Effect of Structure on Charge Distribution in the Isatin Anions in Aprotic Environment: Spectral Study

Abstract
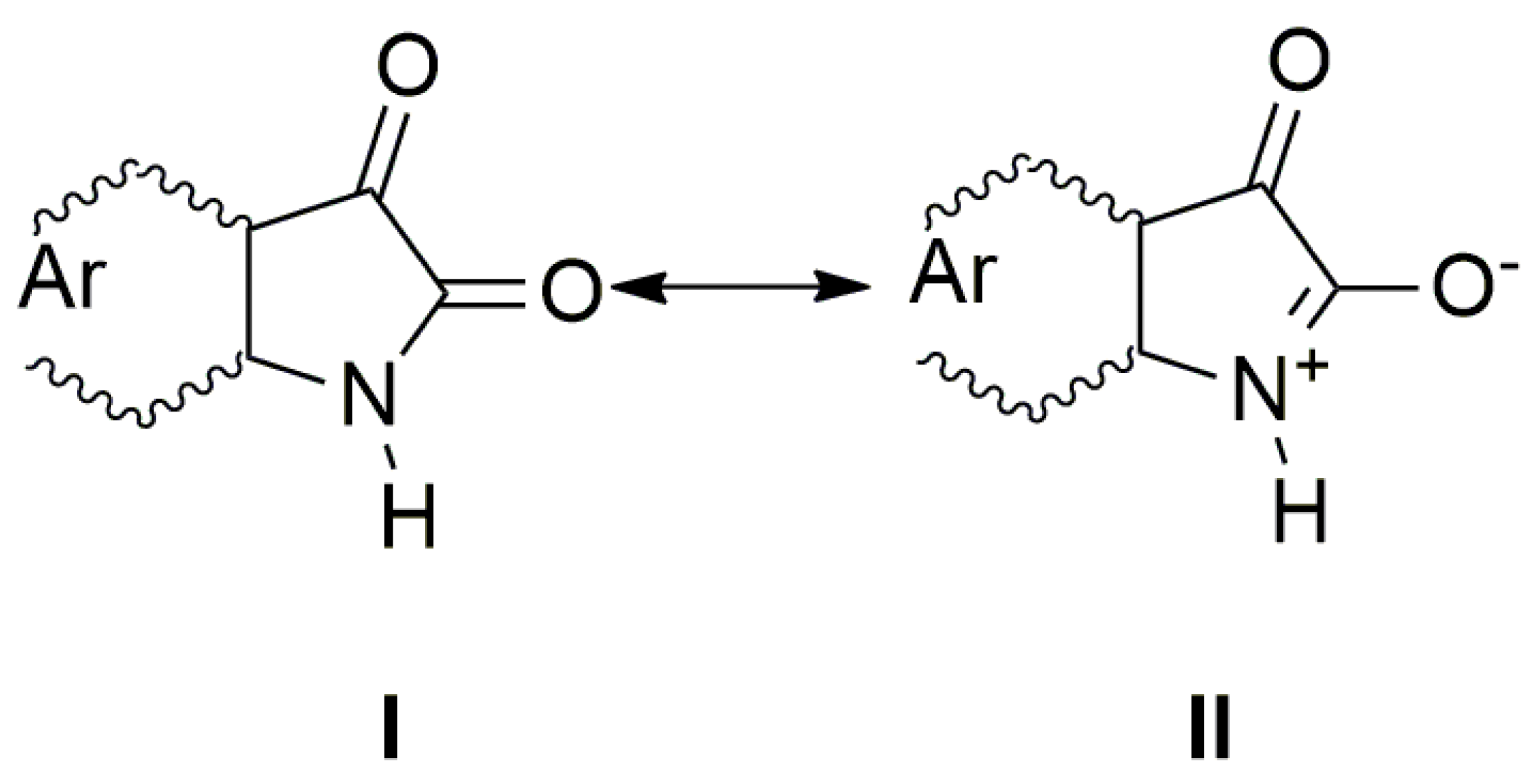
:1. Introduction
2. Results and Discussion
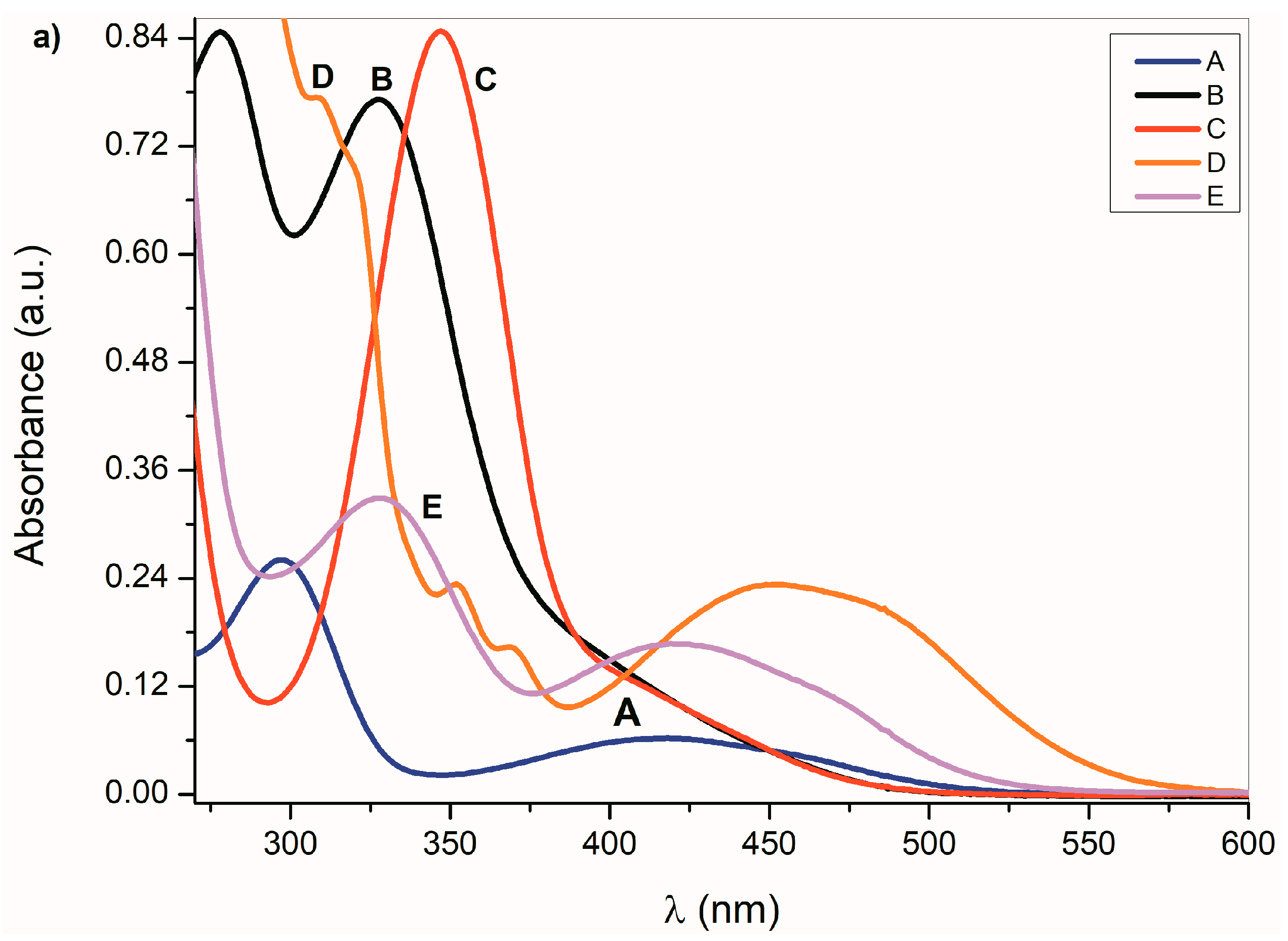
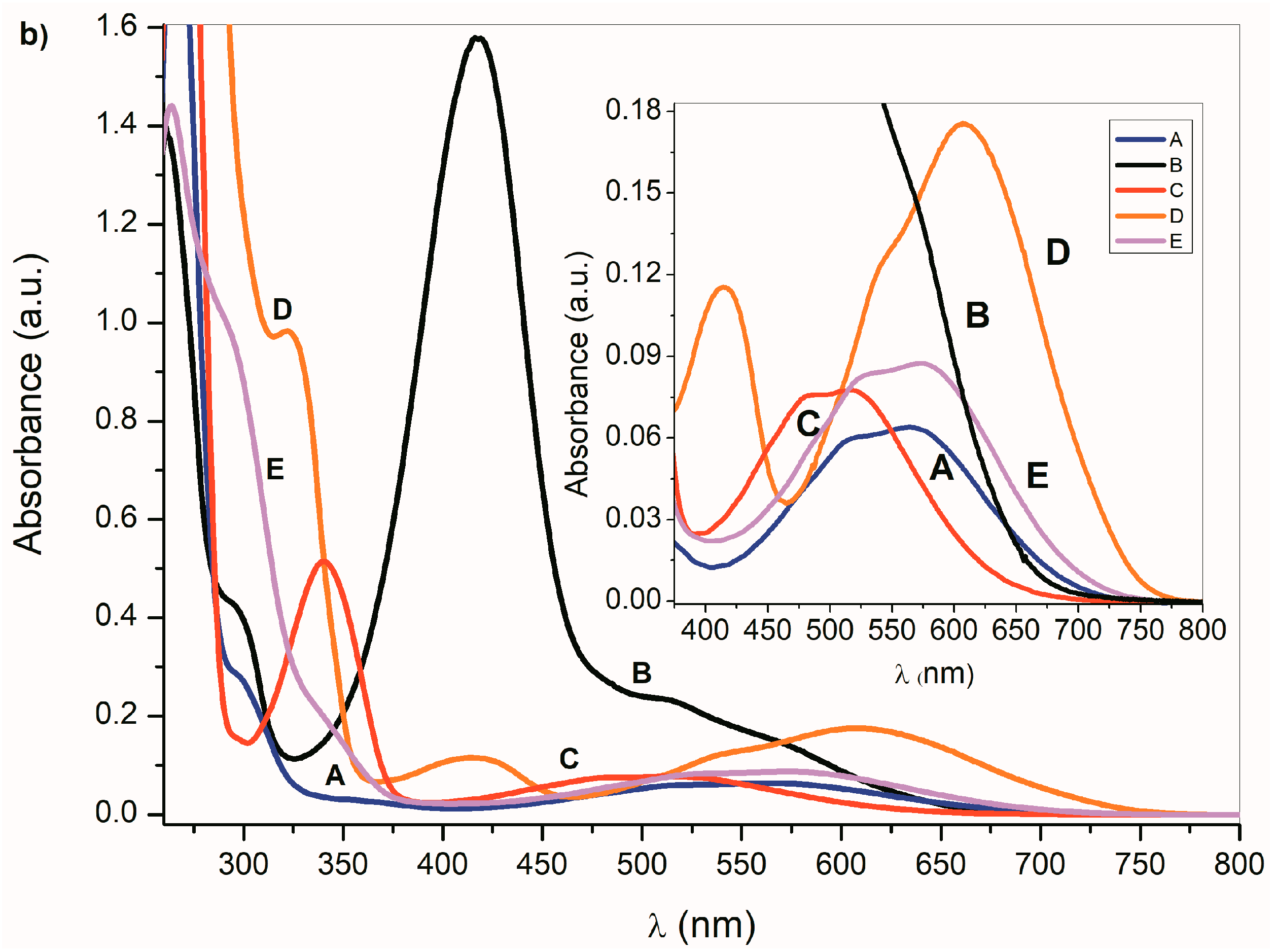
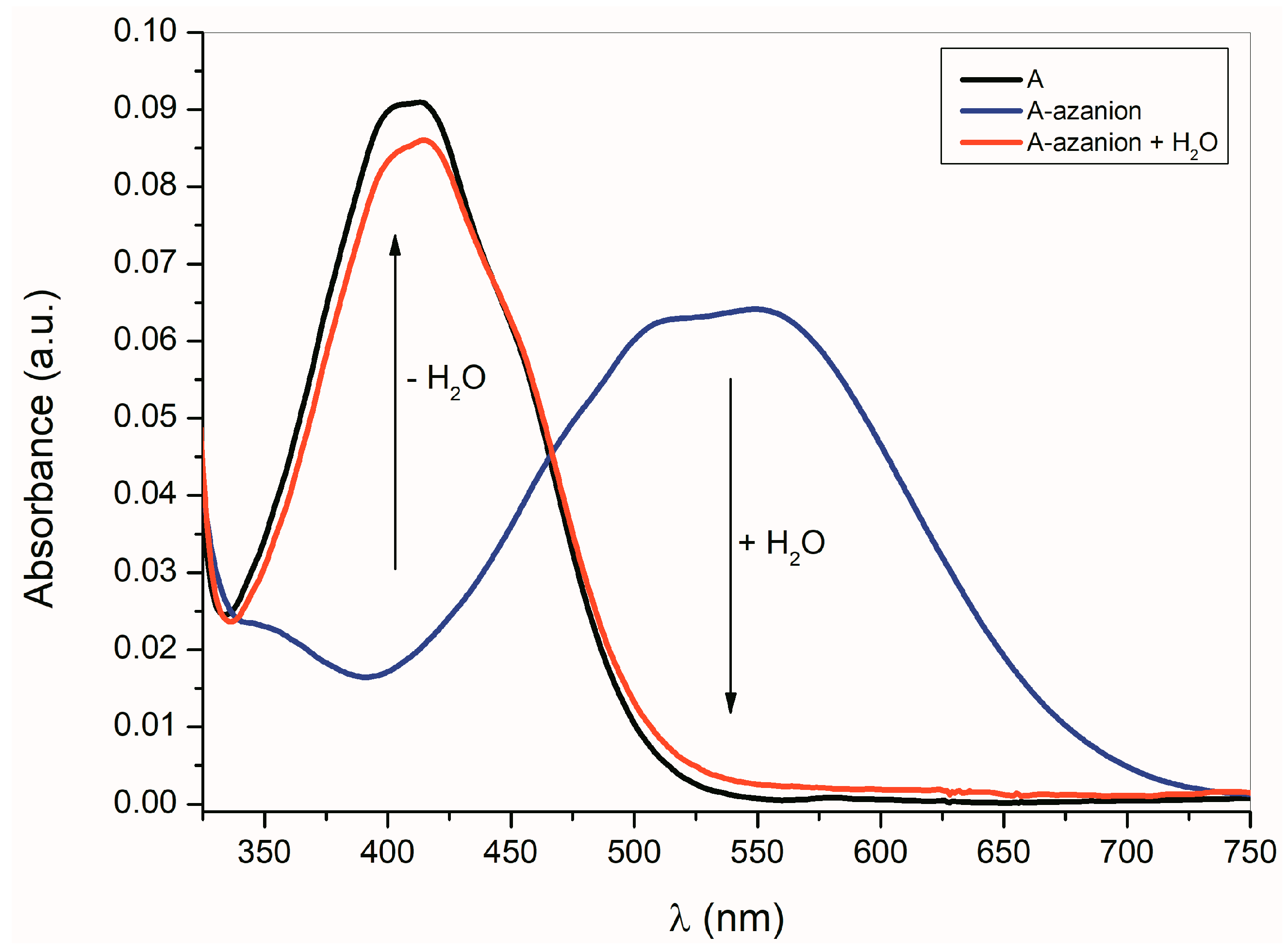
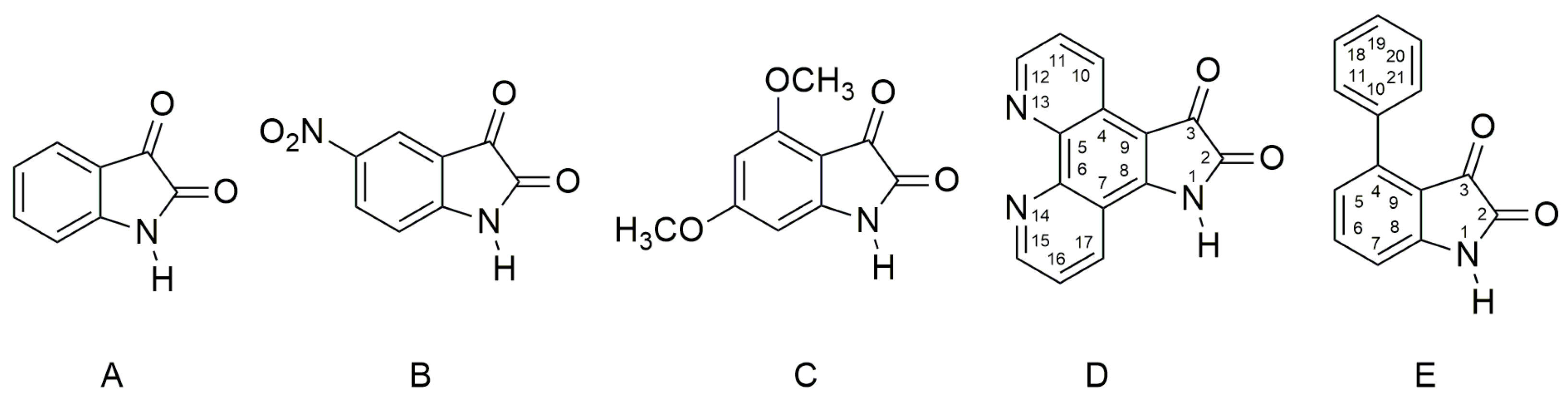
2.1. Ultraviolet–Visible Spectra of the Compounds A–E and Their Azanions
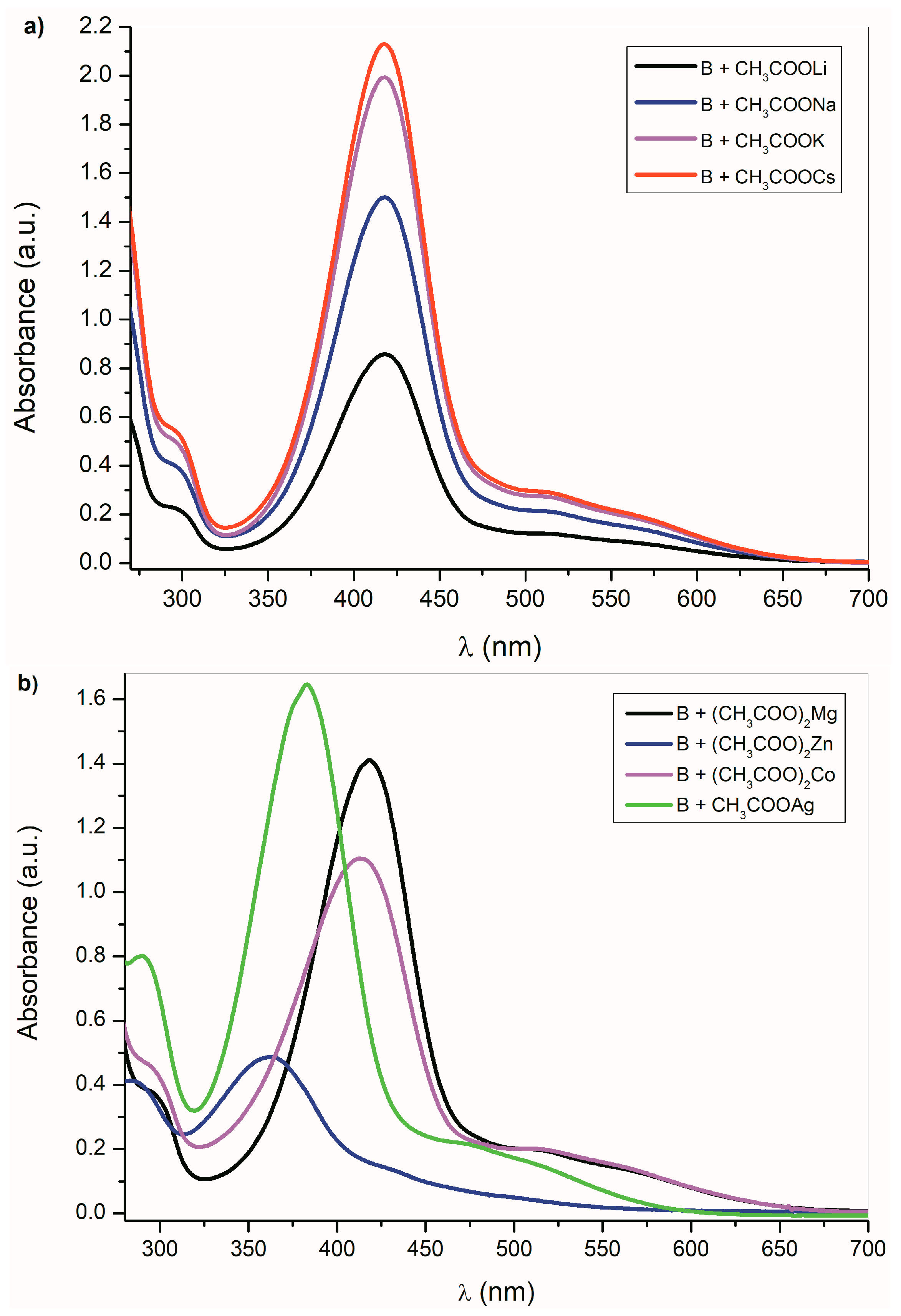
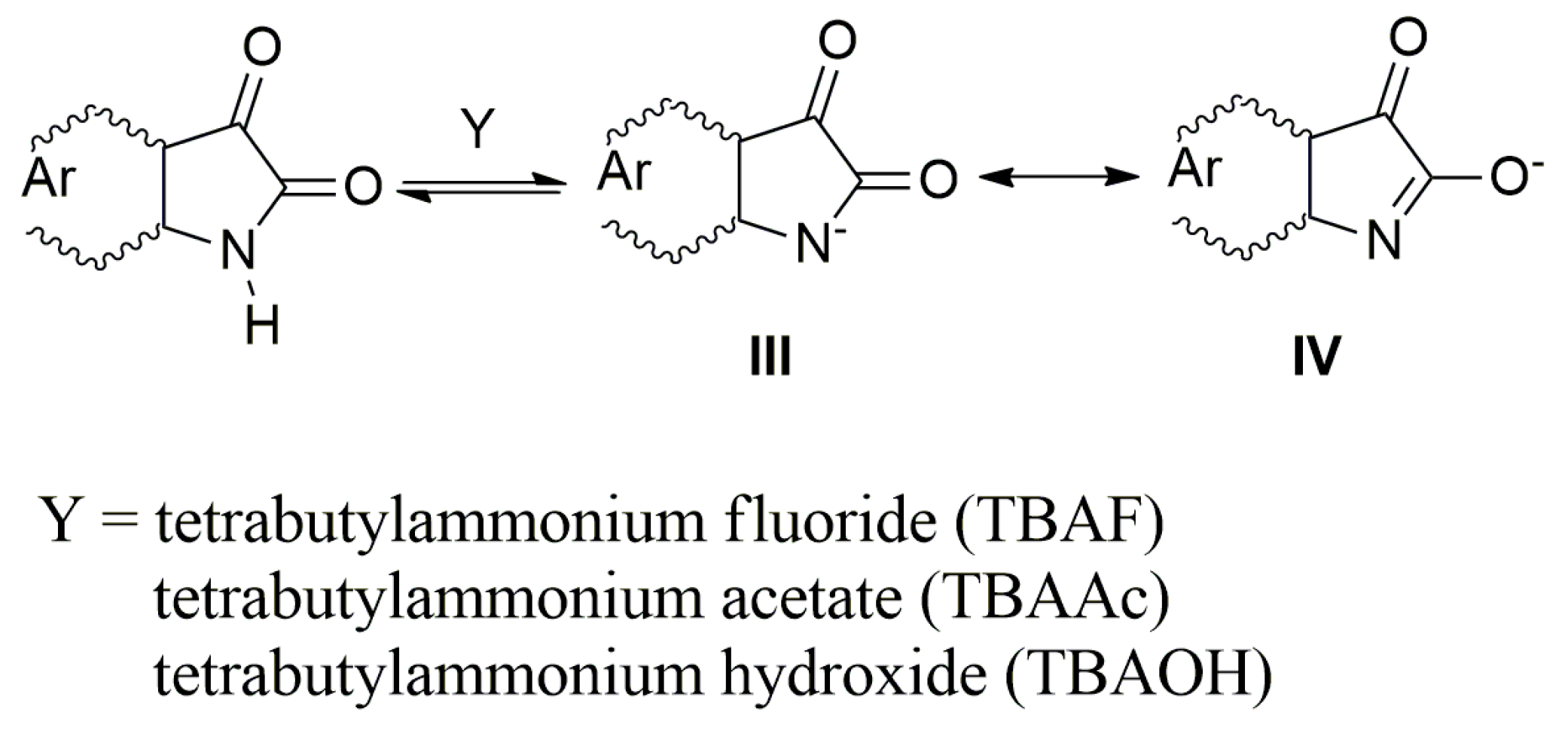
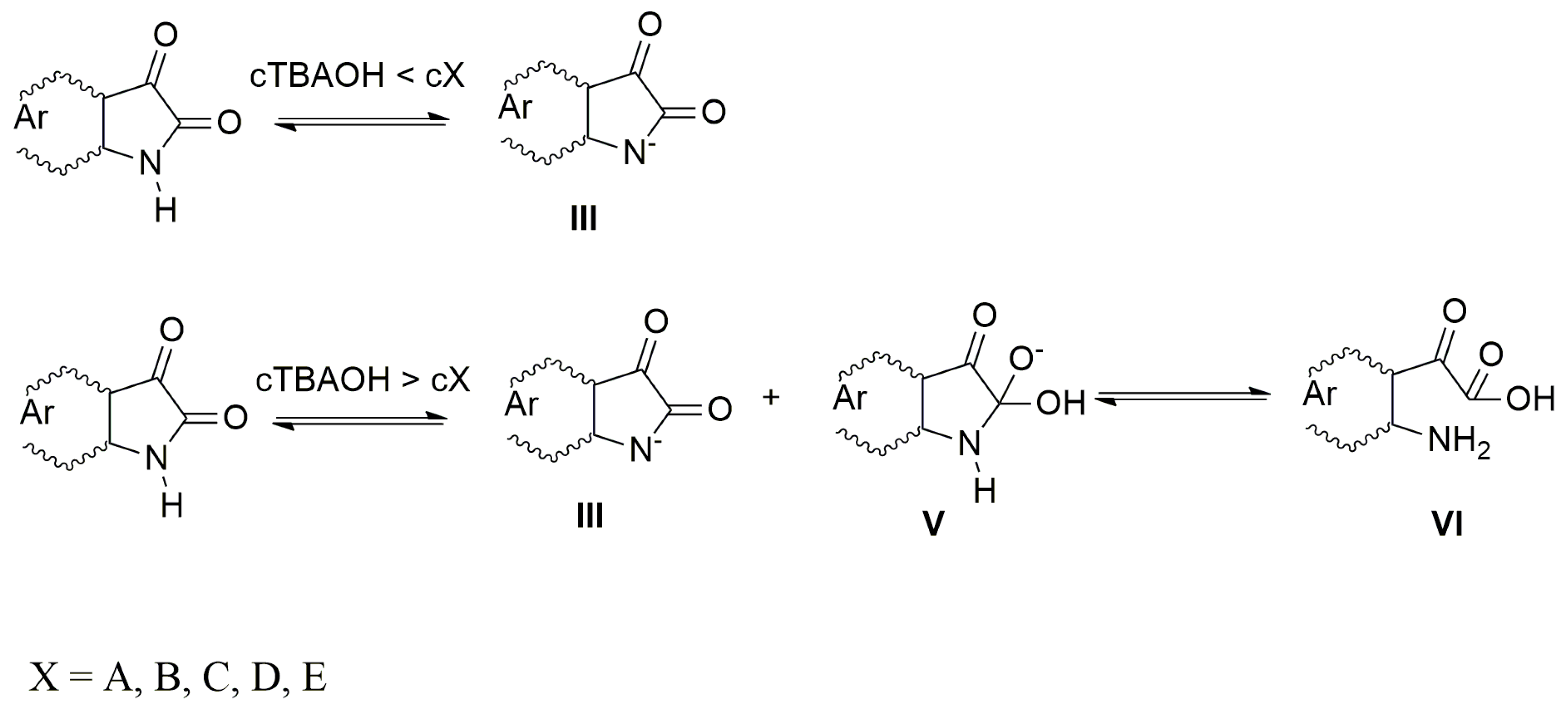
2.2. The Influence of the Acid Anions (F−, CH3COO−, Cl−, Br−, NO3−, HSO4−) and Counter Ions on the Ultraviolet–Visible Spectra of A–E and Their Azanions
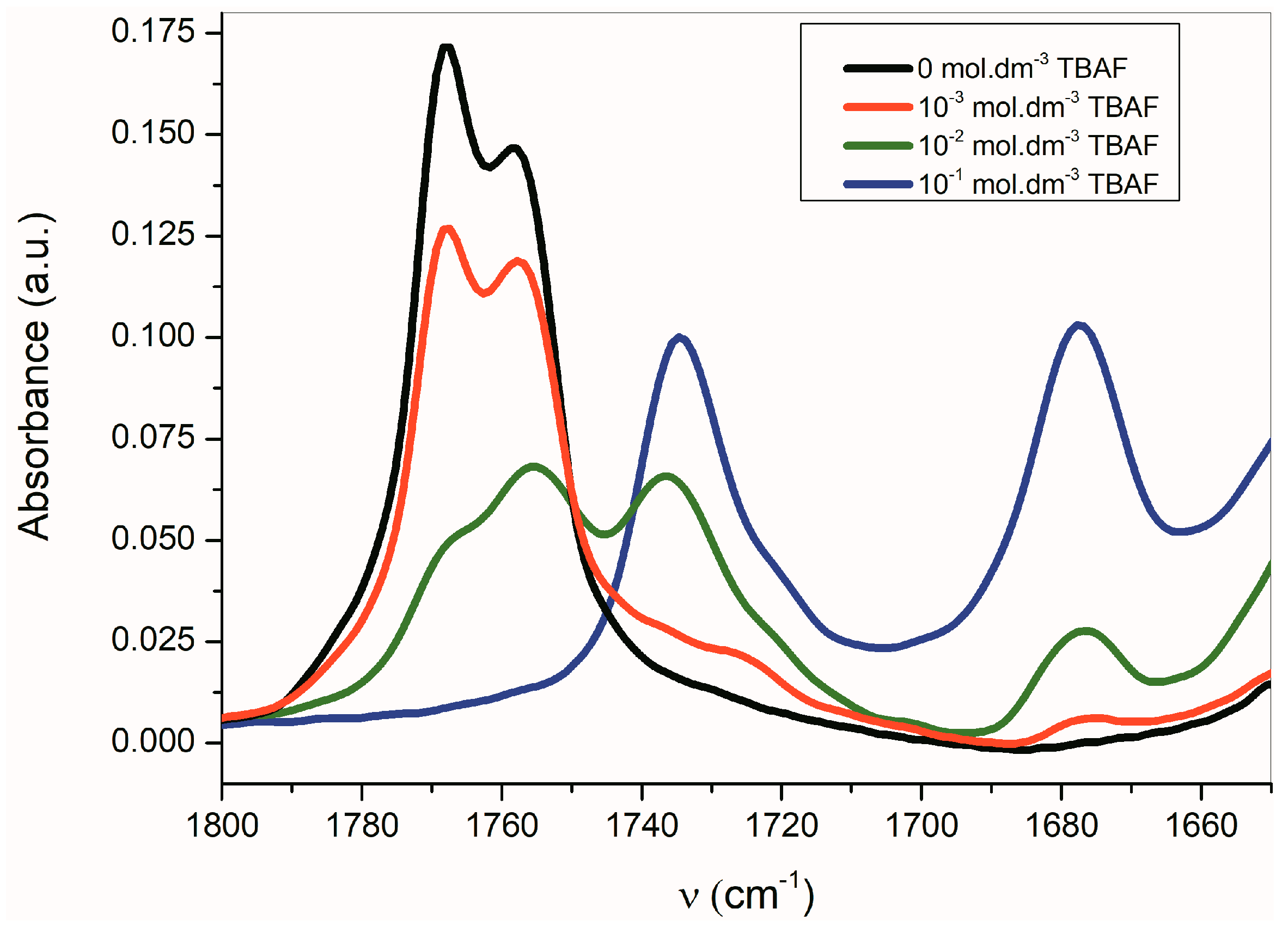
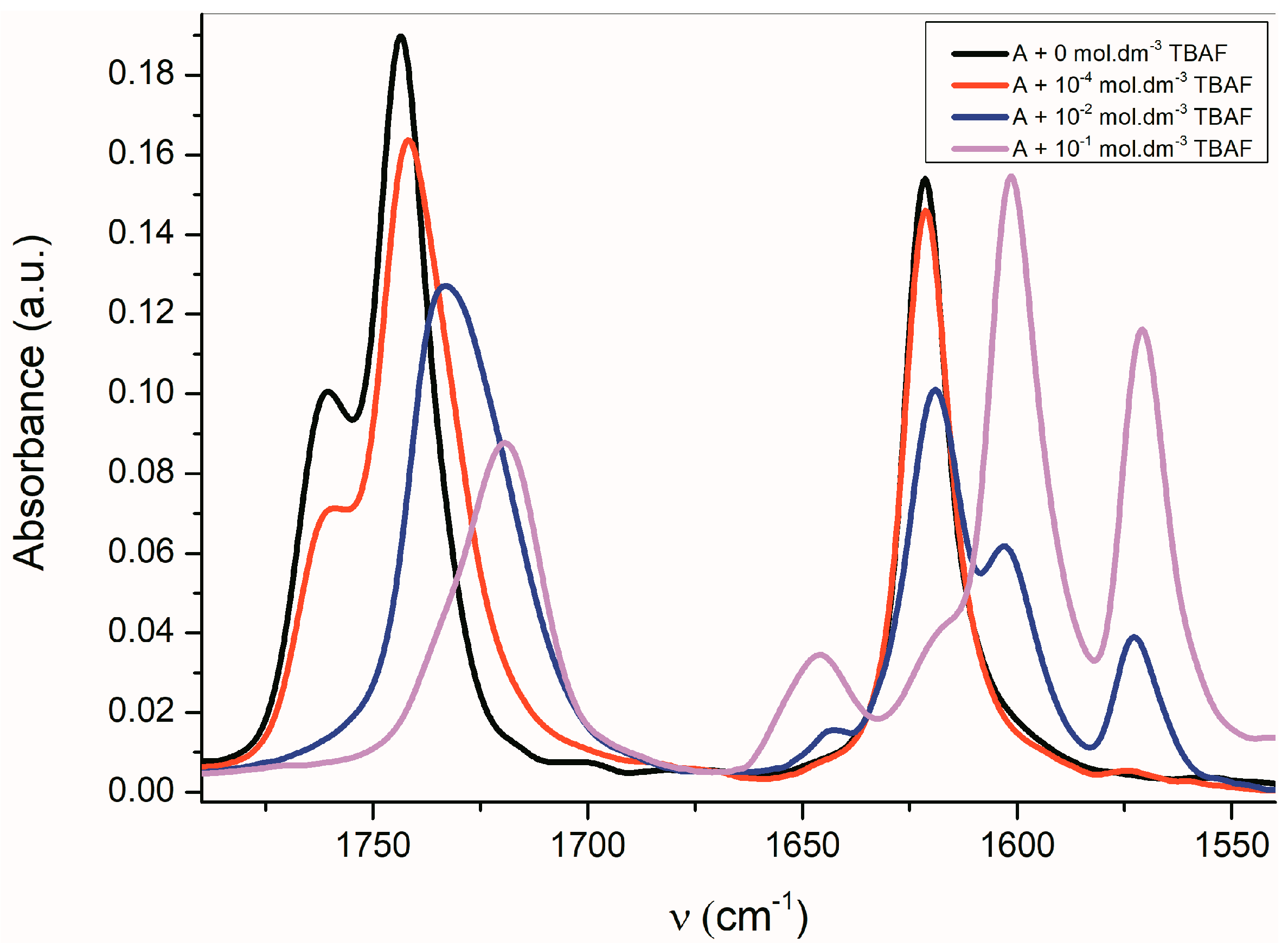
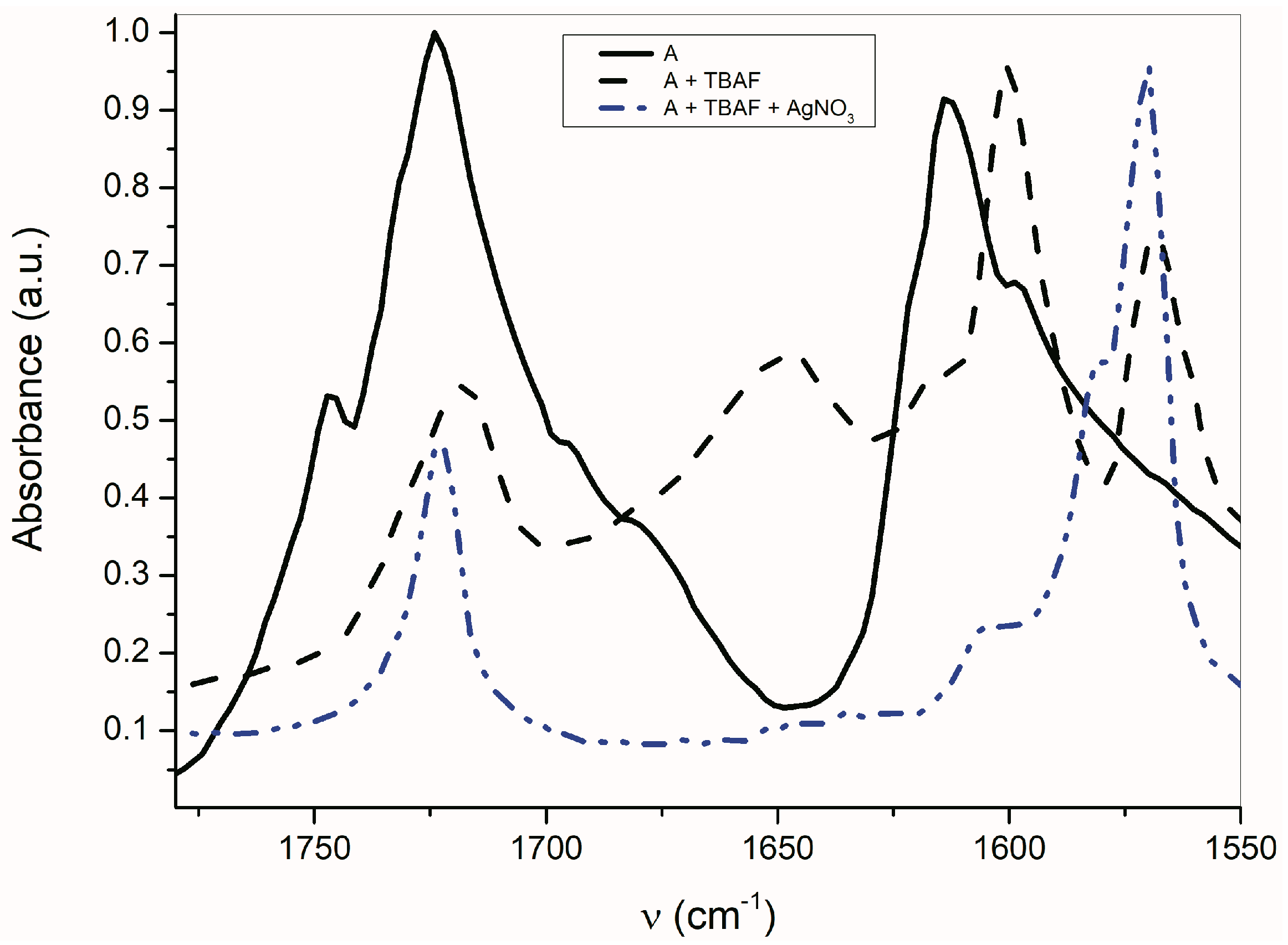
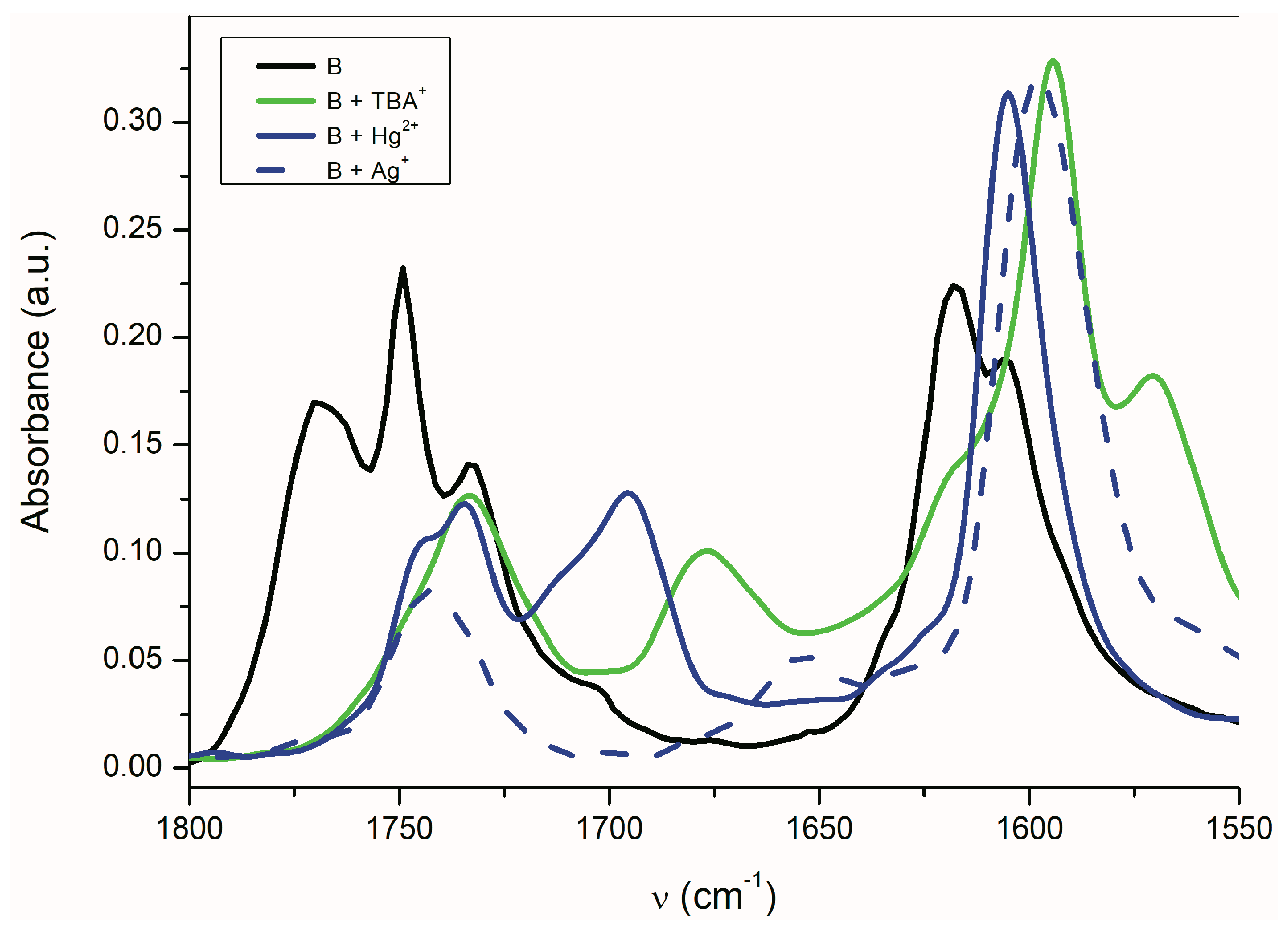
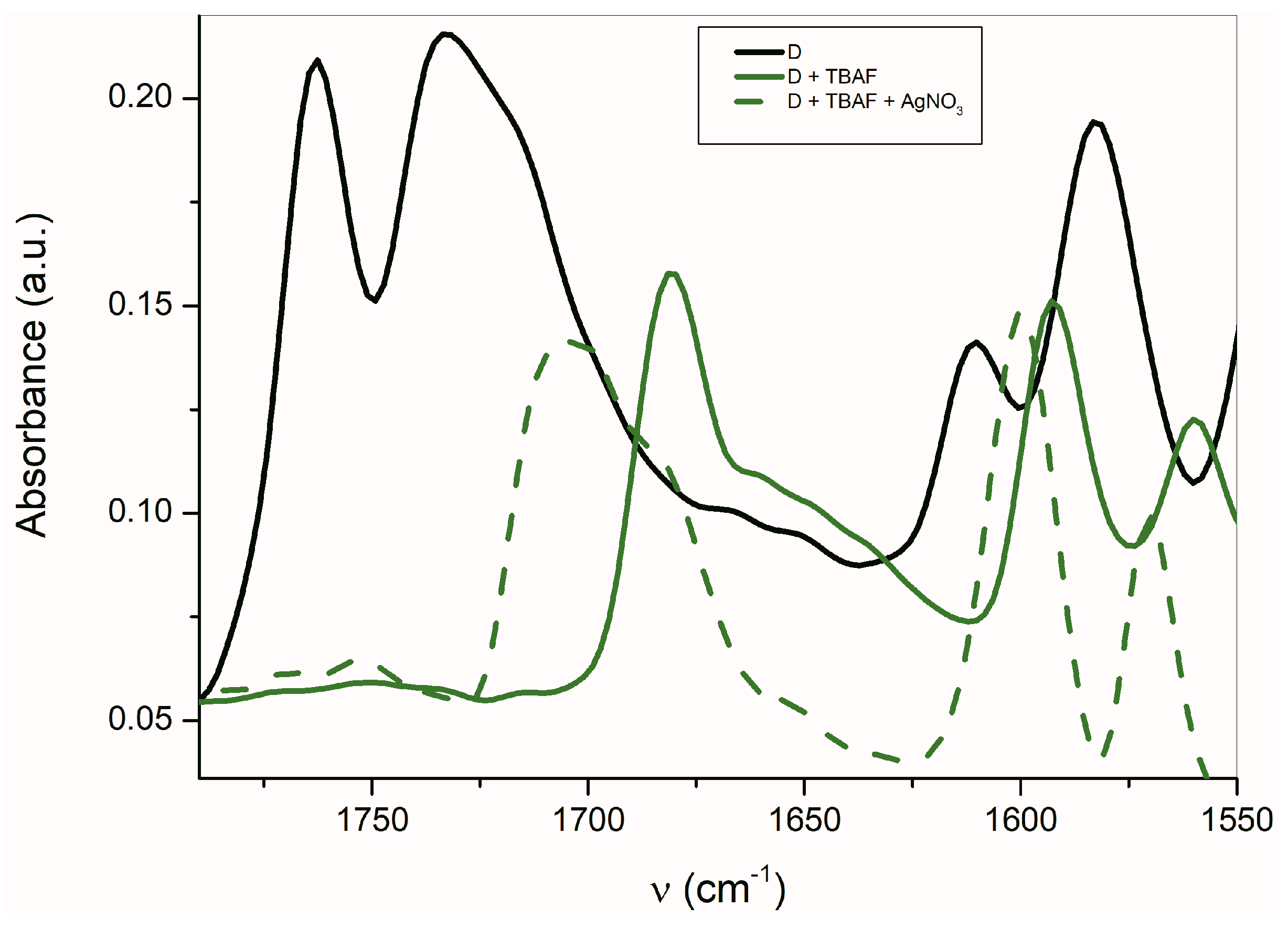
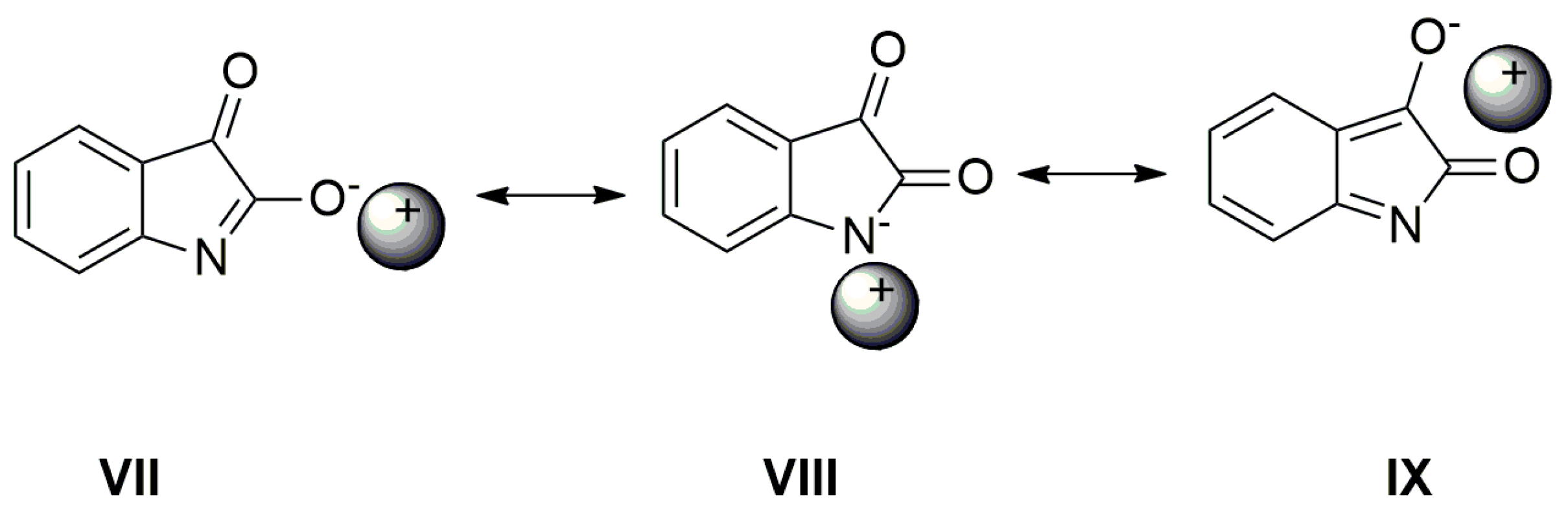
2.3. The Azanions Fourier Transform Infrared Study
2.4. 1H and 13C Nuclear Magnetic Resonance Study
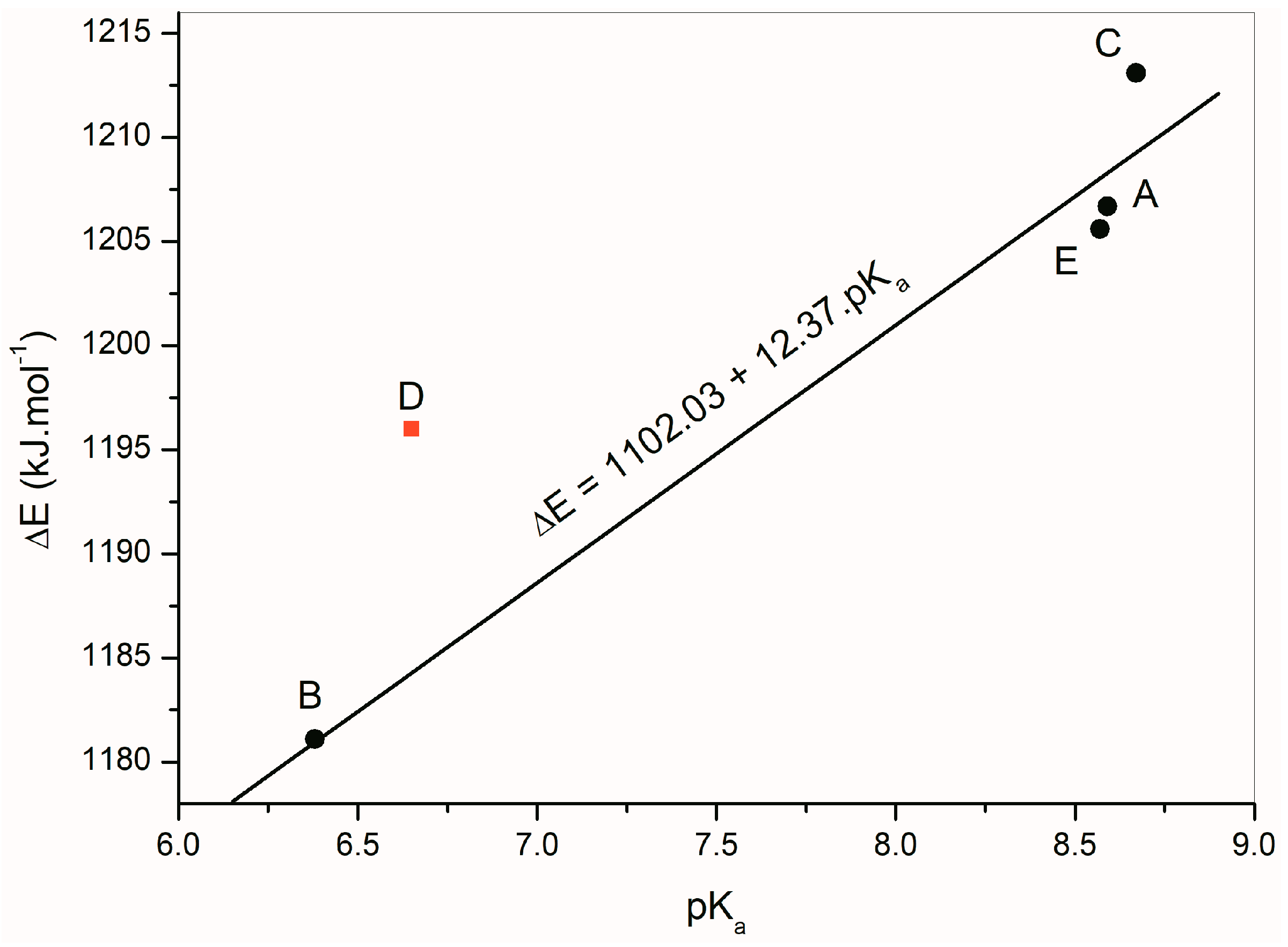
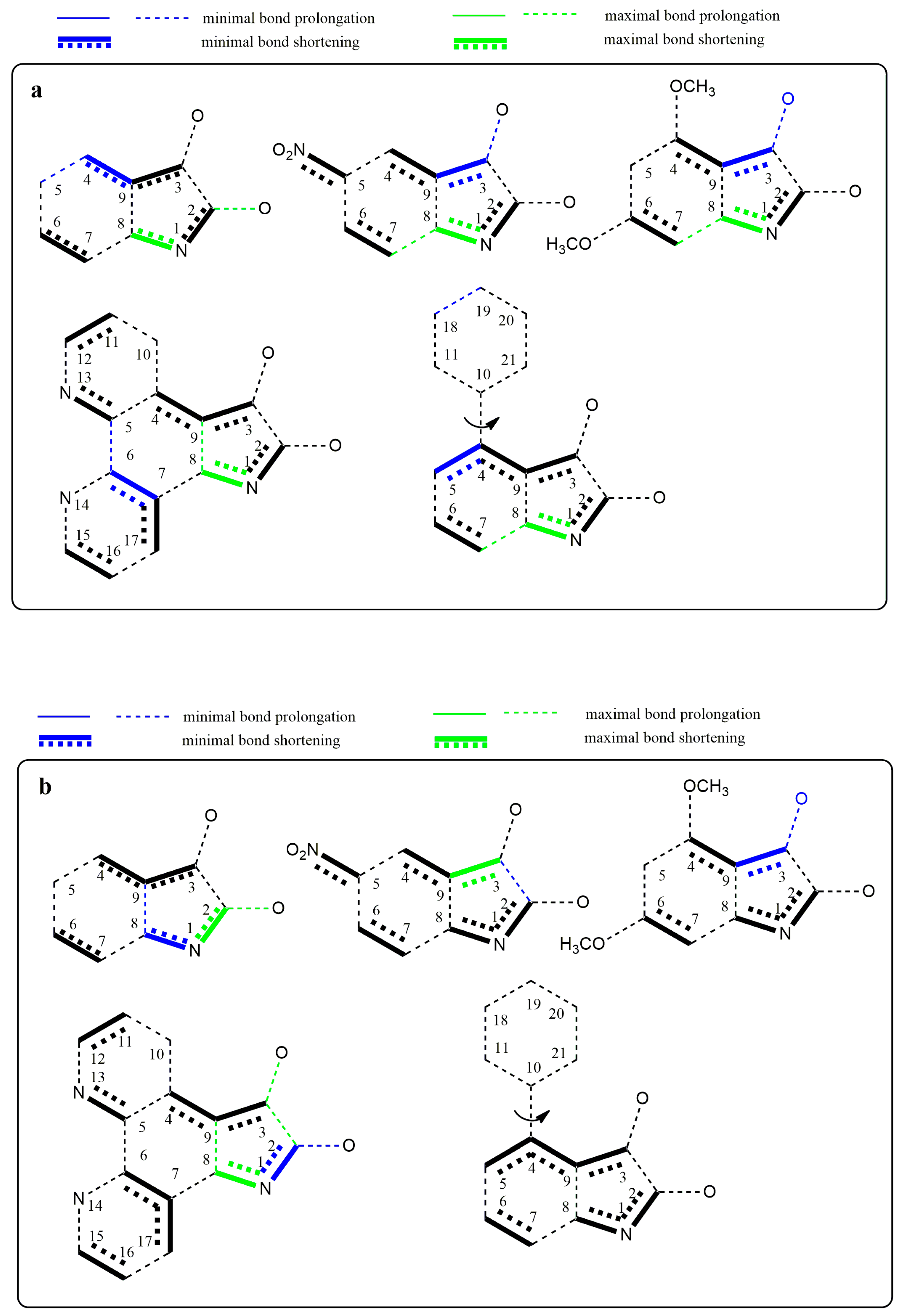
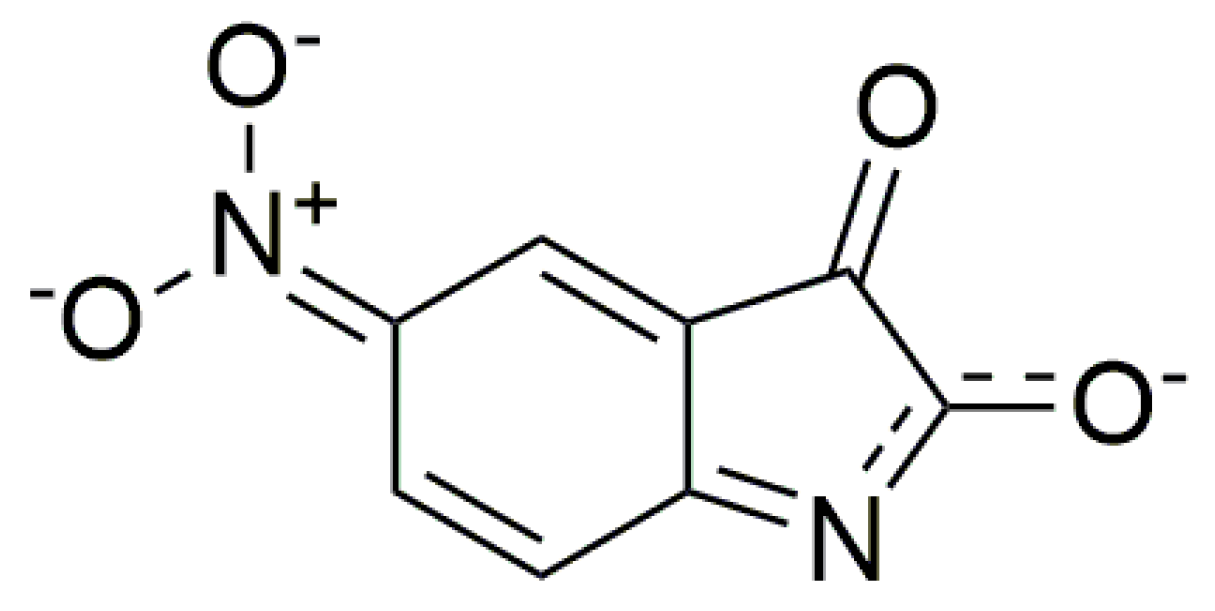
2.5. Quantum-Chemical Calculation
3. Materials and Methods
3.1. General Information
3.2. Synthesis
3.2.1. Synthesis of Compound D
3.2.2. General Preparation of Isatin Azanions
3.3. Quantum-Chemical Calculations
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Raj, V. Review on CNS activity of isatin derivatives. Int. J. Curr. Pharm. Res. 2012, 4, 1–9. [Google Scholar]
- Borad, M.A.; Bhoi, M.N.; Prajapati, N.P.; Patel, H.D. Review of Synthesis of Spiro Heterocyclic Compounds from Isatin. Synth. Commun. 2014, 44, 897–922. [Google Scholar] [CrossRef]
- Kumari, G.; Nutan; Modi, M.; Gupta, S.K.; Singh, R.K. Rhodium(II) acetate-catalyzed stereoselective synthesis, SAR and anti-HIV activity of novel oxindoles bearing cyclopropane ring. Eur. J. Med. Chem. 2011, 46, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Vintonyak, V.V.; Warburg, K.; Kruse, H.; Grimme, S.; Hubel, K.; Rauth, D.; Waldmann, H. Identification of Thiazolidinones Spiro-Fused to Indolin-2-ones as Potent and Selective Inhibitors of the Mycobacterium tuberculosis Protein Tyrosine Phosphatase B. Angew. Chem. Int. Ed. 2010, 49, 5902–5905. [Google Scholar] [CrossRef] [PubMed]
- Yeung, B.K.S.; Zou, B.; Rottmann, M.; Lakshminarayana, S.B.; Ang, S.H.; Leong, S.Y.; Tan, J.; Wong, J.; Keller-Maerki, S.; Fischli, C.; et al. Spirotetrahydro β-Carbolines (Spiroindolones): A new class of potent and orally efficacious compounds for the treatment of malaria. J. Med. Chem. 2010, 53, 5155–5164. [Google Scholar] [CrossRef] [PubMed]
- Rottmann, M.; McNamara, C.; Yeung, B.K.S.; Lee, M.C.S.; Zhou, B.; Russell, B.; Seitz, P.; Plouffe, D.M.; Dharia, N.V.; Tan, J.; et al. Spiroindolones, a potent compound class for the treatment of malaria. Science 2010, 329, 1175–1180. [Google Scholar] [CrossRef] [PubMed]
- Glover, V.; Bhattacharya, S.K.; Sandler, M. Isatin—A new biological factor. Indian J. Exp. Biol. 1991, 29, 1–5. [Google Scholar] [PubMed]
