Infrared spectra were obtained using a Perkin-Elmer 1600 series FTIR spectrometer (Perkin-Elmer, Walluf, Germany) and are given in cm−1 units. Solid samples are measured as CsI or KBr discs while liquids are measured as neat between two NaCl plates; the 1H-NMR spectra were recorded on Bruker Avance 300, Bruker DPX 300 spectrometers (Bruker, Ettlingen, Germany) operating at 300 MHz, or on Bruker Avance 500 spectrometer (Bruker, Ettlingen, Germany) instruments operating at 500 MHz. Chemical shifts are reported as δ in ppm and the coupling constants J in Hz units. In all spectra, the solvent peaks were used as the internal standard. Solvents used are CDCl3 (δ = 7.24 ppm), DMSO-d6 (δ = 2.49 ppm), acetone-d6 (δ = 2.04 ppm), and MeOH-d4 (δ = 3.35, 4.78 ppm). Splitting patterns are designated as follows: s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad; the 13C-NMR spectra were recorded either on a Bruker Avance 300 spectrometer instrument operating at 75 MHz or on a Bruker Avance 500 spectrometer instrument operating at 125 MHz; high resolution mass spectra (HR-MS) were recorded on a Finnigan MAT 900 spectrometer(Scientific Instrument Services, Ringoes, NJ, USA) and are measured for the molecular ion peak (M+); CHN-combustion analyses were measured using an Elementar Vario EL Instrument (Elementar, Langenselbold, Germany).
General Procedures (GP)
GP-1: Horner–Wadsworth–Emmons reaction. In a flame-dried 200 mL three-necked flask in a nitrogen atmosphere, sodium hydride (60% dispersion in oil) was washed with hexane and suspended in THF. Triethylphosphonoacetate was dropped to the suspension at 0 °C over 10 min and stirred at r.t. for 30 min. After that, a solution of the ketone in THF was added in 10 min and the reaction mixture heated to reflux for 10 h. After cooling to r.t., an aqueous NH4Cl solution was added and extracted with 4× diethylether. The organic phases were washed with brine and dried over Na2SO4. After solvent evaporation, the product was purified by column chromatography.
GP-2: Reduction with DIBAL-H (diisobutylaluminium hydride). A solution of DIBAL-H (1.5 M in toluene) was slowly added to a solution of the ester 2 in diethylether cooled to 0 °C and subsequently stirred at r.t. for 2 h. After dilution with diethylether (twice the solvent volume of the starting reaction) and cooling to 0 °C, saturated aqueous NaCl solution was added slowly, the pH was adjusted to 3 with 4 M HCl, and the aqueous phase was extracted with 3× diethylether. The organic phases were washed with brine and dried over Na2SO4. After solvent evaporation, the product was purified by column chromatography.
GP-3: Oxidation with MnO2. A suspension of MnO2 in a toluene solution of the alcohol 3 was stirred overnight at RT. Afterwards, the reaction mixtures were filtered over celite and the solvent was evaporated. The crude product was purified by column chromatography.
GP-4: Aldol reactions with ethyl acetate. A solution of n-BuLi in hexane (19.2 mmol, 2 M) was added at −78 °C to a solution of diisopropylamine (19.2 mmol) in 40 mL of THF. After stirring for 5 min, ethyl acetate (16 mmol) was added and the mixture stirred at −78 °C for 2 h. Afterwards, the aldehyde 4 in 10 mL of THF was added and stirred for 5 min, warmed to −20 °C and quenched with 20 mL of 1 N HCl. The aqueous phase was extracted with 3× diethylether. The organic phases were washed with brine and dried over Na2SO4. After solvent evaporation, the product was purified by column chromatography.
GP-5: Reduction of aldols with NaBH4. In a flame-dried 200 mL three-necked flask in a nitrogen atmosphere, the aldol was dissolved in THF, cooled to 0 °C and NaBH4 was added in one portion. Afterwards, a solution of iodine in THF was slowly added and the reaction mixture heated to reflux for 3 h. After cooling to RT, methanol was carefully added until a clear solution resulted, stirred for 30 min and the solvent evaporated. The colorless residue was dissolved in a 1:1 water-diethylether solvent mixture, stirred for 20 min, separated and the aqueous phase extracted with 3× diethylether. The organic phases were washed with brine and dried over Na2SO4. After solvent evaporation, the product was purified by column chromatography.
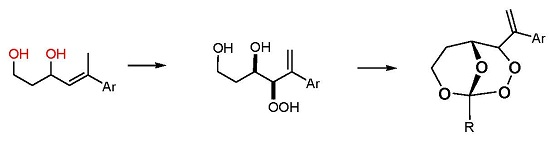
GP-6a: Photooxygenation under homogeneous conditions. The substrate (1 mmol) was dissolved in 30 mL of CCl4 and 3–5 mg (2–4 × 10−4 M) of the sensitizer TPP (meso-tetraphenylporphyrin) were added. The solution was cooled to 10 °C and irradiated with a white LED under oxygen atmosphere until complete conversion (TLC = thin layer chromatography control). Subsequently, the solvent was evaporated and the residue analyzed by NMR. The crude hydroperoxides were directly used for peroxyacetalization.
GP-6b: Photooxygenation under polymer matrix conditions. Commercially available polystyrene-divinylbenzene copolymer beads (2.5 g) are distributed over a Petri dish (19 cm diameter) and swollen by CH2Cl2 (20 mL). The substrate (ca. 10 mmol) and the nonpolar sensitizer (TPP or TTP, ca. 3–6 mg) in ethyl acetate (20 mL) are subsequently added and the excess solvent is evaporated by leaving the Petri dish in a well ventilated hood. The Petri dish is then covered with a glass plate and the sandy solid is irradiated with a halogen lamp or a sodium street lamp. The polymer beads are subsequently rinsed with ethanol (3 × 30 mL) and filtered (the beads are kept for regeneration and reuse). The solvent is evaporated under reduced pressure (caution: water bath temperature should not exceed 30 °C).
GP-7a: Peroxyacetalization to give bicyclic perorthoesters. To a stirred solution of the β-hydroxy hydroperoxide 7 (1 mmol) and 3 equivalents of an orthoester in dry CH2Cl2 (12 mL) was added at room temperature a catalytic amount of pyridinium-p-toluenesulfonic acid (PPTS) (ca. 10 mg) and the mixture was further stirred for about 12 h (overnight) at the same temperature. The reaction mixture was partitioned between CH2Cl2 and saturated NaHCO3 solution and the phases were separated. The aqueous phase was extracted with CH2Cl2 (3 × 30 mL) and the combined organic phases were washed with brine and water, and dried over Na2SO4. Solvent evaporation (caution: water bath temperature should not exceed 30 °C) followed by chromatographic purification afforded the bicyclic perorthoesters as pure products.
GP-7b: Peroxyacetalization to give monocyclic 1,2,4-trioxanes. To a stirred solution of the β-hydroxy hydroperoxide and 1.5 equivalents of the carbonyl component in dry CH2Cl2 (100 mL) was added at room temperature a catalytic amount of boron trifluoride etherate (ca. 0.2 mL) and the mixture was further stirred for about 12 h (overnight) at the same temperature. The reaction mixture was partitioned between CH2Cl2 and saturated NaHCO3 solution and the phases were separated. The aqueous phase was extracted with CH2Cl2 (3 × 30 mL) and the combined organic phases were washed with brine and water, and dried over Na2SO4. Solvent evaporation (caution: water bath temperature should not exceed 30 °C), followed by chromatographic purification, afforded the spiro-bistrioxanes as pure products.
