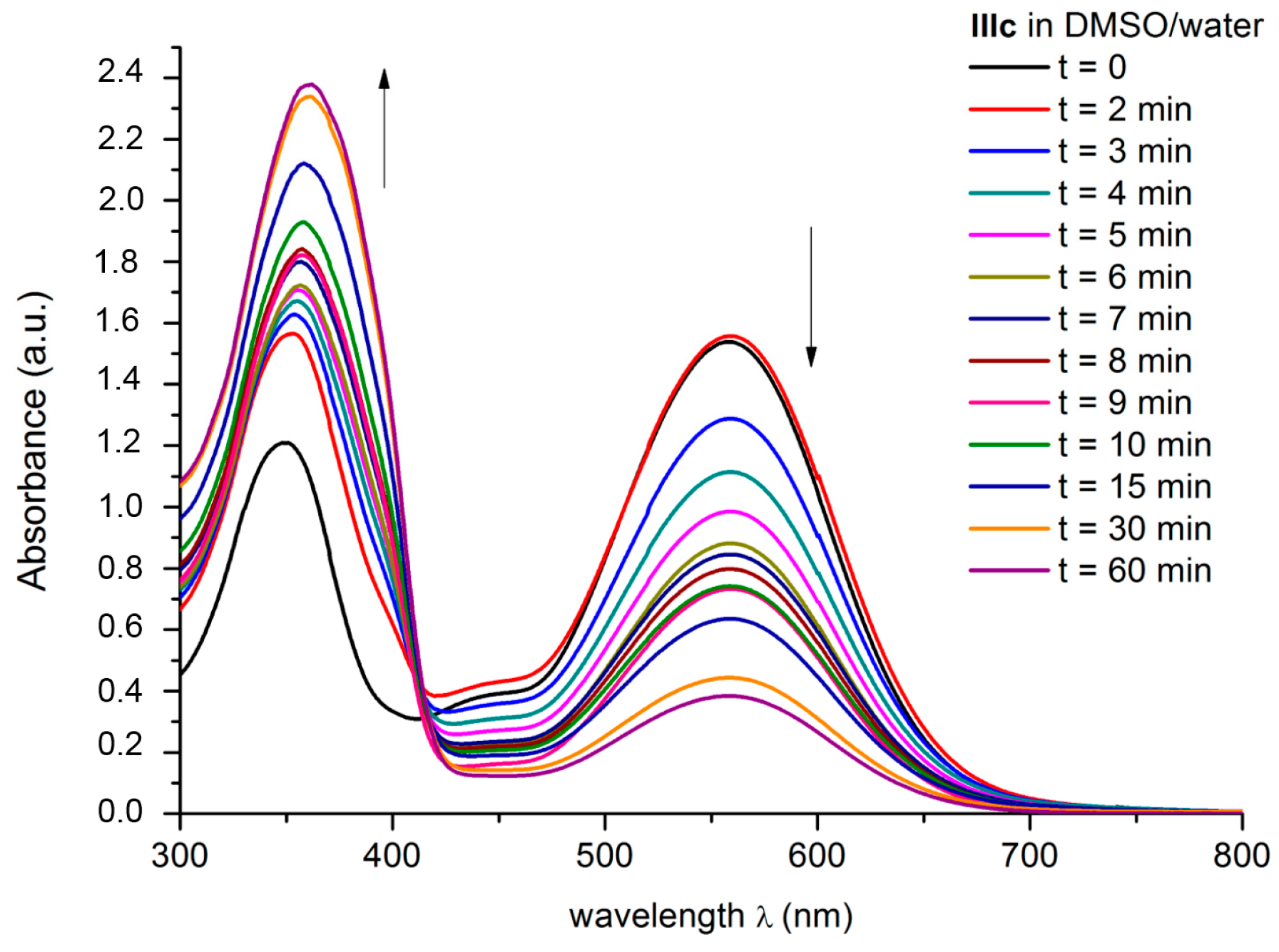
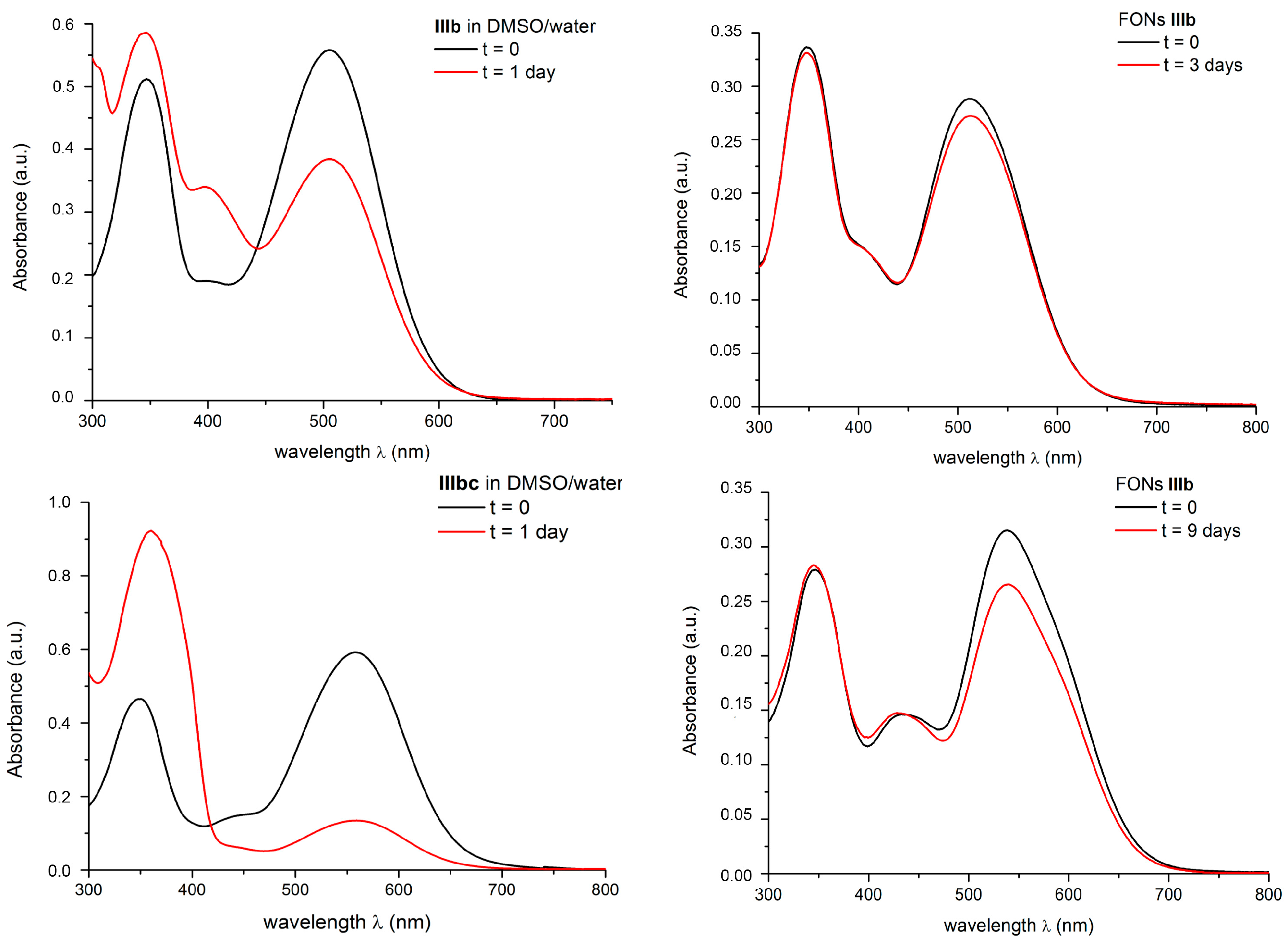
2.6. FONs Stability
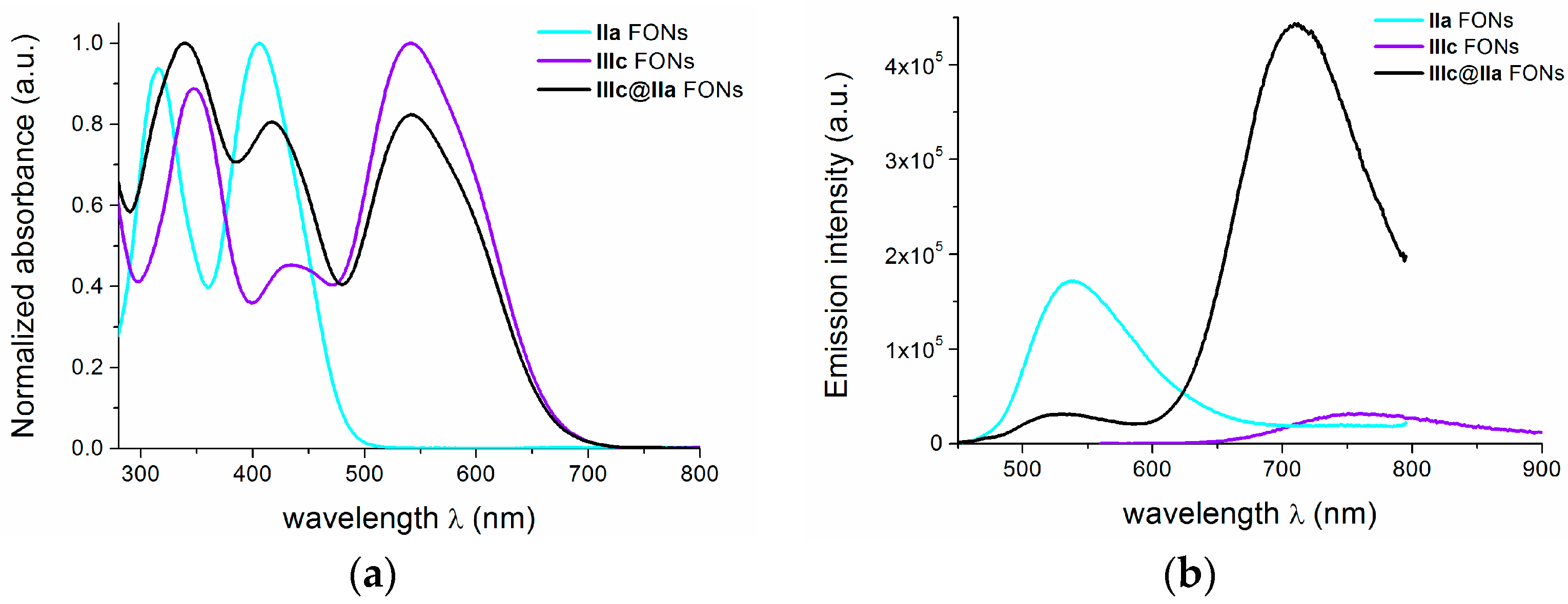
2.7. Core-Shell Nanoparticles
3. Materials and Methods
3.1. Chemical Characterizations
3.2. Experimental Part
3.3. Photophysical Study
3.4. Two-Photon Absorption
3.5. Structural and Morphological Characterization
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
Abbreviations
| CHO | formyl |
| DCV | dicyanovinyl |
| DETB | 1,3-diethyl-2-thiobarbiturate |
| ICT | internal charge transfer |
| NIR | near infrared |
| 1PA | one-photon absorption |
| 2PA | two-photon absorption |
| TPEF | two-photon excited fluorescence |
| FONs | fluorescent organic nanoparticles |
| TEM | transmission electron microscopy |
References
- Haase, M.; Shafer, H. Upconverting nanoparticles. Angew. Chem. Int. Ed. 2011, 50, 5808–5829. [Google Scholar] [CrossRef] [PubMed]
- Medintz, I.L.; Uyeda, H.T.; Goldman, E.R.; Mattoussi, H. Quantum dot bioconjugates for imaging, labelling and sensing. Nat. Mater. 2005, 4, 435–446. [Google Scholar] [CrossRef] [PubMed]
- Robel, I.; Subramanian, V.; Kuno, M.; Kamat, P. Quantum dot solar cells. harvesting light energy with CdSe nanocrystals molecularly linked to mesoscopic TiO2 films. J. Am. Chem. Soc. 2006, 128, 2385–2393. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lo, Y. Highly efficient quantum-dot-sensitized solar cell based on co-sensitization of CdS/CdSe. Adv. Funct Mater. 2009, 19, 604–609. [Google Scholar] [CrossRef]
- Im, J.H.; Lee, C.R.; Lee, J.W.; Park, S.W.; Park, N.G. 6.5% efficient perovskite quantum-dot-sensitized solar cell. Nanoscale 2011, 3, 4088–4093. [Google Scholar] [CrossRef] [PubMed]
- Erathodiyil, N.; Ying, J. Functionalization of inorganic nanoparticles for bioimaging applications. Acc. Chem. Res. 2011, 44, 925–935. [Google Scholar] [CrossRef] [PubMed]
