Design, Synthesis, and Antitumor Activity of Novel Quinazoline Derivatives


Abstract
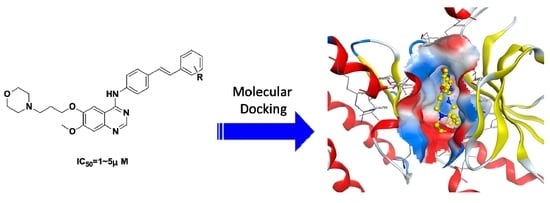
:
1. Introduction
2. Results and Discussion
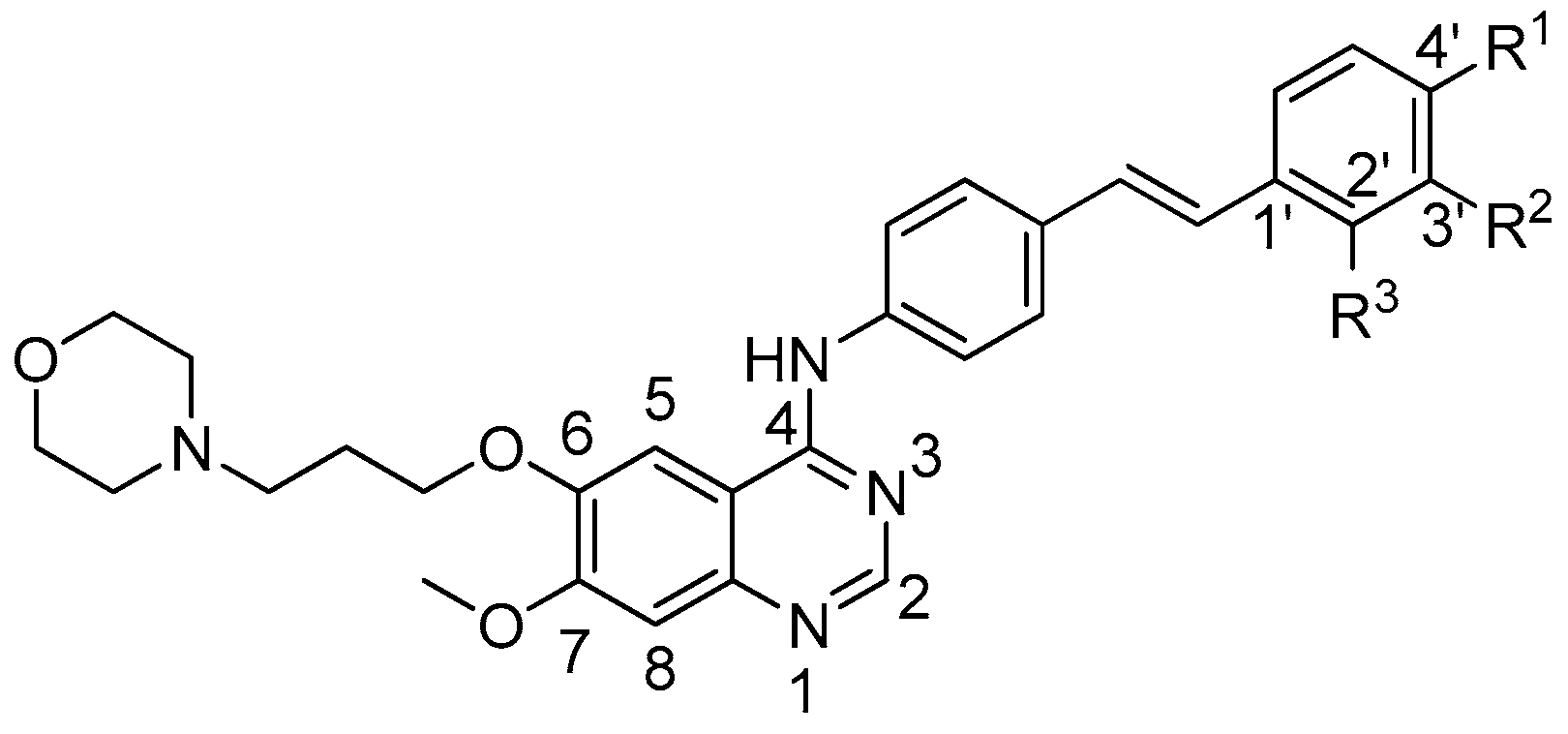
2.1. Chemistry
2.2. Biology
In Vitro Antitumor Evaluation
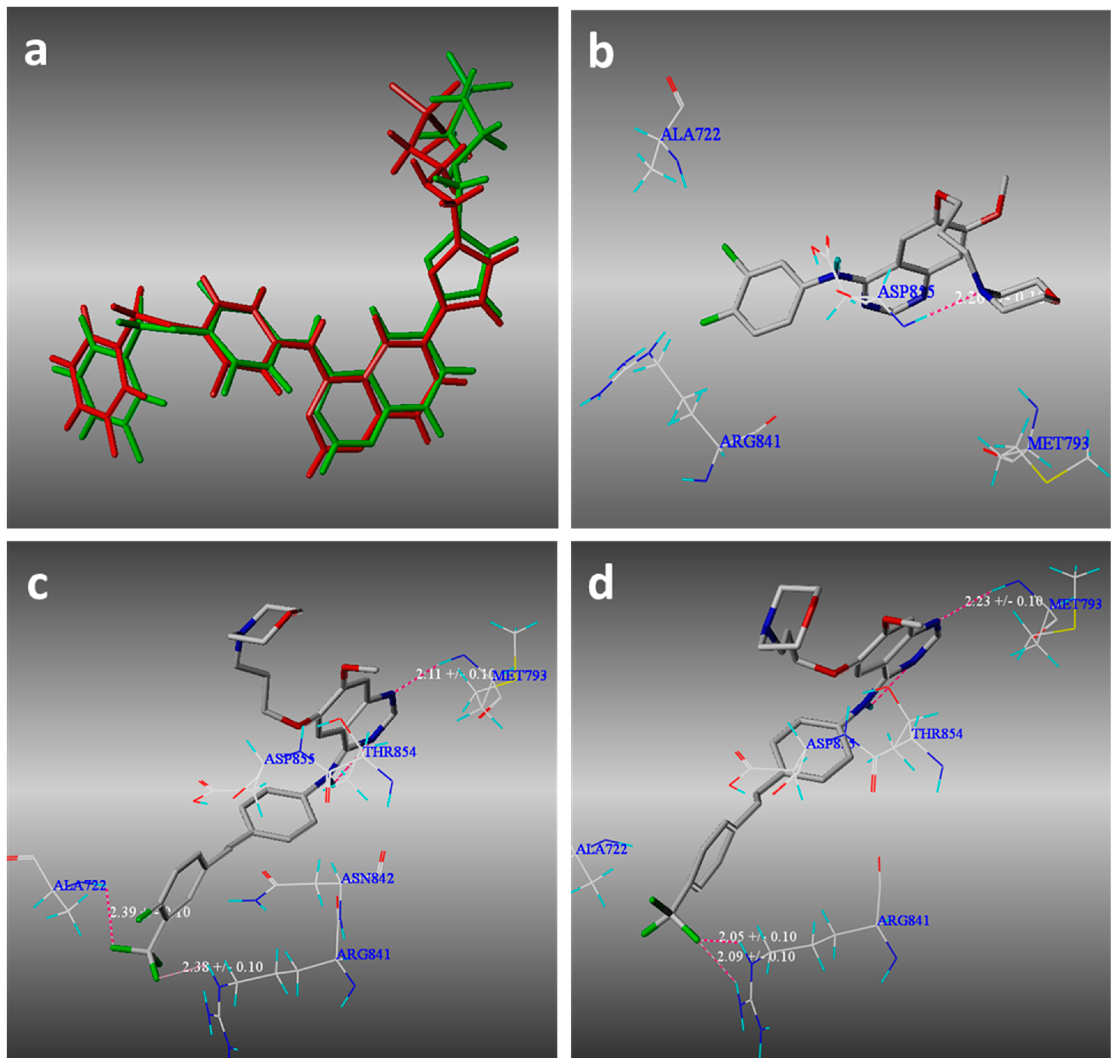
2.3. Molecular Modeling
3. Experiment Section
3.1. General
3.2. Biological Evaluation of Anti-Tumour of the Synthesized Compounds
3.3. Molecular Modeling Methods
4. Conclusions
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Wiley, H.S. Trafficking of the ErbB receptors and its influence on signaling. Exp. Cell Res. 2003, 284, 78–88. [Google Scholar] [CrossRef]
- Herbst, R.S. Review of epidermal growth factor receptor biology. Int. J. Radiat. Oncol. Biol. Phys. 2004, 59, 21–26. [Google Scholar] [CrossRef] [PubMed]
- Pinkaskramarski, R.; Soussan, L.; Waterman, H.; Levkowitz, G.; Alroy, I.; Klapper, L.; Lavi, S.; Seger, R.; Ratzkin, B.J.; Sela, M.; et al. Diversification of Neu differentiation factor and epidermal growth factor signaling by combinatorial receptor interactions. EMBO J. 1996, 15, 2452–2467. [Google Scholar]
- Zhang, H.T.; Berezov, A.; Wang, Q.; Zhang, G.; Drebin, J.; Murali, R.; Greene, M.I. ErbB receptors: From oncogenes to targeted cancer treatment. J. Clin. Investig. 2007, 117, 2051–8205. [Google Scholar] [CrossRef] [PubMed]
- Hynes, N.E.; Lane, H.A. ERBB receptors and cancer: The complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5, 341–354. [Google Scholar] [CrossRef] [PubMed]
- Yarden, Y.; Sliwkowski, M.X. Untangling the ErbB signalling network. Nat. Rev. Mol. Cell Biol. 2001, 2, 127–137. [Google Scholar] [CrossRef] [PubMed]
- Ebner, R.; Derynck, R. Epidermal growth factor and transforming growth factor-alpha: Differential intracellular routing and processing of ligand-receptor complexes. Cell Regul. 1991, 2, 599–612. [Google Scholar] [CrossRef] [PubMed]