- Pakravan, P.; Kashanian, S.; Khodaei, M.M.; Harding, F.J. Biochemical and pharmacological characterization of isatin and its derivatives: From structure to activity. Pharmacol. Rep. 2013, 65, 313–335. [Google Scholar] [CrossRef]
- Lashgari, N.; Ziarani, G.M. Synthesis of heterocyclic compounds based on isatin through 1,3-dipolar cycloaddition reactions. ARKIVOC 2012, 1, 277–320. [Google Scholar] [CrossRef]
- El-Faham, A.; Elzatahry, A.A.; Al-Othman, Z.A.; Elsayed, E.A. Facile method for the synthesis of silver nanoparticles using 3-hydrazino-isatin derivatives in aqueous methanol and their antibacterial activity. Int. J. Nanomed. 2014, 9, 1167–1174. [Google Scholar] [CrossRef] [PubMed]
- Jayapriya, E.; Lalitha, P.; Firdhouse, M.J. Isatin-Mediated synthesis and characterization of silver nanoparticles. Int. J. Pharm. Chem. Res. 2016, 2, 46–54. [Google Scholar] [CrossRef]
- Da Silva, J.F.M.; Garden, S.J.; Pinto, A.C.J. The chemistry of isatins: A review from 1975 to 1999. Braz. Chem. Soc. 2001, 12, 273–324. [Google Scholar] [CrossRef]
- Verma, M.; Pandeya, S.N.; Singh, K.N.; Stables, J.P. Anticonvulsant activity of Schiff bases of isatin derivatives. Acta Pharm. 2004, 54, 49–56. [Google Scholar] [PubMed]
- Raj, A.; Raghunathan, R.; Sridevikumaria, M.R.; Raman, N. Synthesis, antimicrobial and antifungal activity of a new class of spiro pyrrolidines. Bioorg. Med. Chem. 2003, 11, 407–419. [Google Scholar] [CrossRef]
- Patel, A.; Bari, S.; Talele, G.; Patel, J.; Sarangapani, M. Synthesis and antimicrobial activity of some new isatin derivatives. Iran. J. Pharm. Res. 2006, 4, 249–254. [Google Scholar]
- Tripathy, R.; Reiboldt, A.; Messina, P.A.; Iqbal, M.; Singh, J.; Bacon, E.R.; Angeles, T.S.; Yang, S.X.; Albom, M.S.; Robinson, C.; et al. Structure-Guided identification of novel VEGFR-2 kinase inhibitors via solution phase parallel synthesis. Bioorg. Med. Chem. Lett. 2006, 16, 2158–2162. [Google Scholar] [CrossRef] [PubMed]
- Jiang, T.; Kuhen, K.L.; Wolff, K.; Yin, H.; Bieza, K.; Caldwell, J.; Bursulaya, B.; Tuntlad, T.; Zhang, K.; Karanewsky, D.; et al. Design, synthesis, and biological evaluations of novel oxindoles as HIV-1 non-nucleoside reverse transcriptase inhibitors. Part 2. Bioorg. Med. Chem. Lett. 2006, 16, 2109–2112. [Google Scholar] [CrossRef] [PubMed]
- Ratan, B.T.; Anand, B.; Yogeeswari, P.; Sriram, D. Synthesis and evaluation of anti-HIV activity of isatin β-thiosemicarbazone derivatives. Bioorg. Med. Chem. Lett. 2005, 15, 4451–4455. [Google Scholar] [CrossRef]
- Sriram, D.; Yogeeshwari, P.; Meena, K. Synthesis, anti-HIV and antitubercular activities of isatin derivatives. Die Pharm. 2006, 61, 274–277. [Google Scholar] [CrossRef]
- Aboul-Fadl, T.; Bin-Jubair, F.A.S. Anti-tubercular activity of isatin derivatives. Int. J. Res. Pharm. Sci. 2010, 1, 113–126. [Google Scholar]
- Singh, G.S.; Desta, Z.Y. Isatins as privileged molecules in design and synthesis of spiro-fused cyclic frameworks. Chem. Rev. 2012, 112, 6104–6155. [Google Scholar] [CrossRef] [PubMed]