- Zrazhevskiy, P.; Sena, M.; Gao, X. Designing multifunctional quantum dots for bioimaging, detection, and drug delivery. Chem. Soc. Rev. 2010, 39, 4326–4354. [Google Scholar] [CrossRef] [PubMed]
- Sharifi, S.; Behzadi, S.; Laurent, S.; Forrest, L.; Stroeve, P.; Mahmoudi, M. Toxicity of nanomaterials. Chem. Soc. Rev. 2011, 41, 2323–2343. [Google Scholar] [CrossRef] [PubMed]
- Lewinski, N.; Colvin, V.; Drezek, R. Cytotoxicity of Nanoparticles. Small 2008, 4, 26–49. [Google Scholar] [CrossRef] [PubMed]
- Patra, A.; Chandaluri, C.; Radhakrishnan, T.P. Optical materials based on molecular nanoparticles. Nanoscale 2011, 4, 343–359. [Google Scholar] [CrossRef] [PubMed]
- Fery-Forgues, S. Fluorescent organic nanocrystals and non-doped nanoparticles for biological applications. Nanoscale 2013, 5, 8428–8442. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.; Bondu, F.; Adamietz, F.; Blanchard-Desce, M.; Rodriguez, V. Interfacial organization in dipolar dye-based organic nanoparticles probed by second-harmonic scattering. ACS Photonics 2015, 2, 1209–1216. [Google Scholar] [CrossRef]
- Parthasarathy, V.; Fery-Forgues, S.; Campioli, E.; Recher, G.; Terenziani, F.; Blanchard-Desce, M. Dipolar versus octupolar triphenylamine-based fluorescent organic nanoparticles as brilliant one- and two-photon emitters for (bio)imaging. Small 2011, 7, 3219–3229. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.; Godin, A.; Palayret, M.; Lounis, B.; Cognet, L.; Blanchard-Desce, M. Innovative molecular-based fluorescent nanoparticles for multicolor single particle tracking in cells. J. Phys. D Appl. Phys. 2016, 49, 084002. [Google Scholar] [CrossRef]
- Genin, E.; Gao, Z.; Varela, J.; Daniel, J.; Bsaibess, T.; Gosse, I.; Groc, L.; Cognet, L.; Blanchard-Desce, M. “Hyper-bright” near-infrared emitting fluorescent organic nanoparticles for single particle tracking. Adv. Mater. 2014, 26, 2258–2261. [Google Scholar] [CrossRef] [PubMed]