(E)-Ethyl-3-phenylbut-2-enoate (
2a) [
46]. Following GP-1 (General Procedure 1), 1.61 g (13.4 mmol) of acetophenone (
1a) and 6.86 g (34.6 mmol, 2.6 eq) of triethylphosphono acetate and 1.33 g (33.4 mmol, 2.5 eq) of sodium hydride in 40 mL THF were reacted. The product was purified by column chromatography (CH/Et
2O = 9:1,
Rf = 0.22) and isolated as yellow oil: 2.0 g, 10.5 mmol, 78%.
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 1.32 (t, 3H,
J = 7.1 Hz, C
H3CH
2), 2.59 (d, 3H,
J = 1.2 Hz, C
H3CH=), 4.22 (q, 2H,
J = 7.1 Hz, C
H2CH
3), 6.15 (d, 1H,
J = 1.2 Hz, CH
3C
H=), 7.35–7.49 (m, 5H, H
ar.).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 14.3 (
CH
3CH
2), 17.9 (CH
3), 59.8 (CH
3CH
2), 117.1 (H
C=C), 126.2 (C
ar.), 128.4 (C
ar.), 128.9 (C
ar.), 142.2 (C
ar.), 155.4 (CH
3C=), 166.8 (C=O).
(E)-3-Phenylbut-2-enol (
3a) [
47]. Following GP-2 (General Procedure 2), 335 mg (1.76 mmol) of ester
2a was reduced with 3.52 mL DIBAL-H (3.52 mmol, 2 eq, 1H solution in hexane) in 16 mL of diethylether. The product
3a was purified by column chromatography (PE/Et
2O 9:1,
Rf = 0.12) and isolated as colorless oil: 170 mg, 1.23 mmol, 70%.
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 2.09 (s, 3H, CH
3), 2.70 (br s, 1H, OH), 4.38 (d, 2H,
J = 6.63 Hz, C
H2OH), 6.01 (dt, 1H,
J = 1.33 Hz, 6.63 Hz, HC=C), 7.33–7.46 (m, 5H, H
ar.).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 15.8 (CH
3), 59.8 (CH
2OH), 125.6 (C
ar.), 126.5 (H
C=C), 127.0 (C
ar.), 128.1 (C
ar.), 137.3 (CH
3C=) 142.8 (C
ar.).
(E)-3-Phenylbut-2-enal (
4a) [
48]. Following GP-3 (General Procedure 3), 1.50 g (10.1 mmol) of the alcohol
3a and 15 g of MnO
2 in 250 mL of toluene were reacted. The product was purified by column chromatography (PE/Et
2O = 1:1,
Rf = 0.30) and isolated as colorless oil: 0.84 g, 5.7 mmol, 57%.
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 2.55 (s, 3H,
J = 1.2 Hz, CH
3), 6.39 (dq, 1H,
J = 1.2 Hz, 7.8 Hz, HC=C), 7.35–7.55 (m, 5H, H
ar.), 10.17 (d, 1H,
J = 7.8 Hz, CHO).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 16.2 (CH
3), 126.1 (C
ar.), 127.1 (C
ar.), 128.2 (C
ar.), 128.6 (C
ar.), 129.9 (H
C=C), 140.4 (C
ar.), 157.4 (CH
3C=), 191.0 (CHO).
(E)-Ethyl-3-hydroxy-5-phenylhex-4-enoate (5a). Following GP-4 (General Procedure 4), 840 mg (5.7 mmol) of the aldehyde 4a and 0.3 mL (3.8 mmol) of ethyl acetate were reacted. The product 5a was purified by column chromatography (CH/Et2O 3:2, Rf = 0.35) and isolated as yellow oil: 604 mg, 2.6 mmol, 68%. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.30 (t, 3H, J = 7.2 Hz, CH3CH2), 2.13 (d, 3H, J = 1.5 Hz, CH3), 2.38 (m, 2H, CH2), 3.18 (br, 1H, OH), 4.20 (q, 2H, J = 7.2 Hz, CH2CH3), 5.00 (m, 1H, CHOH), 5.82 (dd, 1H, J = 7.2 Hz, 1.2 Hz, C=CH), 7.34–7.41 (m, 5H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 14.0 (CH3CH2), 16.2 (CH3), 41.5 (CH2), 60.3 (CH2CH3), 65.4 (CHOH), 125.7 (HC=C), 127.3 (Car.), 128.1 (Car.), 128.4 (Car.), 137.7 (C=CH), 142.6 (Car.), 172.1 (C=O). IR (Film): ν (cm−1) = 3402 (s), 2979 (m), 2928 (m), 1730 (s), 1646 (w), 1492 (w), 1369 (s), 1277 (s), 1157 (s), 1020 (s), 948 (m), 860 (m). MS (EI, 70 eV): m/z (%) = 216 (M+ − H2O, 7), 171 (4), 143 (74), 128 (100), 115 (53).
(E)-5-Phenylhex-4-ene-1,3-diol (6a). Following GP-5 (General Procedure 5), 600 mg (2.56 mmol) of the aldol 5a was reduced with 240 mg (6.4 mmol, 2.5 eq) of NaBH4 and 200 mg (0.78 mmol, 0.3 eq) of I2. The product was purified by column chromatography (Et2O, Rf = 0.45) and isolated as yellow solid: 396 mg, 2.1 mmol, 80%. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.84 (m, 2H, CH2), 2.10 (s, 3H, CH3), 2.57 (br s, 2H, OH), 3.88 (m, 1H, CH2OH), 4.83 (m, 1H, CHOH), 5.00 (m, 1H, CHOH), 5.84 (dd, 1H, J = 1.2 Hz, 8.4 Hz, C=CH), 7.30–7.40 (m, 5H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 15.9 (CH3), 38.7 (CH2), 60.4 (CH2OH), 68.1 (CHOH), 125.6 (HC=C), 127.0 (Car.), 128.0 (Car.), 130.3 (Car.), 136.1 (C=CH), 142.7 (Car.). IR (Film): ν (cm−1) = 3323 (s), 2943 (m), 2880 (m), 1492 (w), 1379 (s), 1121 (w), 1044 (s), 955 (m), 895 (w), 755 (s), 695 (s). MS (EI, 70 eV): m/z (%) = 177 (M+ − CH3, 9), 159 (177 − H2O, 4), 147 (M+ − OCH2CH3, 87), 129 (100), 115 (53).
(E)-4-Methoxyethyl-3-phenylbut-2-enoate (
2b) [
49]. Following GP-1, 2.00 g (13.4 mmol) of 4-methoxy acetophenone (
1b) and 6.86 g (34.6 mmol, 2.6 eq) of triethylphosphono acetate and 1.33 g (33.4 mmol, 2.5 eq) of sodium hydride in 40 mL THF were reacted. The product was purified by column chromatography (CH/Et
2O = 9:1,
Rf = 0.24) and isolated as yellow oil: 2.3 g, 10.4 mmol, 77%.