- Sharbati, M.T.; Soltani Rad, M.N.; Behrouz, S.; Gharavi, A.; Emami, F. Near infrared organic light-emitting diodes based on acceptor-donor-acceptor (ADA) using novel conjugated isatin Schiff bases. J. Lumin. 2011, 131, 553–558. [Google Scholar] [CrossRef]
- Som, P.K.; Banerjee, A.N. Polymerization of methyl methacrylate using isatin and benzoyl peroxide combination as photoinitiator. Eur. Polym. J. 1993, 29, 889–892. [Google Scholar] [CrossRef]
- Košturiak, A.; Domanský, R. Polarographic study of the hydrolysis of 3-anil-(p-amino) isatin in alkaline medium. Chem. Zvesti 1973, 27, 227–231. [Google Scholar]
- Košturiak, A.; Stankovianský, S.; Zacharová-Kalavská, D. A study of alakline hydrolysis of 3′- and 4′-substituted 3-phenyliminoxindole derivatives. Collect. Czechoslov. Chem. Commun. 1976, 41, 2582–2591. [Google Scholar] [CrossRef]
- Stünzi, H. Derivatives of isatin in aqueous solution. II. Z-E isomerism in isatin β-thiosemicarbazones. Aust. J. Chem. 1981, 34, 373–381. [Google Scholar] [CrossRef]
- Casey, L.A.; Galt, R.; Page, M.I. The mechanisms of hydrolysis of the γ-lactam isatin and its derivatives. J. Chem. Soc. Perkin Trans. 2 1993, 23–28. [Google Scholar] [CrossRef]
- Abu El-nader, V.H.M.; Moussa, M.N.H. Solvent effect on the kinetics of the basic hydrolysis of isatin. Chem. Pharm. Bull. 1996, 44, 1641–1646. [Google Scholar] [CrossRef]
- Ismail, A.M.; Zaghloul, A.A. Kinetics and mechanism of isatin ring opening in aqueous binary mixtures of methanol and acetonitrile cosolvents. Int. J. Chem. Kinet. 1998, 30, 463–469. [Google Scholar] [CrossRef]
- Brum, J.; Dell’Orco, P.; Lapka, S.; Muske, K.K.K.; Sisko, J. Monitoring organic reactions with on-line atmospheric pressure ionization mass spectrometry: The hydrolysis of isatin. Rapid Commun. Mass. Spectrom. 2001, 15, 1548–1553. [Google Scholar] [CrossRef]
- Al-Ayed, A.S.; Ali, M.S.; Al-Lohedan, H.A.; Al-Sulaim, A.M.; Al-Sulaim, A.M.; Kabir-ud-Din, Z.A.I. Micellar effects on the alkaline hydrolysis of isatin and its derivatives. J. Colloid Interface Sci. 2011, 357, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Srikanth, V.; Shyamala, P.; Satyanarayana, A.; Subba Rao, P.V. Alkaline hydrolysis of isatin in the presence of mixed CTAB and Triton X-100 micellar systems: Micellisation and kinetic investigations. Indian J. Chem. 2012, 51A, 1701–1705. [Google Scholar]
- Breugst, M.; Tokuyasu, T.; Mayr, H. Nucleophilic reactions of imide and amide anions. J. Org. Chem. 2010, 75, 5250–5258. [Google Scholar] [CrossRef] [PubMed]
- Bunnett, J.F.; Beale, J.H. Reactivity of some imide and sulfonamide anions with methyl iodide in methanol. J. Org. Chem. 1971, 36, 1659–1661. [Google Scholar] [CrossRef]
- Beale, J.H. Reactivity of N-chloro- and N-methylbenzenesulfonamide anions with methyl methanesulfonate in methanol. J. Org. Chem. 1972, 37, 3871–3872. [Google Scholar] [CrossRef]
- Velcheva, E.A.; Vassileva-Boyadjieva, P.J.; Binev, I.G. Experimental and DFT studies on IR spectral and structural changes arising from the conversion of 1H-indole-2,3-dione (isatin) into azanion. Bulg. Chem. Commun. 2008, 40, 433–439. [Google Scholar]