- Amro, K.; Daniel, J.; Clermont, G.; Bsaibess, T.; Pucheault, M.; Genin, E.; Vaultier, M.; Blanchard-Desce, M. A new route towards fluorescent organic nanoparticles with red-shifted emission and increased colloidal stability. Tetrahedron 2014, 70, 1903–1909. [Google Scholar] [CrossRef]
- Katan, C.; Terenziani, F.; Mongin, O.; Werts, M.; Porrès, L.; Pons, T.; Mertz, J.; Tretiak, S.; Blanchard-Desce, M. Effects of (Multi)branching of dipolar chromophores on photophysical properties and two-photon absorption. J. Phys. Chem. 2005, 109, 3024–3037. [Google Scholar] [CrossRef] [PubMed]
- Droumaguet, C.; Sourdon, A.; Genin, E.; Mongin, O.; Blanchard-Desce, M. Two-photon polarity probes built from octupolar fluorophores: Synthesis, structure–properties relationships, and use in cellular imaging. Chem. Asian J. 2013, 8, 2984–3001. [Google Scholar] [CrossRef] [PubMed]
- Terenziani, F.; Katan, C.; Badaeva, E.; Tretiak, S.; Blanchard-Desce, M. Enhanced two-photon absorption of organic chromophores: Theoretical and experimental assessments. Adv. Mater. 2008, 20, 4641–4678. [Google Scholar] [CrossRef]
- Mishra, A.; Behera, R.K.; Behera, P.K.; Mishra, B.K.; Behera, G.B. Cyanines during the 1990s: A review. Chem. Rev. 2000, 100, 1973–2012. [Google Scholar] [CrossRef] [PubMed]
- Barzoukas, M.; Runser, C.; Fort, A.; Blanchard-Desce, M. A two-state description of (hyper) polarizabilities of push-pull molecules based on a two-form model. Chem. Phys. Lett. 1996, 257, 531–537. [Google Scholar] [CrossRef]
- Strickler, S.J.; Berg, R. Relationship between absorption intensity and fluorescence lifetime of molecules. J. Chem. Phys. 1962, 37, 814–822. [Google Scholar] [CrossRef]
- Xu, C.; Webb, W.W. Measurement of two-photon excitation cross sections of molecular fluorophores with data from 690 to 1050 nm. J. Opt. Soc. Am. B 1996, 13, 481–491. [Google Scholar] [CrossRef]
- Atkins, P.; Friedman, R. Molecular Quantum Mechanics, 4th ed.; Oxfort Universtity Press: Oxford, UK, 2006. [Google Scholar]