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 1.31 (t, 3H,
J = 7.2 Hz, C
H3CH
2), 2.56 (d, 3H,
J = 1.2 Hz, CH
3), 3.82 (s, 3H, CH
3O), 4.20 (q, 2H,
J = 7.2 Hz, CH
3C
H2), 6.11 (d, 1H,
J = 1.2 Hz, C=CH), 6.89 (m, 2H, H
ar.), 7.45 (m, 2H, H
ar.).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 14.3 (
CH
3CH
2), 17.6 (CH
3), 55.3 (CH
3O), 59.6 (CH
3CH
2), 113.8 (C
ar.), 115.3 (
CH=C), 127.6 (C
ar.), 134.3 (C
ar.), 154.8 (CH=
C), 160.4 (C
ar.), 167.0 (C=O). IR (Film): ν (cm
−1) = 2979 (s), 2914 (m), 1709 (s), 1625 (w), 1603 (m), 1513 (s), 1344 (s), 1157 (m), 1082 (m). MS (EI, 70 eV):
m/
z (%) = 221 (M
+, 15), 192 (M
+ − CH
2CH
3, 12), 175 (M
+ − OCH
2CH
3, 100), 148 (M
+ − O=COH
2CH
3, 83). CHN-analysis: (C
13H
16O
3, M = 220.11 g/mol)—calcd.: 70.89% C 7.32% H, found: 71.04% C 7.63% H.
(E)-3-(4-Methoxyphenyl)but-2-ene-1-ol (
3b) [
50]. Following GP-2, 2.00 g (10.5 mmol) of ester
2b was reduced with 21 mL DIBAL-H (21 mmol, 2 eq, 1 M solution in hexane) in 60 mL of diethylether. The product
3b was purified by column chromatography (PE/Et
2O 9:1,
Rf = 0.10) and isolated as colorless oil: 1.79 g, 10.1 mmol, 96%.
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 2.06 (s, 3H, CH
3), 3.81 (s, 3H, CH
3O), 4.35 (d, 2H,
J = 6.9 Hz, CH
2), 5.92 (dt, 1H,
J = 1.2 Hz, 6.9 Hz, C=CH), 6.87 (m, 2H, H
ar.), 7.36 (m, 2H, H
ar.).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 16.0 (CH
3), 55.3 (CH
3O), 59.9 (CH
2OH), 113.6 (C
ar.), 124.8 (C=
CH), 126.8 (C
ar.), 135.3 (CH
2=
C), 137.3 (C
ar.), 158.9 (C
ar.).
(E)-3-(4-Methoxyphenyl)but-2-enal (
4b) [
51]. Following GP-3, 1.79 g (10.1 mmol) of the alcohol
4a and 18 g of MnO
2 in 250 mL of toluene were reacted. The product was purified by column chromatography (PE/Et
2O = 1:1,
Rf = 0.28) and isolated as colorless oil: 1.79 g (6.9 mmol, 68%).
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 2.54 (s, 3H, CH
3), 3.84 (s, 3H, CH
3O), 6.38 (dd, 1H,
J = 1.2 Hz, 7.8 Hz, C=CH), 6.93 (d, 2H,
J = 9 Hz, H
ar.), 7.53 (d, 2H,
J = 9 Hz, H
ar.), 10.15 (d, 1H,
J = 7.8 Hz, CHO).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 16.0 (CH
3), 55.4 (CH
3O), 114.0 (C
ar.), 125.5 (C=
CH), 127.8 (C
ar.), 132.4 (C
ar.), 156.9 (
C=CH), 161.3 (C
ar.), 191.2 (CHO). IR (film): ν (cm
−1) = 2957 (w), 2837 (m), 1649 (s), 1593 (s), 1506 (m), 824 (s).
(E)-Ethyl-3-hydroxy-5-(4-methoxyphenyl)hex-4-enoate (5b). Following GP-4, 325 mg (1.8 mmol) of the aldehyde 4b and 0.13 mL (1.25 mmol) of ethyl acetate were reacted. The product 5a was purified by column chromatography (PE/Et2O 1:1, Rf = 0.18) and isolated as yellow oil: 360 mg, 1.4 mmol, 77%. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.28 (t, 3H, J = 7.2 Hz, CH3CH2O), 2.08 (s, 3H, CH3), 2.60 (m, 2H, CH2), 2.95 (br s, 1H, OH), 3.80 (s, 3H, CH3O), 4.18 (q, 2H, J = 7.2 Hz, CH3CH2O), 4.96 (m, 1H, CHO), 5.72 (dd, 1H, J = 0.9 Hz, 8.4 Hz, C=CH), 6.85 (d, 2H, J = 8.7 Hz, Har.), 7.33 (m, 2H, J = 8.7 Hz, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 14.1 (CH3CH2), 16.3 (CH3), 41.6 (CH2), 55.2 (CH3O), 60.7 (CH3CH2), 65.8 (CHOH), 113.6 (Car.), 126.7 (C=CH), 126.9 (Car.), 135.1 (Car.), 137.3 (C=CH), 159.0 (Car.), 172.3 (C=O). IR (Film): ν (cm−1) = 3429(s), 2982 (s), 2831(m), 1731 (m), 1606 (m), 1506 (s), 826 (s). MS (EI, 70 eV): m/z (%) = 264 (M+, 21), 249 (M+ − CH3, 12), 246 (M+ − H2O, 27), 177 (90), 159 (76), 135 (CH3O(C6H4)CHCH3, 100), 115 (39).
(E)-5-(4-Methoxyphenyl)hex-4-ene-1,3-diol (6b). Following GP-5, 175 mg (0.66 mmol) of the aldol 6a was reduced with 63 mg (1.65 mmol, 2.5 eq) of NaBH4 and 51 mg (0.22 mmol, 0.3 eq) of I2. The product was purified by column chromatography (CHCl3/MeOH 9:1, Rf = 0.27) and isolated as colorless solid: 140 mg, 0.63 mmol, 96%. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.79 (m, 2H, CH2), 2.04 (s, 3H, CH3), 3.34 (br s, 2H, 2 × OH), 3.78 (s, 3H, OCH3), 3.84 (m, 2H, CH2OH), 4.77 (m, 1H, CHOH), 5.75 (dd, 1H, J = 1.2 Hz, 8.4 Hz, C=CH), 6.83 (d, 2H, J = 8.7 Hz, Har.), 7.32 (d, 2H, J = 8.7 Hz, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 16.0 (CH3), 38.8 (CH2), 55.1 (OCH3), 60.7 (CH2OH), 68.4 (CHOH), 113.5 (C=CH), 126.7 (Car.), 128.5 (Car.), 135.1 (Car.), 135.9 (C=CH), 158.8 (Car.). IR (Film): ν (cm−1) = 3332 (s), 2938 (s), 1605 (m), 1510 (s), 1245 (s), 1032 (s), 826 (m). MS (EI, 70 eV): m/z (%) = 222 (M+, 12), 204 (M+ − H2O, 11), 177 (C11H13O2+, 45), 159 (49), 135 (100), 115 (39).