- Berci-Filho, P.; Gehlen, F.H.; Politi, M.J.; Neumann, M.G.; Barros, T.C. Photophysics of ambident organic anions I. J. Photochem. Photobiol. A Chem. 1995, 92, 155–161. [Google Scholar] [CrossRef]
- Shao, J.; Wanga, Y.; Lin, H.; Li, J.; Lina, L. A novel indole phenylhydrazone receptor: Synthesis and recognition for acetate anion. Sens. Actuators B 2008, 134, 849–853. [Google Scholar] [CrossRef]
- Wang, Y.; Lin, H.; Shao, J.; Cai, Z.S.; Lin, H.K. A phenylhydrazone-based indole receptor for sensing acetate. Talanta 2008, 74, 1122–1125. [Google Scholar] [CrossRef] [PubMed]
- Shaikh, A.M.; Sharma, B.K.; Kamble, R.M. Photophysical, electrochemical and thermal studies of 5-methyl-5H-Benz[g]indolo[2,3-b]quinoxaline derivatives: Green and yellow fluorescent materials. Can. Chem. Trans. 2015, 3, 158–170. [Google Scholar] [CrossRef]
- Biamchi, A.; Bowman-James, K.; García-España, E. Supramolecular Chemistry of Anions, 1st ed.; Wiley-VCH: New York, NY, USA, 1997. [Google Scholar]
- Gale, P.A.; Quesada, R. Anion coordination and anion templated assembly: Highlights from 2002 to 2004. Coord. Chem. Rev. 2006, 250, 3219–3244. [Google Scholar] [CrossRef]
- Wenzel, M.; Hiscock, J.R.; Gale, P.A. Anion receptor chemistry: Highlights from 2010. Chem. Soc. Rev. 2012, 41, 480–520. [Google Scholar] [CrossRef] [PubMed]
- Veale, E.B.; Gunnlaugsson, T. Fluorescent Sensors for Ions based on Organic Structures. Annu. Rep. Prog. Chem. Sect. B Org. Chem. 2010, 106, 376–406. [Google Scholar] [CrossRef]
- Boiocchi, M.; Bose, L.D.; Gómez, D.E.; Fabbrizzi, L.; Licchelli, M.; Monzani, E. Nature of urea-fluoride interaction: Incipient and definitive proton transfer. J. Am. Chem. Soc. 2004, 126, 16507–16514. [Google Scholar] [CrossRef] [PubMed]
- Li, A.F.; Wang, J.H.; Jiang, Y.B. Anion complexation and sensing using modified urea and thiourea-based receptors. Chem. Soc. Rev. 2010, 39, 3729–3745. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.J.; Edwards, P.R.; Gale, P.A.; Light, M.E. Carboxylate complexation by a family of easy-to-make ortho-phenylenediamine based bis-ureas: Studies in solution and the solid state. New J. Chem. 2006, 30, 65–70. [Google Scholar] [CrossRef]
- Camiolo, S.; Gale, P.; Hursthouse, M.B.; Light, M.E. Nitrophenyl derivatives of pyrrole 2,5-diamides: Structural behaviour, anion binding and colour change signalled deprotonation. Org. Biomol. Chem. 2003, 1, 741–744. [Google Scholar] [CrossRef] [PubMed]
- Pfeffer, F.M.; Lim, K.F.; Sedgwick, K.J. Indole as a scaffold for anion recognition. Org. Biomol. Chem. 2007, 5, 1795–1799. [Google Scholar] [CrossRef] [PubMed]
- Bates, G.W.; Triyanti; Light, M.E.; Albrecht, M.; Gale, P.A. 2,7-Functionalized indoles as receptors for anions. J. Org. Chem. 2007, 72, 8921–8927. [Google Scholar] [CrossRef] [PubMed]
- Amendola, V.; Esteban-Gómez, D.; Fabbrizzi, L.; Licchelli, M. What anions do to N-H-Containing receptors. Acc. Chem. Res. 2006, 39, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Sreekanth, A.; Kurup, M.R.P. Synthesis, EPR and Mössbauer spectral studies of new iron(III) complexes with 2-benzoylpyridine-N(4), N(4)-(butane-1, 4-diyl) thiosemicarbazone (HBpypTsc): X-ray structure of [Fe (BpypTsc)2]FeCl4·2H2O and the free ligand. Polyhedron 2004, 23, 969–978. [Google Scholar] [CrossRef]