- Masuhara, H.; Nakanishi, H.; Sasaki, K. Single Organic Nanoparticles; Springer-Verlag: New York, NY, USA, 2003. [Google Scholar]
- Sugiyama, T.; Asahi, T.; Takeuchi, H.; Masuhara, H. Size and phase control in quinacridone nanoparticle formation by laser ablation in water. Jpn. J. Appl. Phys. 2006, 45, 384–388. [Google Scholar] [CrossRef]
- Tamaki, Y.; Asahi, T.; Masuhara, H. Nanoparticle formation of vanadyl phthalocyanine by laser ablation of its crystalline powder in a poor solvent. J. Phys. Chem. 2002, 106, 2135–2139. [Google Scholar] [CrossRef]
- Zhao, Y.; Xiao, D.; Yang, W.; Peng, A.; Yao, J. 2,4,5-Triphenylimidazole nanowires with fluorescence narrowing spectra prepared through the adsorbent-assisted physical vapor deposition method. Chem. Mater. 2006, 18, 2302–2306. [Google Scholar] [CrossRef]
- Gomar-Nadal, E.; Puigmartí-Luis, J.; Amabilino, D. Assembly of functional molecular nanostructures on surfaces. Chem. Soc. Rev. 2007, 37, 490–504. [Google Scholar] [CrossRef] [PubMed]
- Nakanishi, H.A. Novel preparation method of organic microcrystals. Jpn. J. Appl. Phys. 1992, 31, L1132–L1134. [Google Scholar]
- Terenziani, F.; Morone, M.; Gmouh, S.; Blanchard-Desce, M. Linear and two-photon absorption properties of interacting polar chromophores: standard and unconventional effects. Chem. Phys. Chem. 2006, 7, 685–696. [Google Scholar] [CrossRef] [PubMed]
- Terenziani, F.; Parthasarathy, V.; Pla-Quintana, A.; Maishal, T.; Caminade, A.; Majoral, J.; Blanchard-Desce, M. Cooperative two-photon absorption enhancement by through-space interactions in multichromophoric compounds. Angew. Chem. Int. Ed. 2009, 48, 8691–8694. [Google Scholar] [CrossRef] [PubMed]
- Robin, A.C.; Parthasarathy, V.; Pla-Quintana, A.; Mongin, O.; Terenziani, F.; Caminade, A.M.; Majoral, J.P.; Blanchard-Desce, M. Cooperative TPA enhancement via through-space interactions in organic nanodots built from dipolar chromophores. 2010, 77740N-77740N-8. [Google Scholar] [CrossRef]