(E)-3-(2-Naphthyl)but-2-enylacetate (
2c) [
52]. Following GP-1, 2.21 g (13.4 mmol) of acetonaphthone (
1c) and 6.86 g (34.6 mmol, 2.6 eq) of triethylphosphono acetate and 1.33 g (33.4 mmol, 2.5 eq) of sodium hydride in 40 mL THF were reacted. The product was purified by column chromatography (CH/Et
2O = 9:1,
Rf = 0.39) and isolated as colorless solid, m.p. 45–46 °C, 2.5 g, 10.5 mmol, 80%.
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 1.36 (t, 3H,
J = 7.2 Hz, C
H3CH
2), 2.71 (d, 3H,
J = 1.2 Hz, CH
3), 4.28 (q, 2H,
J = 7.2 Hz, CH
3C
H2), 6.32 (d, 1H,
J = 1.2 Hz, C=CH), 7.4–7.9 (m, 7H, H
ar.).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 14.3 (CH
3), 17.8 (CH
3), 59.8 (
CH
2CH
3), 117.4 (C=
CH), 123.9 (C
ar.), 125.8 (C
ar.), 126.4 (C
ar.), 126.6 (C
ar.), 127.5 (C
ar.), 128.1 (C
ar.), 128.4 (C
ar.), 133.0 (C
ar.), 133.4 (C
ar.), 139.3 (C
ar.), 155.1 (C=), 166.8 (C=O).
(E)-3-(2-Naphthyl)but-2-ene-1-ol (
3c) [
53]. Following GP-2, 2.50 g (10.5 mmol) of ester
2c was reduced with 1.11 g (29.2 mmol, 2.8 eq) of LiAlH
4 in 16 mL of diethylether. The product
3c was purified by recrystallization from benzene/hexane (1:1) resulting in 1.34 g (6.7 mmol, 64%) of colorless crystals, m.p. 85–86 °C.
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 1.67 (br s, 1H, OH), 2.20 (s, 3H, CH
3) 4.44 (t, 2H,
J = 5.6 Hz, C
H2OH), 6.15 (t, 1H,
J = 6.7 Hz, C=CH), 7.41–7.90 (m, 7H, H
ar.).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 16.0 (CH
3), 60.0 (CH
2OH), 124.1 (C
ar.), 124.5 (C
ar.), 125.8 (C
ar.), 126.1 (C
ar.), 127.0 (C
ar.), 127.5 (C
ar.), 128.1 (C
ar.), 128.4 (C
ar.), 133.3 (C=
CH), 137.6 (C
ar.), and 139.3 (
C=CH).
(E)-3-(2-Naphthyl)but-2-enal (
4c) [
51]. Following GP-3, 1.29 g (6.5 mmol) of the alcohol
3c and 13 g of MnO
2 in 250 mL of toluene were reacted. The product was purified by column chromatography (PE/Et
2O = 1:1,
Rf = 0.56) and isolated as colorless oil: 0.88 g, 4.5 mmol, 69%.
1H-NMR (300 MHz, CDCl
3): δ (ppm) = 2.64 (s, 3H, CH
3), 6.55 (d, 1H,
J = 7.8 Hz, C=CH), 7.51–8.01 (m, 7H, H
ar.), 10.23 (d, 1H,
J = 7.8 Hz, CHO).
13C-NMR (75 MHz, CDCl
3): δ (ppm) = 16.2 (CH
3), 123.3 (C
ar.), 126.2 (C
ar.), 126.6 (C
ar.), 127.1 (C
ar.), 127.3 (C
ar.), 127.5 (C
ar.), 128.3 (C
ar.), 128.6 (C
ar.), 132.8 (C
ar.), 133.9 (C=
CH), 137.4 (C
ar.), 157.1 (
C=CH), 191.1 (CHO).
(E)-Ethyl-3-hydroxy-5-(2-naphthyl)hex-4-enoate (5c). Following GP-4, 875 mg (4.5 mmol) of the aldehyde 4c and 0.30 mL (3.8 mmol) of ethyl acetate were reacted. The product was purified by column chromatography (CH/Et2O 3:2, Rf = 0.38) and isolated as yellow oil: 470 mg, 1.65 mmol, 37%. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.30 (t, 3H, J = 7.2 Hz, CH3CH2) 2.23 (s, 3H, CH3), 2.66 (m, 2H, CH2), 4.11 (q, 2H, J = 7.2 Hz CH3CH2), 5.04 (m, 1H, CHOH), 5.94 (d, 1H, J = 7.2 Hz, C=CH), 7.45–7.82 (m, 7H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 14.2 (CH3), 16.4 (CH2CH3), 41.6 (CH2), 60.9 (CH2CH3), 65.7 (CHOH), 124.2 (Car.), 124.7 (Car.), 125.9 (Car.), 126.2 (C=CH2), 127.5 (Car.), 127.8 (Car.), 128.2 (Car.), 128.8 (Car.), 132.8 (Car.), 133.3 (Car.), 137.8 (C=CH2), 139.1 (Car.). IR (film): ν (cm−1) = 3418 (s), 3054 (m), 2979 (m), 1730 (s), 1369 (m), 1277 (m), 1158 (s), 1019 (s), 853 (m), 816 (s), 747(s). MS (EI, 70 eV): m/z (%) = 284 (M+, 7), 266 (M+ − H2O, 13), 196 (M+ − C(=O)OCH2CH3, 56), 179 (100), 155 (77), 128 (52).
(E)-5-(2-Naphthyl)hex-4-ene-1,3-diol (6c). Following GP-5, 470 mg (1.65 mmol) of the aldol 5c was reduced with 160 mg (4.13 mmol, 2.5 eq) of NaBH4 and 130 mg (0.5 mmol, 0.3 eq) of I2. The product was purified by column chromatography (CHCl3/MeOH 9:1, Rf = 0.23) and isolated as colorless solid: 300 mg, 1.24 mmol, 75%. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.89 (m, 2H, CH2) 2.20 (s, 3H, CH3), 2.29 (br s, 2H, OH), 3.94 (m, 2H, CH2OH), 4.90 (m, 2H, CHOH), 6.00 (dd, 1H, J = 1.2 Hz, 7.2 Hz, C=CH), 7.25–7.82 (m, 7H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 16.3 (CH3), 38.8 (CH2), 61.4 (CH2O), 69.2 (CHOH), 124.1 (Car.), 124.6 (Car.), 127.5 (Car.), 127.6 (Car.), 127.8 (Car.), 128.0 (Car.), 128.1 (Car.), 128.7 (Car.), 130.6 (C=CH), 133.3 (Car.), 136.9 (C=CH), 138.7 (Car.). IR (Film): ν (cm−1) = 3304 (s), 2921 (s), 2851 (m), 1420 (m), 1272 (m), 1046 (s), 904 (s), 854 (m), 815 (s), 727(s). MS (EI, 70 eV): m/z (%) = 242 (M+, 5), 192 (M+ − H2O, 5), 175 (M+ − OCH2CH3, 100), 148 (M+ − O=COH2CH3, 83). Tm = 38–39 °C (aus CHCl3).