- Popov, E.M.; Želtova, V.N. Electronic structure and properties of the peptide group. J. Mol. Struct. 1971, 10, 221–230. [Google Scholar] [CrossRef]
- Kleinpeter, E.; Bölke, U.; Koch, A. Subtle trade-off existing between (anti) aromaticity, push−pull interaction, keto−enol tautomerism, and steric hindrance when defining the electronic properties of conjugated structures. J. Phys. Chem. A 2010, 114, 7616–7623. [Google Scholar] [CrossRef] [PubMed]
- Rekhter, M.A.; Rekhter, B.A. The complementarity principle in chemical reactions. Modifications of the indolinedione-indole rearrangement and enamine synthesis. Chem. Heterocycl. Compd. 2012, 48, 1–4. [Google Scholar] [CrossRef]
- Bigatto, A.; Galasso, V. Infrared and Raman spectra of phthalimide and isatin. Spectrochim. Acta 1979, 35, 725–732. [Google Scholar] [CrossRef]
- Narziev, B.N.; Mulloev, N. Proton donor properties of pyrroles studied by IR spectroscopy. J. Struct. Chem. 1999, 40, 481–484. [Google Scholar] [CrossRef]
- Naumov, P.; Atanasova, F. Experimental and theoretical vibrational study of isatin, its 5-(NO2, F, Cl, Br, I, CH3) analogues and the isatinato anion. Spectrochim. Acta 2001, 57, 469–481. [Google Scholar] [CrossRef]
- Strat, M.; Umreiko, D.S.; Khovratovich, N.N. Spectroscopic detection of an intramolecular hydrogen bond in 2-hydroxybenzophenone derivatives. Zhurnal Prikladnoi Spektroskopii 1973, 19, 103–108. [Google Scholar] [CrossRef]
- Prasad, R.L.; Kushwaha, A.; Suchita; Kumar, M.; Yadav, R.A. Infrared and ab initio studies of conducting molecules: 2,5-Diamino-3,6-dichloro-1,4-benzoquinone. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2008, 69, 304–311. [Google Scholar] [CrossRef] [PubMed]
- Polat, T.; Yurdakul, Ş. Quantum chemical and spectroscopic (FT-IR and FT-Raman) investigations of 3-methyl-3h-imidazole-4-carbaldehyde. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2014, 133, 683–696. [Google Scholar] [CrossRef] [PubMed]
- Arivazhagan, M.; Jeyavijayan, S. FTIR and FT-Raman spectra, assignments, ab initio HF and DFT analysis of xanthine. Spectrochim. Acta A Mol. Biomol. Spectrosc. 2011, 79, 161–168. [Google Scholar] [CrossRef] [PubMed]
- Feuer, H.; Savides, O.; Rao, C.N.R. The infrared spectra of the salts of nitro compounds. Characteristic frequencies of the carbonitronate group, =C=NO2−. Spectrochim. Acta 1963, 19, 431–434. [Google Scholar] [CrossRef]
- Brokes, M.J.; Jonathan, N. Concerning the spectroscopic evidence for the existence of geometric isomers of the sodium salts of nitroparaffins. Spectrochim. Acta 1969, 25, 187–191. [Google Scholar] [CrossRef]
- Bancroft, D.P.; Cotton, F.A.; Falvello, L.R.; Schwotzer, W. Tetramethyldiplatinum (III) (Pt-Pt) complexes with 2-hydroxypyridinato bridging ligands. Inorg. Chem. 1986, 25, 763–770. [Google Scholar] [CrossRef]