- Verlhac, J.-B.; Daniel, J.; Pagano, P.; Clermont, G.; Blanchard-Desce, M. Enhanced two-photon brightness in molecular-based organic nanoparticles built from articulated-dipoles. C. R. Chim. 2016, 19, 28–38. [Google Scholar] [CrossRef]
- Larson, D.R.; Zipfel, W.R.; Williams, R.M.; Clark, S.W.; Bruchez, M.P.; Wise, F.W.; Webb, W.W. Water-soluble quantum dots for multiphoton fluorescence imaging in vivo. Science 2003, 300, 1434–1436. [Google Scholar]
- Lim, S.; Zahid, M.; Le, P.; Ma, L.; Entenberg, D.; Harney, A.S.; Condeelis, J.; Smith, A.M. Brightness-equalized quantum dots. Nat. Commun. 2015, 6, 8210. [Google Scholar] [CrossRef] [PubMed]
- Daniel, J.; Godin, A.; Clermont, G.; Lounis, B.; Cognet, L.; Blanchard-Desce, M. NIR-emitting molecular-based nanoparticles as new two-photon absorbing nanotools for single particle tracking. Int. Soc. Opt. Photonics 2015, 95230M-95230M-8. [Google Scholar] [CrossRef]
- Campioli, E.; Rouxel, C.; Campanini, M.; Nasi, L.; Blanchard-Desce, M.; Terenziani, F. Enforcing luminescence at organic nanointerfaces: Luminescence spatial confinement and amplification in molecular-based core–shell nanoparticles. Small 2013, 9, 1982–1988. [Google Scholar] [CrossRef] [PubMed]
- Campioli, E.; Nikolaidou, D.; Hugues, V.; Campanini, M.; Nasi, L.; Blanchard-Desce, M.; Terenziani, F. Amplified two-photon brightness in organic multicomponent nanoparticles. J. Mater. Chem. C 2015, 3, 7483–7491. [Google Scholar] [CrossRef]
- Resch-Genger, U.; Rurack, K. Determination of the photoluminescence quantum yield of dilute dye solutions (IUPAC Technical Report). Pure Appl. Chem. 2013, 85, 2005–2013. [Google Scholar] [CrossRef]
- Resch-Genger, U.; DeRose, P. Fluorescence standards: Classification, terminology, and recommendations on their selection, use, and production (IUPAC Technical Report). Pure Appl. Chem. 2010, 82, 2315–2335. [Google Scholar] [CrossRef]