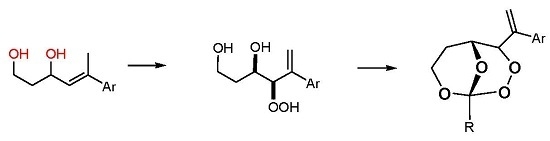
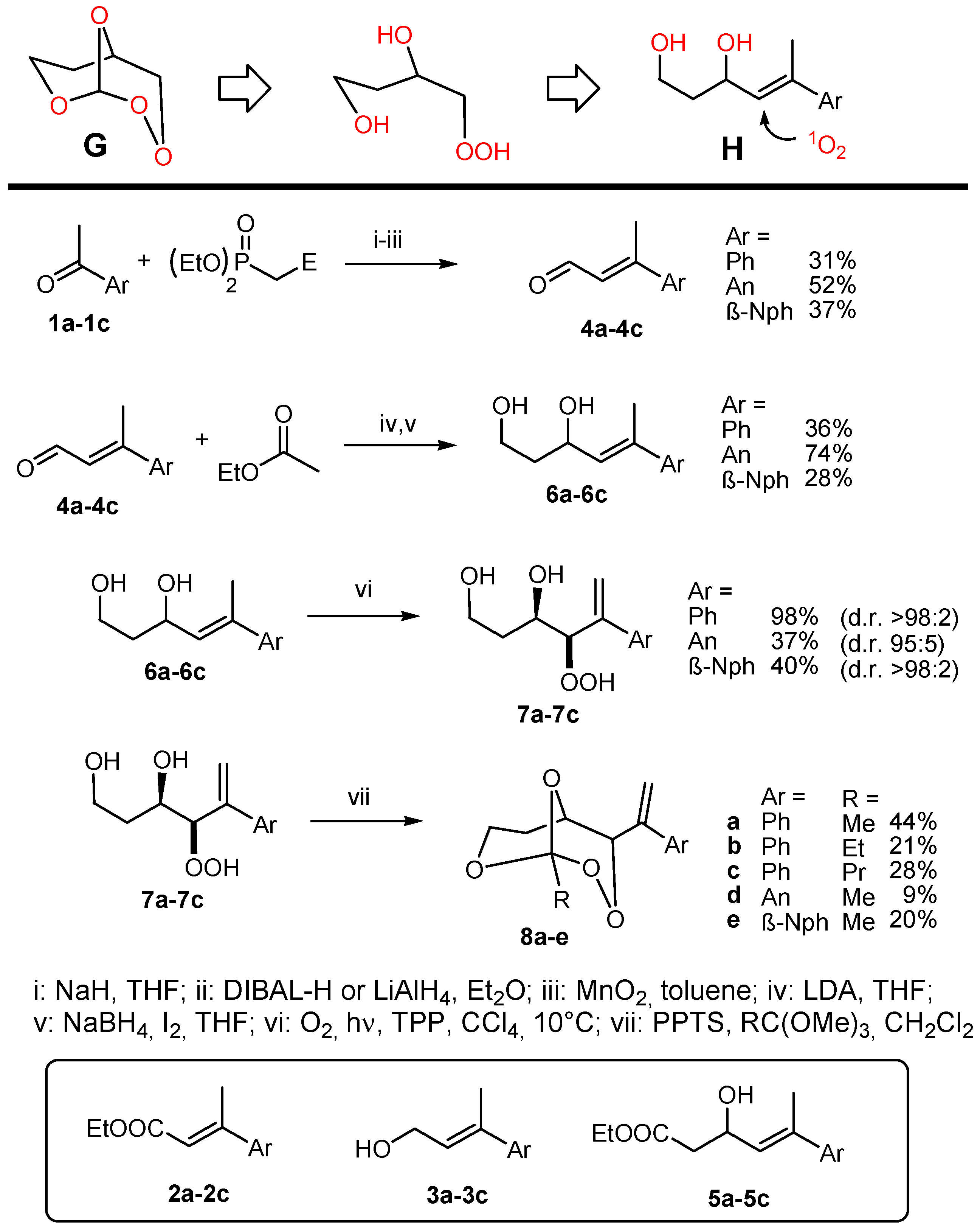
(R*R*)-4-Hydroperoxy-5-phenylhex-5-ene-1,3-diol (7a). Following GP-6a, 100 mg (0.52 mmol) of the diol 6a and 2.5 mg of TPP in 30 mL of CCl4 was irradiated for 5 h (complete conversion). Evaporation of the solvent delivered the crude product. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.83 (m, 2H, CH2), 3.54 (m, 2H, CH2O), 4.42 (d, 1H, J = 3.9 Hz, CHOO), 4.99 (m, 1H, CHO), 5.09 (dt, 2H, J = 1.2 Hz, 12.6 Hz, CH2=C), 7.30–7.60 (m, 5H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 34.1 (CH2), 60.6 (CH2OH), 72.3 (CHOH), 91.3 (CHOOH), 118.0 (C=CH2), 126.9 (Car.), 128.0 (Car.), 128.5 (Car.), 139.5 (C=CH2), 144.9 (Car.).
(R*R*)-4-Hydroperoxy-5-(4-methoxyphenyl)hex-5-ene-1,3-diol (7b). Following GP-6a (General Procedure 6a), 126 mg (0.57 mmol) of the diol 6b and 4 mg of TPP in 30 mL of CCl4 was irradiated for 5 h (37% conversion). Evaporation of the solvent delivered the crude product as a mixture of diastereoisomers (dr 95:5). 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.67 (m, 2H, CH2), 3.74 (s, 3H, OCH3), 3.88 (m, 2H, CH2O), 4.26 (d, 1H, J = 3.5 Hz, CHOO), 4.62 (m, 1H, CHO), 5.14 (d, 2H, J = 12.3 Hz, CH2=C), 6.7–7.4 (m 4H, Car.).
(R*R*)-4-Hydroperoxy-5-(2-naphthyl)hex-5-ene-1,3-diol (7c). Following GP-6b (General Procedure 6b), 200 mg (0.83 mmol) of the diol 6c was irradiated for 5h (40% conversion). Evaporation of the solvent delivered the crude product. 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.89 (m, 2H, CH2) 3.97 (m, 2H, CH2OH), 4.34 (m, 1H, CHOO), 4.65 (m, 2H, CHOH), 5.81 (m, 2H, C=CH2), 7.25–7.82 (m, 7H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 35.8 (CH2), 60.2 (CH2O), 69.8 (CHOH), 124.1 (Car.), 124.6 (Car.), 127.5 (Car.), 127.6 (Car.), 127.8 (Car.), 128.0 (Car.), 128.1 (Car.), 128.7 (Car.), 129.4 (C=CH2), 133.3 (Car.), 138.7 (Car.), 139.7 (C=CH2).