- Bancroft, D.P.; Cotton, F.A. Tetramethyldiplatinum(III) (Pt-Pt) complexes with 2-hydroxypyridinato bridging ligands. 2. Reversals of ligand orientations. Inorg. Chem. 1988, 27, 1633–1637. [Google Scholar] [CrossRef]
- Rawson, J.M.; Winpenny, R.E.P. The coordination chemistry of 2-pyridone and its derivatives. Coord. Chem. Rev. 1995, 139, 313–374. [Google Scholar] [CrossRef]
- Breugst, M.; Mayr, H. Ambident Reactivities of Pyridone Anions. J. Am. Chem. Soc. 2010, 132, 15380–15389. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Raso, A.; Fiol, J.J.; Molins, E.; Calafat, A.M.; Marzilli, P.A.; Marzilli, L.G. Metallation of Isatin (2,3-Indolinedione). X-Ray Structure and Solution Behavior of Bis(Isatinato)Mercury(II). Met. Based Drugs 1995, 2, 81–90. [Google Scholar] [CrossRef] [PubMed]
- Kaeser, A.; Delavaux-Nicot, B.; Duhayon, C.; Coppel, Y.; Nierengarten, J.-F. Heteroleptic silver(I) complexes prepared from phenanthroline and bis-phosphine ligands. Inorg. Chem. 2013, 52, 14343–14354. [Google Scholar] [CrossRef] [PubMed]
- Goldschmidt, G.H.; Llewellyn, F.J. The crystalline structure of isatin. Acta Crystallogr. 1950, 3, 294–305. [Google Scholar] [CrossRef]
- Palmer, M.H.; Blake, A.J.; Gould, R.O. 14N nuclear quadrupole coupling in cyclic amides and thioamides. Ab initio simulations of the solid state environment as interpretation of the NQR spectra of 2-pyridinone, isatin and benzothiazole-2-one. A new X-ray structure for isatin. Chem. Phys. 1987, 115, 219–227. [Google Scholar] [CrossRef]
- Güngör, T.; Chen, Y.; Golla, R.; Ma, Z.; Corte, J.R.; Northrop, J.P.; Bin; Dickson, J.K.; Stouch, T.; Zhou, R.; et al. Synthesis and characterization of 3-Arylquinazolinone and 3-Arylquinazolinethione derivatives as selective estrogen receptor beta modulators. J. Med. Chem. 2006, 49, 2440–2455. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.-C.; Ye, C.-J.; Chen, Q.; Yang, G.-F. Efficient synthesis of bulky 4-substituted-isatins via microwave-promoted Suzuki cross-coupling reaction. Tetragedron Lett. 2013, 54, 949–955. [Google Scholar] [CrossRef]
Sample Availability: Samples of the compound D are available from the authors. |


















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | DMSO | CH3CN | ||||||
|---|---|---|---|---|---|---|---|---|
| λmax/logε | λmax/logε | λmax/logε | λmax/logε | λmax/logε | λmax/logε | λmax/logε | λmax/logε | |
| A | 259/3.43 | 298/3.53 | 415/2.93 | - | 242/4.37 | 295/3.55 | 408/2.91 | - |
| Aazanion | 266/4.30 | 297P/3.46 | - | 569/2.82 | 255/4.41 | 292P/3.41 | - | 555/2.75 |
| B | 278/3.93 | 329/3.89 | - | - | 275/4.24 | 316/4.02 | - | - |
| Bazanion | 294P/3.63 | 296P/3.61 | 417/4.20 | 531P/3.34 | 290P/3.74 | - | 410/4.36 | 508/3.48 |
| C | - | 348/3.92 | 409P/3.11 | - | - | 343/3.92 | 400P/3.11 | - |
| Cazanion | 269/4.43 | 340/3.72 | - | 509/2.89 | 265/4.41 | 338/3.67 | - | 512/2.82 |
| D | - | 309/3.89 | 461/3.38 | - | 278P/3.56 | 320P/3.12 | 443/2.90 | - |
| Dazanion * | 277/4.46 | 322/3.99 | 421/3.08 | 610/3.24 | 274/3.86 | 320/3.35 | 405/2.47 | 597/2.60 |
| E | - | 329/3.52 | 421/3.22 | - | - | 327/3.41 | 416/3.18 | - |