- Sample Availability: Not available.
















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compd. | R1, R2 | Eq 1 | Solvents | Duration (h) | Yield (%) |
|---|---|---|---|---|---|
| Ib | H, H | 2.0 | Tol:EtOH = 1.5:1.0 | 24 | 91 |
| IIb | Br, H | 2.2 | EtOH | 48 | 87 |
| II’b | Br, Br | 1.1 | EtOH | 72 | 66 |
| IIIb | tert-butyl-C6H4, H | 9.3 | Tol:EtOH = 1.7:1.0 | 24 | 75 |
| Ic | H, H | 1.1 | EtOH | 24 | 83 |
| IIc | Br, H | 1.0 | EtOH | 24 | 86 |
| II’c | Br, Br | 1.1 | EtOH | 24 | 81 |
| IIIc | tert-butyl-C6H4, H | 1.1 | Tol:EtOH = 1.0:1.3 | 24 | 77 |
| Compd. | λmax1PA (nm) | εmax (104 M−1·cm−1) | λmaxem (nm) | Φf | Stokes Shift (103 cm−1) | εmaxΦf (104 M−1·cm−1) | τ (ns) | kr (109 s−1) | knr (109 s−1) | λmax2PA (nm) | σ2max (GM) |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Ia | 417 | 3.9 | 534 | 0.84 | 5.2 | 3.3 | 2.8 | 0.30 | 0.06 | 830 | 240 |
| IIa | 409 | 3.7 | 526 | 0.84 | 5.4 | 3.1 | 2.5 | 0.34 | 0.06 | 820 | 330 |
| II’a | 401 | 2.9 | 538 | 0.76 | 6.3 | 2.2 | 2.6 | 0.29 | 0.09 | 820 | 314 |
| IIIa | 429 | 3.9 | 582 | 0.73 | 6.1 | 2.8 | 3.3 | 0.22 | 0.08 | 890 | 480 |
| Ib | 511 | 4.2 | 652 | 0.47 | 4.2 | 2.0 | 2.6 | 0.18 | 0.20 | 1050 | 1164 |
| IIb | 495 | 5.4 | 629 | 0.31 | 4.3 | 1.7 | 1.5 | 0.21 | 0.46 | 960 | 934 |
| II’b | 482 | 4.5 | 638 | 0.42 | 5.1 | 1.9 | 2.4 | 0.18 | 0.24 | 960 | 862 |
| IIIb | 525 | 4.5 | 700 | 0.24 | 4.8 | 1.1 | 1.9 | 0.13 | 0.40 | 1060 | 1402 |
| Ic | 563 | 6.0 | 712 | 0.37 | 3.7 | 2.2 | 1.9 | 0.19 | 0.33 | 1110 | 1194 |
| IIc | 547 | 6.8 | 681 | 0.28 | 3.6 | 1.9 | 1.2 | 0.23 | 0.60 | 1110 | 1072 |
| II’c | 522 | 5.5 | 694 | 0.30 | 4.7 | 1.65 | 1.6 | 0.19 | 0.44 | 1060 | 802 |
| IIIc | 576 | 5.5 | 755 | 0.06 | 4.1 | 0.33 | 0.6 | 0.10 | 1.57 | 1120 | 2002 |
| Compd. | dTEM (nm) | ζ-Potential (mV) | N 1 (104) |
|---|---|---|---|
| Ia | 42 | −70 | 5.7 |
| IIa | 37 | −85 | 2.8 |
| II’a | 35 | −74 | 2.0 |
| IIIa | 27 | −72 | 0.9 |
| Ib | 21 | −74 | 0.6 |