- Longva, K.E.; Blystad, F.D.; Stang, E.; Larsen, A.M.; Johannessen, L.E.; Madshus, I.H. Ubiquitination and proteasomal activity is required for transport of the EGF receptor to inner membranes of multivesicular bodies. J. Cell Biol. 2002, 156, 843–854. [Google Scholar] [CrossRef] [PubMed]
- Downward, J.; Parker, P.; Waterfield, M.D. Autophosphorylation sites on the epidermal growth factor receptor. Nature 1984, 311, 483–485. [Google Scholar] [CrossRef] [PubMed]
- Oda, K.; Matsuoka, Y.; Funahashi, A.; Kitano, H. A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol. 2005, 1. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.M.; Yeung, T.K.; Wang, Z.X. Enhanced Drug Resistance in Cells Coexpressing ErbB2 with EGF Receptor or ErbB3. Biochem. Biophys. Res. Commun. 2000, 277, 757–763. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, S.S.; Malerczyk, C.; Aigner, A.; Czubayko, F.; Hsieh, S.S.; Malerczyk, C.; Aigner, A. Czubayko FERbB-2 expression is rate-limiting for epidermal growth factor-mediated stimulation of ovarian cancer cell proliferation. Int. J. Cancer 2000, 86, 644–651. [Google Scholar] [CrossRef]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W.; Harris, P.L.; Haserlat, S.M.; Supko, J.G.; Haluska, F.G.; et al. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar] [CrossRef] [PubMed]
- Kuan, C.T.; Wikstrand, C.J.; Bigner, D.D. EGF mutant receptor vIII as a molecular target in cancer therapy. Endocr.-Relat. Cancer 2001, 8, 83–96. [Google Scholar] [CrossRef] [PubMed]
- Walker, F.; Abramowitz, L.; Benabderrahmane, D.; Duval, X.; Descatoire, V.; Hénin, D.; Lehy, T.; Aparicio, T. Growth factor receptor expression in anal squamous lesions: Modifications associated with oncogenic human papillomavirus and human immunodeficiency virus. Hum. Pathol. 2009, 40, 1517–1527. [Google Scholar] [CrossRef] [PubMed]