| Eazanion | 290P/4.01 | 341P/3.26 | - | 576/2.95 | 284P/3.98 | 331P/3.22 | - | 554/2.90 |
| DMSO | 10−2 kTBAOH (s−1) |
|---|---|
| B ** | 5.98 |
| E | 2.84 |
| D | 1.77 |
| A | 1.34 |
| C | 0 |
| Compound * | ATR/(cm−1) | CH3CN/(cm−1) | CHCl3/(cm−1) | |
|---|---|---|---|---|
| A | νs(C=O) | 1724 | 1744 | 1743 |
| νas(C=O) | 1746 | 1761 | 1760 | |
| Aazanion/TBAF | νs(C=O) | 1716 | 1720 | 1719 |
| νas(C=O) | 1645 | 1632 | 1645 | |
| Aazanion/Ag+ | νs(C=O) | 1723 | - | - |
| νas(C=O) | 1570 | - | - | |
| Aazanion/Hg2+ | νs(C=O) | 1733 | - | - |
| νas(C=O) | 1676 | - | - | |
| B | νs(C=O) | 1749, 1732 | 1757 | 1754 |
| νas(C=O) | 1770 | 1768 | 1774p | |
| Bazanion/TBAF | νs(C=O) | 1735 | 1735 | 1734 |
| νas(C=O) | 1674 | 1677 | 1672 | |
| Bazanion/Ag+ | νs(C=O) | 1743 | - | - |
| νas(C=O) | 1654, 1597 | - | - | |
| Bazanion/Hg2+ | νs(C=O) | 1745p,1734 | - | - |
| νas(C=O) | 1696, 1605 | - | - | |
| C | νs(C=O) | 1730 | - | - |
| νas(C=O) | 1748 | - | - | |
| Cazanion/TBAF | νs(C=O) | 1716 | - | - |
| νas(C=O) | 1646 | - | - | |
| Cazanion/Ag+ | νs(C=O) | 1714 | - | - |
| νas(C=O) | 1634 | - | - | |
| D | νs(C=O) | 1732 | - | - |
| νas(C=O) | 1763 | - | - | |
| Dazanion/TBAF | νs(C=O) | 1681 | - | - |
| νas(C=O) | 1650p | - | - | |
| Dazanion/Ag+ | νs(C=O) | 1705 | - | - |
| νas(C=O) | 1685p | - | - | |
| E | νs(C=O) | 1731 | - | - |
| νas(C=O) | 1743 | - | - | |
| Eazanion/TBAF | νs(C=O) | 1727 | - | - |
| νas(C=O) | 1652 | - | - | |
| Eazanion/Ag+ | νs(C=O) | 1710 | - | - |
| νas(C=O) | 1634 | - | - |
| Compound/Hydrogen | A (ppm) | Aazanion (ppm) | B (ppm) | Bazanion (ppm) | C (ppm) | Cazanion (ppm) | D (ppm) | Dazanion (ppm) | E (ppm) | Eazanion (ppm) |
|---|---|---|---|---|---|---|---|---|---|---|
| H1 | 11.03 | - | 11.63 | - | 10.94 | - | 12.25 | - | 11.10 | - |
| H4 | 7.48 | 7.11 | 8.20 | 7.71 | - | - | - | - | - | - |
| H5 | 7.05 | 6.64 | - | - | 6.15 | 6.06 | - | - | 6.99 | 6.29 |
| H6 | 7.57 | 7.27 | 8.43 | 8.04 | - | - | - | - | 7.58 | 7.11 |
| H7 | 6.89 | 6.62 | 7.06 | 6.55 | 5.98 | 6.06 | - | - | 6.87 | 6.36 |
| - | - | - | - | 3.85 | 3.83 | - | - | - | - | |
| - | - | - | - | 3.83 | 3.82 | - | - | - | - | |
| H10 | - | - | - | - | - | - | 9.14 | 8.45 | - | - |
| H11 | - | - | - | - | - | - | 8.04 | 7.43 | 7.53 | 7.40 |
| H12 | - | - | - | - | - | - | 9.03 | 8.49 | - | - |
| H15 | - | - | - | - | - | - | 9.38 | 9.06 | - | - |
| H16 | - | - | - | - | - | - | 8.09 | 7.65 | - | - |
| H17 | - | - | - | - | - | - | 8.97 | 8.64 | - | - |
| H18 | - | - | - | - | - | - | - | - | 7.43 | 7.35 |
| H19 | - | - | - | - | - | - | - | 7.43 | 7.30 | |
| H20 | - | - | - | - | - | - | - | - | 7.43 | 7.35 |
| H21 | - | - | - | - | - | - | - | - | 7.53 | 7.40 |
| Compound/Carbon | A (ppm) | Aazanion (ppm) | B (ppm) | Bazanion (ppm) | C (ppm) | Cazanion (ppm) | D (ppm) | Dazanion (ppm) | E (ppm) | Eazanion (ppm) |
|---|---|---|---|---|---|---|---|---|---|---|
| C2 | 159.79 | 166.16 | 160.32 | 172.76 | 162.00 | 162.87 | 160.90 | 175.05 | 159.46 | 170.67 |
| C3 | 184.81 | 192.92 | 182.80 | 194.93 | 178.86 | 179.88 | 182.16 | 193.00 | 183.40 | 197.10 |