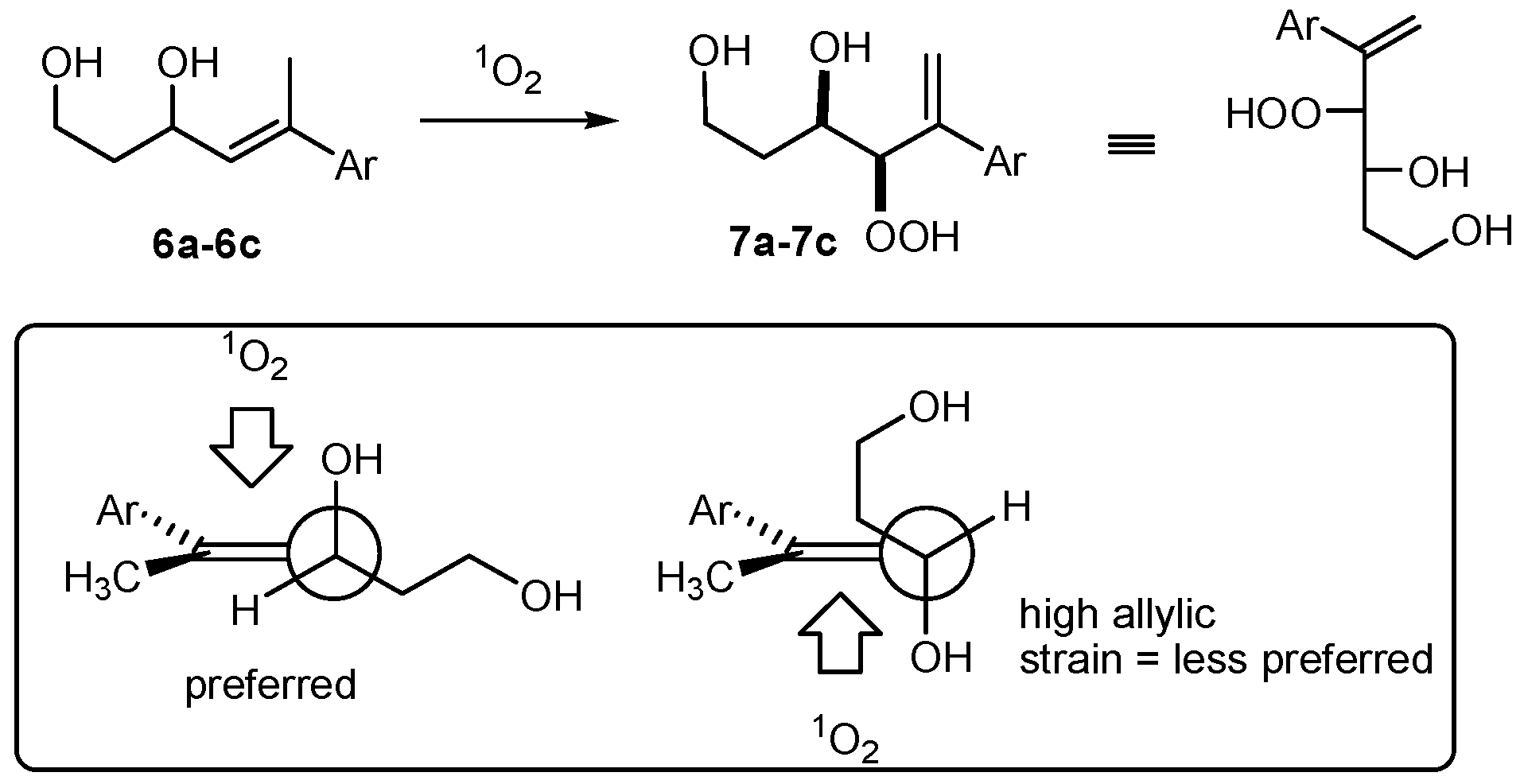
(R*R*)-1-Methyl-4-(1-phenylvinyl)-2,3,8,9-tetraoxabicyclo[3.3.1]nonane (8a). Following GP-7a (General Procedure 7a), 224 mg (0.5 mmol) of the hydroperoxide 7a was reacted with 0.08 mL (1.5 mmol, 3 eq) of trimethyl orthoacetate and catalytic amounts of PPTS in 6 mL of dichloromethane. The product was purified by column chromatography (PE/Et2O 9:1, Rf = 0.43) and isolated as colorless oil (dr 94:6): 57 mg (0.23 mmol, 46%). 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.50 (s, 3H, CH3), 1.74 (m, 1H, CH2), 2.45 (m, 2H, CH2), 3.94 (m, 1H, CH2O), 4.21 (d, 1H, J = 6.3 Hz, CHO), 4.74 (m, 1H, CH2O), 4.95 (s, 1H, CHOO), 5.58 (s, 1H, C=CH2), 5.73 (s, 1H, C=CH2), 7.35 (m, 5H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 24.1 (CH3), 27.5 (CH2), 60.0 (CH2O), 65.7 (CHO), 82.4 (CHOO), 113.5 (OCOO), 117.4 (C=CH2), 126.9 (Car.), 127.9 (Car.), 128.5 (Car.), 139.2 (Car.), 144.2 (C=CH2). IR (Film): ν (cm−1) = 2962 (m), 2923 (m), 2853 (m), 1493 (w), 1442 (m), 1380 (s), 1256 (s), 1105 (s), 966 (m), 900 (m), 752 (s), 702 (s).
(R*R*)-1-Ethyl-4-(1-phenylvinyl)-2,3,8,9-tetraoxabicyclo[3.3.1]nonane (8b). Following GP-7a, 224 mg (0.5 mmol) of the hydroperoxide 7a was reacted with 0.08 mL (1.5 mmol, 3 eq) of triethyl orthopropionate and catalytic amounts of PPTS in 6 mL of dichloromethane. The product was purified by column chromatography (PE/Et2O 4:1, Rf = 0.38) and isolated as colorless oil (dr 95:5): 27 mg (0.10 mmol, 21%). 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.00 (t, 3H, J = 7.5 Hz, CH3CH2), 1.75 (m, 3H, CH3CH2, CH2), 2.44 (m, 1H, CH2), 3.91 (m, 1H, CH2O), 4.21 (d, 1H, J = 6.6 Hz, CHO), 4.68 (m, 1H, CH2O), 4.96 (s, 1H, CHOO), 5.57 (s, 1H, C=CH2), 5.71 (s, 1H, C=CH2), 7.35 (m, 5H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 6.7 (CH3CH2), 27.8 (CH2), 30.8 (CH3CH2), 59.6 (CH2O), 65.8 (CHO), 82.9 (CHOO), 114.7 (OCOO), 117.6 (C=CH2), 127.0 (Car.), 128.0 (Car.), 128.5 (Car.), 139.1 (Car.), 144.4 (C=CH2). IR (Film): ν (cm−1) = 2975 (m), 1493 (w), 1360 (m), 1250 (s), 1105 (s), 914 (s), 777 (s), 702 (s).
(R*R*)-4-(1-Phenylvinyl)-1-propyl-2,3,8,9-tetraoxabicyclo[3.3.1]nonane (8c). Following GP-7a, 224 mg (0.5 mmol) of the hydroperoxide 7a was reacted with 0.24 mL (1.5 mmol, 3 eq) of trimethyl orthobutyrate and catalytic amounts of PPTS in 6 mL of dichloromethane. The product was purified by column chromatography (PE/Et2O 4:1, Rf = 0.48) and isolated as colorless oil (dr 97:3): 39 mg (0.14 mmol, 28%). 1H-NMR (300 MHz, CDCl3): δ (ppm) = 0.92 (t, 3H, J = 7.5 Hz, CH3CH2), 1.54 (m, 2H, CH3CH2), 1.71 (m, 3H, CH3CH2CH2, CH2), 2.42 (m, 1H, CH2), 3.21 (m, 1H, CH2O), 4.21 (dd, 1H, J = 1.8 Hz, 6.6 Hz, CHO), 4.68 (m, 1H, CH2O), 4.96 (s, 1H, CHOO), 5.57 (s, 1H, C=CH2), 5.71 (s, 1H, C=CH2), 7.35 (m, 5H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 14.0 (CH3CH2), 15.7 (CH3CH2CH2), 27.8 (CH2), 39.6 (CH3CH2), 59.6 (CH2O), 65.6 (CHO), 82.8 (CHOO), 114.4 (OCOO), 117.5 (C=CH2), 126.9 (Car.), 127.9 (Car.), 128.5 (Car.), 139.1 (Car.), 144.4 (C=CH2). IR (Film): ν (cm−1) = 2963 (s), 2928 (s), 2872 (s), 1732 (m), 1493 (w), 1378 (m), 1247 (s), 1119 (s), 928 (s), 777 (s), 701 (s).