- Ranson, M.; Hammond, L.A.; Ferry, D.; Kris, M.; Tullo, A.; Murray, P.I.; Miller, V.; Averbuch, S.; Ochs, J.; Morris, C.; et al. ZD1839, a selective oral epidermal growth factor receptor-tyrosine kinase inhibitor, is well tolerated and active in patients with solid, malignant tumors: Results of a phase I trial. J. Clin. Oncol. 2002, 20, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Cohen, M.H.; Williams, G.A.; Sridhara, R.; Chen, G.; Pazdur, R. FDA Drug Approval Summary: Gefitinib (ZD1839) (Iressa®) Tablets. Oncologist 2003, 8, 303–306. [Google Scholar] [CrossRef] [PubMed]
- Ciardiello, F.; Tortora, G. A novel approach in the treatment of cancer: Targeting the epidermal growth factor receptor. Clin. Cancer Res. 2001, 7, 2958–2970. [Google Scholar] [PubMed]
- Burris, H.A. Dual kinase inhibition in the treatment of breast cancer: Initial experience with the EGFR/ErbB-2 inhibitor lapatinib. Oncologist 2004, 9, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Higa, G.M.; Abraham, J. Lapatinib in the treatment of breast cancer. Expert Rev. Anticancer 2007, 7, 1183–1192. [Google Scholar] [CrossRef] [PubMed]
- Sun, B.; Hoshino, J.; Jermihov, K.; Marler, L.; Pezzuto, J.M.; Mesecar, A.D.; Cushman, M. Design, synthesis, and biological evaluation of resveratrol analogues as aromatase and quinone reductase 2 inhibitors for chemoprevention of cancer. Bioorg. Med. Chem. 2010, 18, 5352–5366. [Google Scholar] [CrossRef] [PubMed]
- Baur, J.A.; Sinclair, D.A. Therapeutic potential of resveratrol: The in vivo evidence. Nat. Rev. Drug Discov. 2006, 5, 493–506. [Google Scholar] [CrossRef] [PubMed]