| C4 | 125.12 | 123.86 | 120.02 | 119.07 | 160.71 | 160.55 | 124.88 | 126.94 | 141.94 | 140.13 |
| C5 | 123.19 | 119.76 | 143.05 | 137.92 | 92.44 | 92.08–92.19 | 138.28 | 140.75 | 124.76 | 118.93 |
| C6 | 138.80 | 138.30 | 133.50 | 134.08 | 170.17 | 170.13 | 146.58 | 151.16 | 138.31 | 137.52 |
| C7 | 112.63 | 114.65 | 112.94 | 115.88 | 91.95 | 92.08–92.19 | 118.76 | 124.48 | 111.52 | 115.48 |
| C8 | 151.15 | 164.16 | 155.62 | 178.84 | 155.08 | 156.93 | 155.90 | 182.50 | 151.90 | 174.99 |
| C9 | 118.26 | 119.65 | 118.59 | 119.34 | 100.97 | 101.08 | 104.76 | 102.42 | 114.65 | 116.87 |
| C4-OCH3 | - | - | - | - | 57.98 | 57.96 | - | - | - | - |
| C6-OCH3 | - | - | - | - | 56.69 | 56.33 | - | - | - | - |
| C10 | - | - | - | - | - | - | 134.84 | 127.89 | 136.77 | 138.32 |
| C11 | - | - | - | - | - | - | 126.72 | 124.65 | 129.28 | 128.91 |
| C12 | - | - | - | - | - | - | 146.15 | 144.23 | - | - |
| C13 | - | - | - | - | - | - | - | - | - | - |
| C14 | - | - | - | - | - | - | - | - | - | - |
| C15 | - | - | - | - | - | - | 154.79 | 153.58 | - | - |
| C16 | - | - | - | - | - | - | 125.72 | 123.23 | - | - |
| C17 | - | - | - | - | - | - | 134.00 | 133.04 | - | - |
| C18 | - | - | - | - | - | - | - | - | 128.49 | 128.09 |
| C19 | - | - | - | - | - | - | - | - | 129.06 | 128.09 |
| C20 | - | - | - | - | - | - | - | - | 128.49 | 128.09 |
| C21 | - | - | - | - | - | - | - | - | 129.28 | 128.91 |
| ∆E (kJ·mol−1) * | pKa | λmax | λmax(azanion) | |
|---|---|---|---|---|
| A | 1206.7 | 8.59 | 374; 261; 218 | 488; 237 |
| B | 1181.1 | 6.38 | 357; 256; 211 | 461; 344; 231 |
| C | 1213.1 | 8.67 | 312; 213 | 446; 297; 241 |
| D | 1196.0 | 6.65 | 420; 240 | 529; 352p; 261 |
| E | 1205.6 | 8.57 | 376; 312; 229 | 500; 266; 234 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tisovský, P.; Šandrik, R.; Horváth, M.; Donovalová, J.; Jakusová, K.; Cigáň, M.; Sokolík, R.; Gáplovský, A.; Gáplovský, M.; Filo, J. Effect of Structure on Charge Distribution in the Isatin Anions in Aprotic Environment: Spectral Study. Molecules 2017, 22, 1961. https://doi.org/10.3390/molecules22111961
Tisovský P, Šandrik R, Horváth M, Donovalová J, Jakusová K, Cigáň M, Sokolík R, Gáplovský A, Gáplovský M, Filo J. Effect of Structure on Charge Distribution in the Isatin Anions in Aprotic Environment: Spectral Study. Molecules. 2017; 22(11):1961. https://doi.org/10.3390/molecules22111961
Chicago/Turabian StyleTisovský, Pavol, Róbert Šandrik, Miroslav Horváth, Jana Donovalová, Klaudia Jakusová, Marek Cigáň, Róbert Sokolík, Anton Gáplovský, Martin Gáplovský, and Juraj Filo. 2017. "Effect of Structure on Charge Distribution in the Isatin Anions in Aprotic Environment: Spectral Study" Molecules 22, no. 11: 1961. https://doi.org/10.3390/molecules22111961
APA StyleTisovský, P., Šandrik, R., Horváth, M., Donovalová, J., Jakusová, K., Cigáň, M., Sokolík, R., Gáplovský, A., Gáplovský, M., & Filo, J. (2017). Effect of Structure on Charge Distribution in the Isatin Anions in Aprotic Environment: Spectral Study. Molecules, 22(11), 1961. https://doi.org/10.3390/molecules22111961