(R*R*)-4-(1-(4-Methoxyphenyl)vinyl)-1-methyl-2,3,8,9-tetraoxabicyclo[3.3.1]nonane (8d). Following GP-7a, 120 mg (0.54 mmol) of the hydroperoxide 7b was reacted with 0.21 mL (1.62 mmol, 3 eq) of triethyl orthoacetate and catalytic amounts of PPTS in 6 mL of dichloromethane. The product was purified by column chromatography (PE/Et2O 4:1, Rf = 0.34) and isolated as colorless oil: 14 mg (0.05 mmol, 9%). 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.39 (s, 3H, CH3), 1.70 (m, 1H, CH2), 2.45 (m, 1H, CH2), 3.70 (s, 3H, CH3O), 3.82 (m, 1H, CH2O), 4.09 (dt, 1H, J = 1.9 Hz, 6.3 Hz, CHO), 4.61 (m, 1H, CH2O), 4.83 (s, 1H, CHOO), 5.40 (s, 1H, C=CH2), 5.51 (s, 1H, C=CH2), 6.76 (d, 2H, J = 8.8, Har.), 7.22 (d, 2H, J = 8.8 Hz, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 24.1 (CH3), 27.8 (CH2), 55.3 (CH3O) 59.8 (CH2O), 65.8 (CHO), 82.6 (CHOO), 113.6 (OCOO), 113.9 (Car.), 116.2 (C=CH2), 128.0 (Car.), 131.3 (Car.), 143.5 (C=CH2), 159.3 (Car.). IR (Film): ν (cm−1) = 2961 (s), 1606 (m), 1510 (s), 1464 (w), 1380 (m), 1246 (s), 1107 (m), 966 (m), 835 (s), 804 (m), 744 (w).
(R*R*)-1-Methyl-4-(1-(2-naphthyl)vinyl)-2,3,8,9-tetraoxabicyclo[3.3.1]nonane (8e). Following GP-7a, 220 mg (0.8 mmol) of the hydroperoxide 7c was reacted with 0.21 mL (1.62 mmol, 2 eq) of triethyl orthoacetate and catalytic amounts of PPTS in 8 mL of dichloromethane. The product was purified by column chromatography (PE/Et2O 4:1, Rf = 0.36) and isolated as colorless oil (dr 95:5): 50 mg (0.17 mmol, 20%). 1H-NMR (300 MHz, CDCl3): δ (ppm) = 1.49 (m, 1H, CH2), 1.75 (s, 3H, CH3), 2.28 (m, 1H, CH2), 3.91 (m, 1H, CH2O), 4.29 (m, 1H, CH2O), 4.36 (s, 1H, CHO), 5.28 (s, 1H, CHOO), 5.51 (s, 1H, C=CH2), 5.58 (s, 1H, C=CH2), 7.41–7.80 (m, 7H, Har.). 13C-NMR (75 MHz, CDCl3): δ (ppm) = 22.4 (CH3), 28.1 (CH2), 58.8 (CH2O), 76.9 (CHO), 80.0 (CHOO), 113.7 (OCOO), 120.1 (C=CH2), 124.7 (Car.), 124.8 (Car.), 126.2 (Car.), 126.4 (Car.), 127.6 (Car.), 128.0 (Car.), 128.3 (Car.), 132.9 (Car.), 133.2 (Car.), 136.1 (Car.), 145.8 (C=CH2). IR (film): ν (cm−1) = 3054 (m), 2966 (m), 2925 (m), 1731 (m), 1397 (s), 1300 (s), 1120 (s), 1012 (s), 942 (m), 855 (s), 750 (m). MS (EI, 20 eV): m/z (%) = 282 (M+ − CH3, 5), 266 (M+ − O2, 2), 240 (M+ − O2CCH3, 30), 222 (M+ − CH3CO3, 22), 195 (27), 184 (Naphthyl − C(CH2CH3)=CH2, 82), 165 (Naphthyl − C(=CH2)CH3, 57), 155 (Naphthyl − C=CH2, 100). 141 (Naphthyl − CH2, 27), 128 (Naphthyl, 27), 75 (CH3CO3, 56), 57 (CH3CO2, 89).
3,3-Dimethoxy-5-methyl-6-(1-propen-2-yl)-1,2,4-trioxane (10). Following the general procedure GP-6, 60 mg (0.45 mmol, 1.0 eq) of the hydroperoxide threo-9, 62 mg (0.03 mL, 0.23 mmol, 0.5 eq) of tetramethoxymethane and 12 mg (0.045 mmol, 0.1 eq) PPTS were reacted to give 28 mg (0.14 mmol, 30%) of the trioxane 10 after column chromatography (c-Hex/EtOAc (1:1), Rf = 0.85; 1H-NMR (300 MHz, CDCl3): δ (ppm): 1.17 (d, 3H, 3J = 6.0 Hz, O-CH-CH3), 1.77 (s, 3H, C-CH3), 3.44/3.49 (s, 2 × 3H, O-CH3), 4.26 (d, 1H, 3J = 8.6 Hz, C-CH-OO), 4.28–4.37 (m, 1H, CH3-CH-O), 4.13/4.30 (d, 1H, 3J = 8.6 Hz, CH-OOH). 5.13 (s, 2H, C=CH2); 13C-NMR (75.5 MHz, CDCl3): δ (ppm): 16.2 (q, 1C, O-CH-CH3), 19.5 (q, 1C, C-CH3), 51.4/52.3 (q, 2C, C-O-CH3), 71.0 (d, 1C, CH3-CH-O), 87.5 (d, 1C, C-CH-OOH), 118.7 (t, 1C, C-CH2), 137.0 (s, 1C, C=CH2). CHN-analysis: C12H22O3, (M = 214.30 g/mol), calcd. C 67.26% H 10.35% found: C 66.91% H 9.68%.