- Jang, M.; Cai, L.; Udeani, G.O.; Slowing, K.V.; Thomas, C.F.; Beecher, C.W.W.; Fong, H.H.S.; Farnsworth, N.R.; Kinghorn, A.D.; Mehta, R.G.; et al. Cancer chemopreventiveactivity of resveratrol, a natural product derived from grapes. Science 1997, 275, 218–220. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Go, M.L. Quinone reductase induction activity of methoxylated analogues of resveratrol. Eur. J. Med. Chem. 2007, 42, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Sviripa, V.; Kril, L.M.; Chen, X.; Yu, T.; Shi, J.; Rychahou, P.; Evers, B.M.; Watt, D.S.; Liu, C. Fluorinated N,N-dialkylaminostilbenes for Wnt pathway inhibition and colon cancer repression. J. Med. Chem. 2011, 54, 1288–1297. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Dong, A.; Gao, C.; Tan, C.; Liu, H.; Zu, X.; Jiang, Y. The design, synthesis, and anti-tumor mechanism study of N -phosphoryl amino acid modified resveratrol analogues. Bioorg. Med. Chem. 2008, 16, 10013–10021. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.C.; Li, J.; Zhang, X.Q.; Gu, H.M.; Li, B.L. Microwave-assisted solvent-free synthesis of (E)-stilbenes. J. Chem. Res. 2012, 36, 231–234. [Google Scholar] [CrossRef]
- Ishino, Y.; Zhu, C.; Harris, D.L.; Joyce, A.N.C. Protein tyrosine phosphatase-1B (PTP1B) helps regulate EGF-induced stimulation of S-phase entry in human corneal endothelial cells. Mol. Vis. 2008, 14, 61–70. [Google Scholar] [PubMed]
Sample Availability: Samples of the compounds 6a–6j are available from the authors. |




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compounds | A431 | A549 | Hela | HL-60 | SMMC-7721 | BGC823 | SK-OV-3 | HepG2 |
|---|---|---|---|---|---|---|---|---|
| 6a | 1.55 ± 0.10 | 1.40 ± 0.13 | 3.30 ± 0.06 | 2.281 ± 0.29 | 3.91 ± 0.66 | - | 3.34 ± 0.37 | 5.175 ± 0.95 |
| 6b | 1.81 ± 1.15 | 2.61 ± 0.89 | 2.89 ± 0.38 | not fit | 2.46 ± 0.19 | - | 2.54 ± 0.14 | 4.579 ± 0.83 |
| 6c | 1.23 ± 0.36 | 1.44 ± 0.52 | 1.68 ± 0.32 | 2.675 ± 0.20 | 1.73 ± 0.04 | - | 1.68 ± 0.07 | 1.829 ± 0.28 |