| IIb | 39 | −75 | 3.0 |
| II’b | 43 | −56 | 3.5 |
| IIIb | 32 | −79 | 1.5 |
| Ic | 30 | −67 | 1.4 |
| IIc | 29 | −75 | 1.0 |
| II’c | 26 | −70 | 0.6 |
| IIIc | 32 | −72 | 1.2 |
| Compd. | λabsmax(Δλmax) 1 (nm) | εmax(Δεmax) 2 (104M−1·cm−1) | λmaxem(Δλmax) 3 (nm) | Φf | Stokes Shift (103∙cm−1) | τ1/τ2 (ns) | λmax2PA (nm) | σ2max(Δσ2max) 4 (GM) |
|---|---|---|---|---|---|---|---|---|
| Ia | 410 (−7) | 3.4 (−0.5) | 557 (+23) | 0.05 | 6.4 | 1.2 (0.46)/3.9 (0.54) | 910 | 395 (+155) |
| IIa | 407 (−2) | 3.6 (−0.1) | 541 (+15) | 0.05 | 6.1 | 0.9 (0.57)/2.8 (0.43) | 820 890 | 457 (+127) 557 (+227) |
| II’a | 401 (0) | 2.5 (−0.4) | 540 (+2) | 0.04 | 6.4 | 0.8 (0.71)/2.4 (0.29) | 830 | 165 (−149) |
| IIIa | 424 (−5) | 3.3 (−0.6) | 570 (−12) | 0.06 | 6.0 | 0.9 (0.59)/3.8 (0.41) | 910 | 182 (−298) |
| Ib | 476 (−35) | 3.5 (−0.7) | 719 (+67) | 0.01 | 7.1 | 1.5 (0.58)/4.0 (0.42) | 1070 | 300 (−864) |
| IIb | 478 (−17) | 6.1 (+0.7) | 711 (+82) | 0.01 | 6.9 | 1.1 (0.73)/3.5 (0.27) | 1060 | 921 (−13) |
| II’b | 478 (−4) | / | 702 (+64) | 0.01 | 6.7 | 1.1 (0.84)/3.5 (0.16) | 1060 | 739 (−123) |
| IIIb | 507 (−18) | 5.0 (+0.5) | 714 (+14) | 0.02 | 5.7 | 1.5 (0.50)/4.6 (0.50) | 1080 | 726 (−676) |
| Ic | 517 (−46) | 5.2 (−0.8) | 754 (+42) | 0.004 | 6.1 | 0.9 (0.80)/2.7 (0.20) | / | / |
| IIc | 509 (−38) | 5.5 (−1.3) | 752 (+71) | 0.007 | 6.3 | 1.0 (0.80)/2.9 (0.20) | / | / |
| II’c | 502 (−20) | 5.2 (−0.3) | 725 (+30) | 0.006 | 6.1 | 0.7 (0.84)/2.2 (0.16) | / | / |
| IIIc | 541 (−35) | 4.9 (−0.6) | 749 (−6) | 0.005 | 5.1 | 1.0 (0.76)/3.0 (0.24) | / | / |
| Compd. | λmax1PA (nm) | λmaxem (nm) | FWHMem (103∙cm−1) | Φf | εmaxNPsΦf * (108 M−1∙cm−1) | λmax2PA (nm) | σ2maxNPsΦf * (106 GM) |
|---|---|---|---|---|---|---|---|
| Ia | 410 | 557 | 3.1 | 0.05 | 1.0 | 910 | 1.12 |
| IIa | 407 | 541 | 3.0 | 0.05 | 0.5 | 890 | 0.76 |
| II’a | 401 | 540 | 3.0 | 0.04 | 0.1 | 830 | 0.64 |
| IIIa | 424 | 570 | 3.6 | 0.06 | 0.4 | 910 | 0.22 |