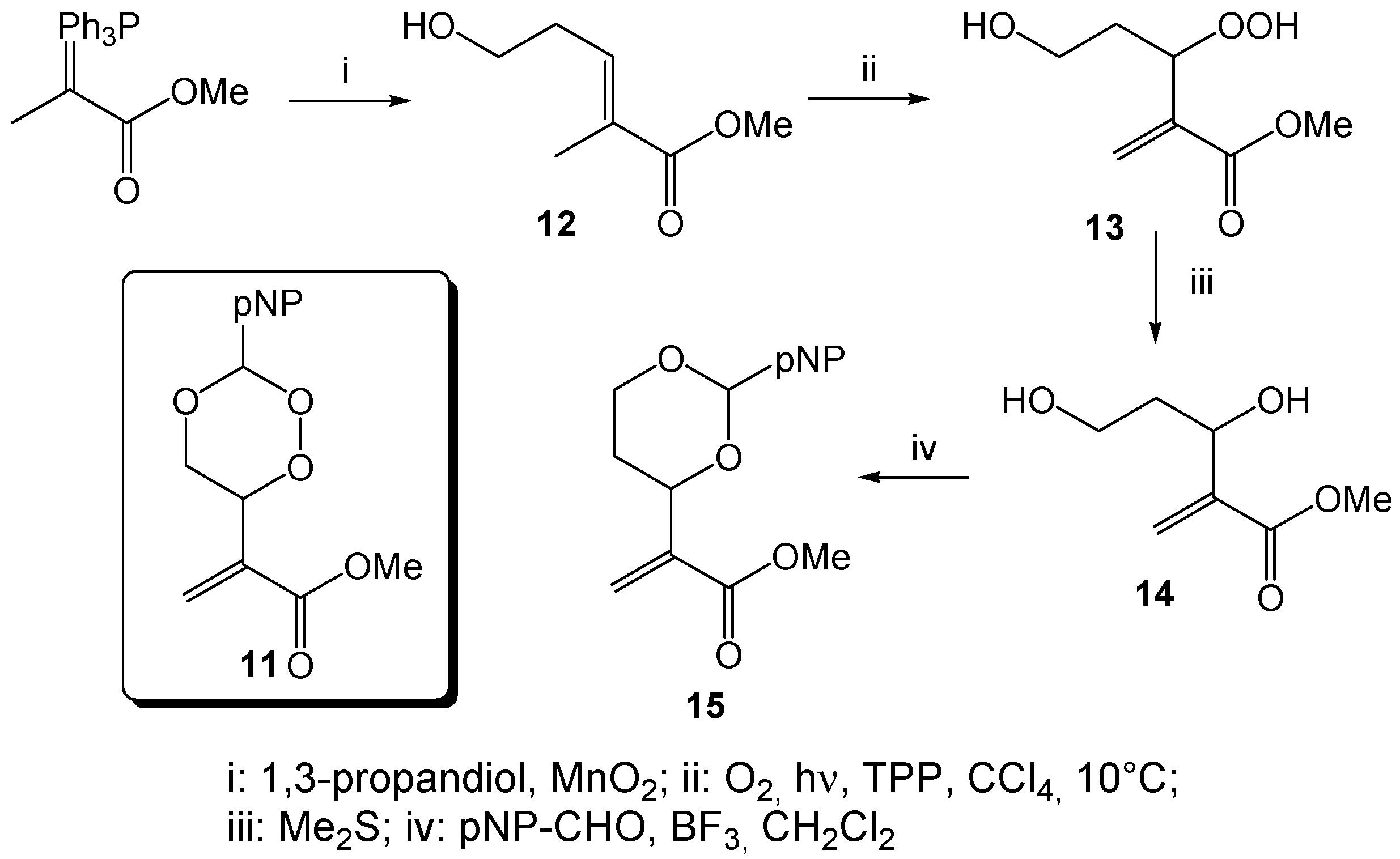
Methyl-5-hydroxy-2-methylpent-2-enonate (
12). A mixture of 0.47 mL of propane-1,3-diol (0.5 g, 6.57 mmol) and 5.0 g of the ylene Methyl-2-(triphenylphosphoranyliden)propanoate [
54] was dissolved in 17 mL of dichloromethane and 10.4 g of activated MnO
2 were added. The mixture was stirred for 3 days at r.t. and filtered. The solid residue was washed with methylene chloride and, after solvent evaporation, purified by column chromatography (hexane/EtOAc 1:4,
Rf = 0.52) to give 447 mg (3.10 mmol, 47%)
12 as a yellow oil.
1H-NMR (300 MHz, CDCl
3): δ = 1.81 (s, 3H), 2.40 (q,
J = 6.7 Hz, 2H), 3.68 (m, 5H), 6.74 (t,
J = 7.3 Hz, 1H);
13C-NMR (75 MHz, CDCl
3): δ = 12.6 (q), 32.1 (t), 51.9 (q), 61.2 (t), 129.6 (s), 138.7 (d), 168.6 (s).
Methyl 3,5-dihydroxy-2-methylenpentanoate (14). A chloroform solution of the hydroperoxide 13 (1.93 g, 10.9 mmol), obtained from the photooxygenation of the homoallylic alcohol 12 (following GP-6a), was treated with 16.5 mL of dimethylsulfide (13.9 g, 223 mmol) and stirred for 18h. After solvent and excess reagent evaporation, the residue was purified by column chromatography (CH/EtOAc 2:3, Rf = 0.30) to give 397 mg (2.25 mmol, 21%) 14 as a colorless oil. 1H-NMR (300 MHz, CDCl3): δ = 1.66 (m, 1H), 1.84 (m, 1H), 3.68 (m, 5H), 4.61 (dd, J = 3.1; 8.5 Hz, 1H), 5.86 (s, 1H), 6.19 (s, 1H); 13C-NMR (75 MHz, CDCl3): δ = 37.7 (t), 51.9 (q), 60.5 (t), 69.6 (d), 125.1 (t), 142.5 (s), 166.8 (s).
Methyl-2-(2-(4-nitrophenyl)-1,3-dioxan-4-yl)acrylate (15). A mixture of 711 mg of the diol 14 (4.00 mmol) and 737 mg of 4-nitrobenzaldehyde (4.80 mmol) in 38 mL of dry CH2Cl2 was treated with a stoechiometric amount (0.5 mL) of BF3 × Et2O and stirred for 18 h at r.t. After addition of 5 mL of aqueous Na2CO3, extraction with diethyl ether and solvent evaporation, the residue was purified by column chromatography (CH/EtOAc 3:1, Rf = 0.45) to give 346 mg (1.18 mmol, 49%) 15 as a yellowish solid, mp 99 °C; 1H-NMR (300 MHz, CDCl3): δ = 1.72 (dd, J = 12.0, 4.7 Hz, 1H), 1.86 (t, J = 21.2 Hz, 1H), 3.72 (s, 3H), 4.01 (t, J = 11.8 Hz, 1H), 4.24 (dd, J = 11.4, 4.5 Hz, 1H), 4.73 (d, J = 10.6 Hz, 1H), 5.64 (s, 1H), 5.95 (s, 1H), 6.28 (s, 1H), 7.61 (d, J = 8.3 Hz, 2H), 8.14 (d, J = 8.4 Hz, 2H); 13C-NMR (75 MHz, CDCl3): δ = 31.9 (t), 52.1 (q), 67.3 (t), 75.0 (d), 99.8 (d), 123.5 (d), 125.8 (t), 127.3 (d), 140.1 (s), 145.0 (s), 148.2 (s), 165.8 (s); IR: ν (cm−1) = 1031 (s), 1093 (s), 1118 (s), 1215 (s), 1273 (m), 1292 (m), 1345 (s), 1437 (m), 1520 (s), 1607 (m), 1630 (m), 1933 (w), 2858 (w), 2951 (w); CHN analysis (C14H15NO6): calcd. C 57.34 H 5.16 N 4.78, found C 57.08 H 5.25 N 4.67.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}