| 6d | 2.35 ± 0.85 | 2.17 ± 0.29 | 2.64 ± 0.93 | 2.62 ± 0.08 | 3.56 ± 1.01 | 1.32 ± 0.29 | 2.65 ± 0.87 | 3.04 ± 0.17 |
| 6e | 2.50 ± 0.25 | 2.19 ± 0.15 | 2.63 ± 0.67 | 2.99 ± 0.55 | 3.11 ± 0.60 | 1.82 ± 0.52 | 3.11 ± 0.58 | 3.15 ± 0.29 |
| 6f | 1.74 ± 0.20 | 2.37 ± 0.29 | 1.96 ± 0.12 | 2.63 ± 0.06 | 2.90 ± 0.73 | 1.59 ± 0.74 | 2.71 ± 0.52 | 3.16 ± 0.42 |
| 6g | 1.96 ± 0.98 | 3.18 ± 1.03 | 3.05 ± 0.10 | 2.59 ± 0.02 | 4.21 ± 0.63 | 1.38 ± 0.16 | 3.70 ± 0.79 | 3.94 ± 1.56 |
| 6h | 2.02 ± 0.80 | 2.22 ± 0.82 | 1.74 ± 0.44 | 2.49 ± 0.19 | 2.34 ± 0.67 | 1.27 ± 0.16 | 2.77 ± 0.68 | 2.79 ± 0.19 |
| 6i | 1.27 ± 0.95 | 1.67 ± 0.38 | 3.77 ± 0.63 | 3.11 ± 0.01 | 5.37 ± 0.02 | 1.66 ± 0.38 | 5.26 ± 1.30 | 3.98 ± 0.88 |
| 6j | 2.71 ± 0.17 | 3.82 ± 0.80 | - | 1.73 ± 0.47 | 3.36 ± 0.93 | 2.13 ± 0.85 | 2.18 ± 0.97 | 2.68 ± 0.59 |
| Gefitinib b | 12.93 ± 4.54 | 13.75 ± 5.73 | 17.92 ± 1.50 | 17.72 ± 1.76 | 23.27 ± 0.66 | >10 | 12.31 ± 0.33 | >10 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, L.; Li, P.; Li, B.; Wang, Y.; Li, J.; Song, L. Design, Synthesis, and Antitumor Activity of Novel Quinazoline Derivatives. Molecules 2017, 22, 1624. https://doi.org/10.3390/molecules22101624
Wang L, Li P, Li B, Wang Y, Li J, Song L. Design, Synthesis, and Antitumor Activity of Novel Quinazoline Derivatives. Molecules. 2017; 22(10):1624. https://doi.org/10.3390/molecules22101624
Chicago/Turabian StyleWang, Liuchang, Pengna Li, Baolin Li, Yawen Wang, Jiangtao Li, and Limei Song. 2017. "Design, Synthesis, and Antitumor Activity of Novel Quinazoline Derivatives" Molecules 22, no. 10: 1624. https://doi.org/10.3390/molecules22101624
APA StyleWang, L., Li, P., Li, B., Wang, Y., Li, J., & Song, L. (2017). Design, Synthesis, and Antitumor Activity of Novel Quinazoline Derivatives. Molecules, 22(10), 1624. https://doi.org/10.3390/molecules22101624