| Ib | 476 | 719 | 2.5 | 0.01 | 0.02 | 1070 | 0.02 |
| IIb | 478 | 711 | 5.6 | 0.01 | 0.2 | 1060 | 0.3 |
| II’b | 478 | 702 | 2.6 | 0.01 | / | 1060 | 0.1 |
| IIIb | 507 | 714 | 2.5 | 0.02 | 0.4 | 1080 | 0.5 |
| Ic | 517 | 754 | 2.5 | 0.004 | 0.03 | / | / |
| IIc | 509 | 752 | 2.6 | 0.007 | 0.04 | / | / |
| II’c | 502 | 725 | 2.7 | 0.006 | 0.04 | / | / |
| IIIc | 541 | 749 | 2.5 | 0.005 | 0.02 | / | / |
| FONs | λmax1PA (nm) | λmaxem (nm) | Φf | τ1/τ2 (ns) |
|---|---|---|---|---|
| IIa | 407 | 541 | 0.05 a | 0.9 (0.57)/2.8 (0.43) |
| IIIc | 541 | 749 | 0.005 b | 1.0 (0.76)/3.0 (0.24) |
| IIIc@IIa | 339 417 542 | 710 | 0.10 (exc@417 nm) b | 0.5 (0.80)/2.1 (0.20) c 2.2 (0.62)/4.6 (0.38) d |
© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mastrodonato, C.; Pagano, P.; Daniel, J.; Vaultier, M.; Blanchard-Desce, M. Molecular-Based Fluorescent Nanoparticles Built from Dedicated Dipolar Thienothiophene Dyes as Ultra-Bright Green to NIR Nanoemitters. Molecules 2016, 21, 1227. https://doi.org/10.3390/molecules21091227
Mastrodonato C, Pagano P, Daniel J, Vaultier M, Blanchard-Desce M. Molecular-Based Fluorescent Nanoparticles Built from Dedicated Dipolar Thienothiophene Dyes as Ultra-Bright Green to NIR Nanoemitters. Molecules. 2016; 21(9):1227. https://doi.org/10.3390/molecules21091227
Chicago/Turabian StyleMastrodonato, Cristiano, Paolo Pagano, Jonathan Daniel, Michel Vaultier, and Mireille Blanchard-Desce. 2016. "Molecular-Based Fluorescent Nanoparticles Built from Dedicated Dipolar Thienothiophene Dyes as Ultra-Bright Green to NIR Nanoemitters" Molecules 21, no. 9: 1227. https://doi.org/10.3390/molecules21091227
APA StyleMastrodonato, C., Pagano, P., Daniel, J., Vaultier, M., & Blanchard-Desce, M. (2016). Molecular-Based Fluorescent Nanoparticles Built from Dedicated Dipolar Thienothiophene Dyes as Ultra-Bright Green to NIR Nanoemitters. Molecules, 21(9), 1227. https://doi.org/10.3390/molecules21091227